

Abstract
Brain-derived neurotrophic factor (BDNF) has been shown to mediate the effects of exercise on synaptic plasticity and cognitive function, in a process in which energy metabolism probably plays an important role. The purpose of the present study was to examine the influence of exercise on rat hippocampal expression of molecules involved in the regulation of energy management and cognitive function, and to determine the role of BDNF in these events. One week of voluntary exercise that enhanced learning and memory performance elevated the expression of molecular systems involved in the metabolism of energy [AMP-activated protein kinase (AMPK), ubiquitous mitochondrial creatine kinase (uMtCK) and uncoupling protein 2] and molecules that work at the interface of energy and synaptic plasticity [BDNF, insulin-like growth factor I (IGF-I) and ghrelin]. The levels of BDNF mRNA were associated with the mRNA levels of AMPK, uMtCK, IGF-I and ghrelin. Inhibiting the action of BDNF during exercise abolished an exercise-mediated enhancement in spatial learning and increased the expression of all of the molecular systems studied. BDNF blocking also disrupted the association between learning speed and levels of AMPK, uMtCK, ghrelin and IGF-I mRNAs. These findings suggest that the effects of exercise on synaptic plasticity and cognitive function involve elements of energy metabolism, and that BDNF seems to work at the interface between the two processes as a metabotrophin.
Keywords: AMP-activated protein kinase, energy, metabolism, rat, synaptic plasticity
Introduction
Energy homeostasis is fundamental for the survival of both the cell and the organism. New evidence indicates that mechanisms of energy metabolism may play a key role in mediating aspects of higher order cognitive function. Physical activity, an event that intrinsically impacts energy management, has repeatedly been demonstrated to enhance cognitive function in both animal and human studies (Fordyce & Wehner, 1993; Kramer et al., 1999; Laurin et al., 2001). The effect of exercise on preserving or enhancing cognition applies across the dynamics of life events, i.e. under normal conditions (Fordyce & Wehner, 1993), following brain injury (Griesbach et al., 2004) and during ageing (Kramer et al., 1999; Laurin et al., 2001). Exercise increases the expression of brain-derived neurotrophic factor (BDNF) in the hippocampus, an integral area for learning and memory (Neeper et al., 1996; Vaynman et al., 2003). BDNF has a central role in the ability of exercise to enhance cognitive function using molecules involved in synaptic plasticity and cognition (Vaynman et al., 2004b). According to the results of new studies (see below), it is possible that the supporting role of BDNF on cognition is associated with its ability to interface energy metabolism and synaptic plasticity.
BDNF has a remarkable capacity for supporting neuronal plasticity in the central nervous system through its diverse actions on axonal and dendritic remodeling (Shimada et al., 1998; Lom & Cohen-Cory, 1999; McAllister et al., 1999; Yacoubian & Lo, 2000), synaptogenesis (Alsina et al., 2001) and synaptic efficacy (Lohof et al., 1993; Kang & Schuman, 1995; Boulanger & Poo, 1999; Kafitz et al., 1999). The function of BDNF has been shown to be deeply involved in learning and memory using several BDNF deletion paradigms (Kesslak et al., 1998; Linnarsson et al., 1997; Minichiello et al., 1999; Broad et al., 2002; Heldt et al., 2007). Importantly, BDNF also functions in a metabotrophic capacity, i.e. to mediate critical aspects of energy metabolism. Prime examples come from studies of transgenic mice heterozygous for BDNF, which suffer from hyperphagia, obesity and hyperinsulinemia (Lyons et al., 1999; Kernie et al., 2000). The peripheral or central administration of BDNF reduces body weight and improves blood glucose control in obese diabetic rodents (Tonra, 1999). BDNF also appears to be a positive regulator of energy expenditure as BDNF treatments have been demonstrated to prevent body temperature reduction during cold exposure or food deprivation (Tsuchida et al., 2001). An increase in oxidative stress, a consequence of aberrant energy metabolism, results in a decrease in BDNF levels (Wu et al., 2004).
The purpose of the present study was to examