

The crystallization and preliminary X-ray diffraction studies of the Skp1–Fbg3 complex are reported. Crystallization by repeated microseeding using selected crystals as a source of microseeds is described.
Keywords: F-box proteins, Fbg3, Skp1–Fbg3 complex, ubiquitin ligases
Abstract
F-box proteins are the substrate-recognition components of Skp1–Cullin1–F-box protein–Rbx1 (SCF) ubiquitin ligase complexes. Fbs1, an F-box protein, binds specifically to proteins modified with high-mannose oligosaccharides. Fbg3, another F-box protein, has 51% sequence identity to Fbs1. Although the residues that are necessary for binding to oligosaccharides are conserved between Fbs1 and Fbg3, Fbg3 does not bind glycoproteins. Skp1 and Fbg3 were co-expressed in Escherichia coli and their complex was purified to homogeneity and crystallized. Microseeding combined with the sandwiched hanging-drop technique improved the quality of the resulting crystals. The plate-shaped crystals belonged to space group P212121, with unit-cell parameters a = 34.1, b = 76.6, c = 193.9 Å and one molecule per asymmetric unit.
1. Introduction
The ubiquitin–proteasome system plays important roles in numerous biological processes such as regulation of cell-cycle progression, apoptosis, modulation of the immune and inflammatory responses and quality control of proteins (Hershko & Ciechanover, 1998 ▶). Ubiquitin, a 76-amino-acid protein, is covalently attached to the system’s target proteins. Protein ubiquitination is catalyzed by a cascade reaction involving three enzymes: ubiquitin-activating (E1), ubiquitin-conjugating (E2) and ubiquitin-ligating (E3) enzymes. Multiple rounds of ubiquitination lead to the formation of polyubiquitin chains; polyubiquitinated proteins are degraded by 26S proteasome. Only the E3 enzymes are involved in substrate recognition (Pickart, 2001 ▶). The E3 enzymes have been classified into several families, of which the SCF complex (Skp1–Cul1–F-box) is the best characterized. The SCF complex contains four subunits: Skp1, Cullin1 (Cul1), Rbx1 (a RING-finger protein) and an F-box protein (Deshaies, 1999 ▶). F-box proteins consist of an N-terminal F-box domain that binds to Skp1 and varying C-terminal substrate-recognition domains. They have been classified into three groups according to the type of substrate-recognition domain (Jin et al., 2004 ▶): Fbw (or FBXW), which contains WD-40 repeats, Fbl (or FBXL), which contains leucine-rich repeats, and Fbx (or FBXO), which does not contain either of these domains. Fbs1, an Fbx-family protein, recognizes the innermost portion of the carbohydrate moieties of high-mannose-type asparagine-linked sugar chains (N-glycans); it is a member of the Fbs subfamily within the Fbx classification (Yoshida et al., 2002 ▶; Yoshida, 2007 ▶). SCFFbs1 is involved in ER quality control (Yoshida, 2007 ▶; Yoshida et al., 2007 ▶) and functions to ubiquitinate ERAD substrates. We have previously reported the X-ray crystal structures of the substrate-binding domain of Fbs1, of Skp1–Fbs1 and of its complex with glycoprotein (Mizushima et al., 2004 ▶, 2007 ▶). The structures revealed that SCFFbs1 recognizes the innermost Man3GlcNAc2 in N-glycans; accessible Man3GlcNAc2 serves as a marker for denatured protein. Fbg3, another F-box protein, has 51% sequence identity to Fbs1 (Yoshida, 2007 ▶) and the residues that are necessary for binding to N-glycans in Fbs1 are conserved in Fbg3. However, Fbg3 does not bind to the innermost chitobiose in N-glycans. Here, we report the expression, purification and crystallization of Skp1–Fbg3 using repeated microseeding. We also report preliminary X-ray analysis as a prelude to the determination of the three-dimensional structure of the complex, with the ultimate goal of understanding the mechanistic details of sugar recognition.
2. Materials and methods
2.1. Protein expression and purification
Human Fbg3 (29.7 kDa; 255 amino acids) cDNA (accession No. NM_033182) was amplified by PCR using upstream primer FBG3-5 (5′-CGGGATCCCCATGGCTGTGGGGAACATCAACGA-3′) and downstream primer FBG3-3 (5′-TAAAGCGGCCGCTCAGGGTTCAGCAGATGGGGG-3′) from the first-strand DNA mixture of HepG2 cells. The resultant 0.9 kb fragment was digested with BamHI and NotI, ligated into pcDNA3.1 (Invitrogen) and verified by sequencing. pET28b (Novagen) containing 6×His-tagged human Skp1 (18.1 kDa; 160 amino acids; accession No. NM_006930) was constructed as described previously (Mizushima et al., 2007 ▶) and human untagged Fbg3 was inserted downstream of the BamHI–NotI site of pET28b-6×His-Skp1 for co-expression. The thrombin-cleavage site for removal of the 6×His tag was replaced by a cleavage site for PreScission protease. The encoded recombinant protein contained an N-terminal MGSSHHHHHHSSGLEVLFQGP tag followed by the amino-acid sequence of human Skp1. Skp1–Fbg3 co-expressing BL21 (RIPL) was incubated in LB medium containing kanamycin (50 µg ml−1) at 310 K until the optical density (OD) at 600 nm reached ∼0.5. The culture was induced with 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 310 K for 4 h. Harvested cells were collected by centrifugation, resuspended in lysis buffer (20 mM Tris–HCl pH 7.4 and 150 mM NaCl) and sonicated at 277 K. After sonication, the cell lysate was centrifuged for 45 min at 20 400g and the supernatant was collected and applied onto a 5 ml Ni–NTA column (Qiagen) previously equilibrated with lysis buffer. The column was washed with 80 ml washing buffer 1 (20 mM imidazole, 20 mM Tris–HCl pH 7.4 and 500 mM NaCl). The protein was eluted with buffer 2 (300 mM imidazole, 20 mM Tris–HCl pH 7.4 and 500 mM NaCl). Imidazole was removed by dialysis overnight against 25 mM Tris–HCl buffer pH 7.5 and 10 mM 2-mercaptoethanol. The histidine tag was cleaved from Skp1 using PreScission protease (GE Healthcare, Piscataway, New Jersey, USA). The resultant protein contained two additional residues (GP) at the N-terminal flanking region derived from the PreScission protease-recognition sequence. The untagged Skp1–Fbg3 was purified by anion-exchange chromatography using a linear 0–0.8 M NaCl gradient in 25 mM Tris–HCl pH 7.5 and 10 mM β-mercaptoethanol. The purified sample was further applied onto a HiLoad 26/60 Superdex 75 gel-filtration column (GE Healthcare, Piscataway, New Jersey, USA) equilibrated with 25 mM Tris–HCl pH 7.5 and 10 mM β-mercaptoethanol. Chromatography was performed at a flow rate of 1 ml min−1.
2.2. Crystallization and production of microseeds
Prior to crystallization, the protein solution was dialyzed against 25 mM Tris–HCl pH 7.5 and 1 mM dithiothreitol and concentrated to 4–10 mg ml−1. Crystallization trials were performed by the hanging-drop vapour-diffusion method at 288 K using VDX 24-well crystallization plates (TPP). Initial screening of crystallization conditions was carried out using Crystal Screen 1 and 2, PEG/Ion Screen (Hampton Research, Laguna Niguel, California, USA) and Wizard I and II (Emerald BioSystems, Bainbridge Island, Washington, USA) crystallization screening kits. 2 µl protein solution (9.96 mg ml−1) was mixed with 2 µl reservoir solution and equilibrated against 300 µl reservoir solution. Subsequent crystallizations were carried out using the same reservoir volume. Small clusters of thin plate-shaped crystals (Fig. 1 ▶ a) were obtained using condition No. 41 of Crystal Screen 1 [0.1 M Na HEPES pH 7.5, 10%(v/v) 2-propanol, 20%(w/v) polyethylene glycol 4000]. After refinement of the crystallization conditions, 0.3 × 0.4 mm crystals (Fig. 1 ▶ b) were obtained by mixing 2 µl protein solution (4–6 mg ml−1) and 2 µl reservoir solution consisting of 0.1 M Na PIPES pH 7.5, 8%(v/v) 2-propanol and 16%(w/v) polyethylene glycol 4000.
Figure 1.

Crystals of the Skp1–Fbg3 complex. (a) shows clusters of plate-shaped crystals obtained from condition No. 41 of Crystal Screen 1. Refinement of this crystallization condition improved the size of the crystals (b) and these crystals were used for the first microseeding. (c)–(f) show crystals obtained from repeated microseeding: (c) second generation, (d) third generation, (e) fourth generation and (f) fifth generation. During microseeding, we selected the finest, most highly birefringent crystals as the source of microseeds. From the third generation (d) onward, 1 M lithium chloride was added to the drop. From the fourth generation (e) onward, we combined microseeding with the sandwiched hanging-drop technique.
In order to obtain single crystals, microseeding (McPherson, 1982 ▶) was employed. Crystals (Fig. 1 ▶ b) were transferred to a 2 µl drop of reservoir solution on a cover glass and were crushed manually using a Micro-Needle (Hampton Research, Laguna Niguel, California, USA) under a microscope. The seed stock was equilibrated against 300 µl reservoir solution for more than a day. The seeds were transferred to new drops using the tip of a nylon loop with no equilibration prior to the introduction of seeds. Although single crystals (Fig. 1 ▶ c) were obtained by one round of seeding, the resulting diffraction data were not sufficient to determine the structure. Therefore, the microseeding was repeated another three times in order to improve the crystal quality and surface morphology (Thaller et al., 1981 ▶). Highly birefringent seeded crystals were selected as the source of microseeds for the next round of seeding. In the second and subsequent rounds of microseeding, 0.1 µl 1 M lithium chloride was added to the drop, resulting in an increased thickness of the crystals (Fig. 1 ▶ d). In the third and subsequent rounds we combined microseeding with the sandwiched hanging-drop method (Adachi et al., 2004 ▶) in order to increase the size and the thickness of the crystals (Fig. 1 ▶ e). The sandwiched hanging drop consisted of 2.5 µl protein solution, 2.5 µl reservoir solution, 0.5 µl additive solution and 0.1 µl Fluorinert (Sigma, St Louis, Missouri, USA) placed over the drop. Judging from the crystals shown in Figs. 1 ▶(c)–1(e), in addition to microseeding, the addition of lithium chloride and crystallization by the sandwiched hanging-drop technique both contributed to the improvement in the size and thickness of the crystals. Finally, crystals with approximate dimensions of 0.2 × 0.1 × 0.02 mm were obtained by repetitive microseeding combined with the sandwiched hanging-drop technique (Fig. 1 ▶ f). The final crystallization drop contained 3 µl protein solution (4.9 mg ml−1) in 25 mM Tris–HCl pH 7.5 and 1 mM DTT, 3 µl reservoir solution [8%(v/v) 2-propanol, 16%(w/v) PEG 4000, 0.1 M Na PIPES pH 7.5] and 0.6 µl 1 M lithium chloride as an additive, with 0.2 µl Fluorinert placed on the crystallization drop. SDS–PAGE of carefully washed and redissolved crystals showed bands for both components of the complex (data not shown). X-ray diffraction data were collected from these crystals.
2.3. Cryoprotection and preliminary X-ray diffraction analysis
Crystals were soaked in cryoprotectant solution [20%(v/v) glycerol, 0.1 M Na PIPES pH 7.5, 8%(v/v) 2-propanol and 16%(w/v) polyethylene glycol 4000] for 30 s. The crystals were mounted on a nylon loop and flash-cooled at 100 K using a nitrogen stream. Data sets were collected at an X-ray wavelength of 0.9 Å on beamline BL44XU at SPring-8, Japan. The diffraction data were processed using the software DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶). The final statistics of data collection are summarized in Table 1 ▶. The Matthews coefficient (V M; Matthews, 1968 ▶) of 2.6 Å3 Da−1 and the solvent content of 52.6% indicated that the crystal contained one Skp1–Fbg3 molecule per asymmetric unit. Structure determination of the Skp1–Fbg3 complex by molecular replacement using the Skp1–Fbs1 structure (PDB code 2e31; Mizushima et al., 2007 ▶) is in progress.
Table 1. Data-collection statistics for Skp1–Fbg3.
Values in parentheses are for the highest resolution shell.
| Temperature (K) | 100 |
| X-ray wavelength (Å) | 0.90 |
| Space group | P212121 |
| Unit-cell parameters | |
| a (Å) | 34.1 |
| b (Å) | 76.6 |
| c (Å) | 193.9 |
| Solvent content (%) | 52.6 |
| Resolution (Å) | 97.1–2.6 (2.65–2.6) |
| No. of observations | 77996 |
| No. of unique reflections | 16474 |
| Completeness (%) | 99.5 (99.6) |
| Redundancy | 4.8 (4.6) |
| Rmerge† (%) | 6.6 (30.8) |
| 〈I/σ(I)〉 | 19.8 |
R
merge = 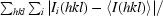
 .
.
3. Results and discussion
3.1. Crystallization using selective microseeding
The crystals obtained initially were small and clustered (Fig. 1 ▶ a). Refinement of the crystallization conditions, involving manipulation of the precipitant and protein concentrations, pH and buffer, resulted in slightly larger crystals. However, these larger crystals diffracted poorly (to 8 Å resolution; Fig. 1 ▶ b). Subsequently, we attempted a microseeding technique and obtained thin single crystals (Fig. 1 ▶ c). While non-birefringent crystals only diffracted to 4.5 Å resolution (Fig. 2 ▶ a), the highly birefringent crystals in the same drop diffracted to 3.5 Å (Fig. 2 ▶ b). We conducted consecutive microseeding a further three times using highly birefringent crystals as the source of microseeds in order to obtain crystals that diffracted to higher resolution (Fig. 1 ▶ d–1f). Through a total of four iterations of repetitive microseeding, the quality of the crystals was gradually improved. The final crystals diffracted to a resolution of 2.6 Å (Figs. 1 ▶ f and 2 ▶ c).
Figure 2.

Diffraction images from crystals of Skp1–Fbg3. (a) and (b) are the diffraction patterns of non-birefringent and highly birefringent crystals in Fig. 1 ▶(c), respectively. X-ray diffraction experiments were performed using an R-AXIS VII image plate with a Rigaku FR-E rotating-anode X-ray generator operating at 45 kV and 45 mA. These diffraction images were collected at a wavelength of 1.54 Å; the crystal-to-detector distance was 200 mm, the oscillation range per image angle was 1° and the exposure time was 3 min. (c) The diffraction image of a crystal in Fig. 1 ▶(f). The diffraction image was collected at a wavelength of 0.9 Å; the crystal-to-detector distance was 500 mm, the oscillation range per image angle was 1° and the exposure time was 10 s on beamline BL44XU at SPring-8, Japan.
In the microseeding, we used microseeds equilibrated for more than a day after crushing the crystal (hereafter referred to as ‘mature seeds’). We chose to do so because most of the crystals obtained from recently crushed microseeds were clustered; in contrast, the crystals obtained from the mature seeds were single crystals.
Our results indicate that repeated microseeding using selected crystals as a source of microseeds might be an effective way to improve the quality of crystals, especially with regard to morphology.
Acknowledgments
We thank all the members of beamline BL44XU for help during data collection at SPring-8. This work was supported in part by Grant-in-Aid for Scientific Research in Priority Areas 90362269 and Grant-in-Aid for Young Scientists (B) 19770080 from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Adachi, H., Matsumura, H., Takano, K., Niino, A., Inoue, T., Mori, Y. & Sasaki, T. (2004). Jpn. J. Appl. Phys.43, L79–L81.
- Deshaies, R. J. (1999). Annu. Rev. Cell Dev. Biol.15, 435–467. [DOI] [PubMed]
- Hershko, A. & Ciechanover, A. (1998). Annu. Rev. Biochem.67, 425–479. [DOI] [PubMed]
- Jin, J., Cardozo, T., Lovering, R. C., Elledge, S. J., Pagano, M. & Harper, J. W. (2004). Genes Dev.18, 2573–2580. [DOI] [PMC free article] [PubMed]
- Matthews, P. C. (1968). Int. J. Soc. Psychiatry, 14, 125–133. [DOI] [PubMed]
- McPherson, A. (1982). Preparation and Analysis of Protein Crystals, 1st ed., pp. 96–97. New York: John Wiley.
- Mizushima, T., Hirao, T., Yoshida, Y., Lee, S. J., Chiba, T., Iwai, K., Yamaguchi, Y., Kato, K., Tsukihara, T. & Tanaka, K. (2004). Nature Struct. Mol. Biol.11, 365–370. [DOI] [PubMed]
- Mizushima, T., Yoshida, Y., Kumanomidou, T., Hasegawa, Y., Suzuki, A., Yamane, T. & Tanaka, K. (2007). Proc. Natl Acad. Sci. USA, 104, 5777–5781. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pickart, C. M. (2001). Annu. Rev. Biochem.70, 503–533. [DOI] [PubMed]
- Thaller, C., Weaver, L. H., Eichele, G., Wilson, E., Karlsson, R. & Jansonius, J. N. (1981). J. Mol. Biol.147, 465–469. [DOI] [PubMed]
- Yoshida, Y. (2007). Biosci. Biotechnol. Biochem.71, 2623–2631. [DOI] [PubMed]
- Yoshida, Y., Chiba, T., Tokunaga, F., Kawasaki, H., Iwai, K., Suzuki, T., Ito, Y., Matsuoka, K., Yoshida, M., Tanaka, K. & Tai, T. (2002). Nature (London), 418, 438–442. [DOI] [PubMed]
- Yoshida, Y., Murakami, A., Iwai, K. & Tanaka, K. (2007). J. Biol. Chem.282, 7137–7144. [DOI] [PubMed]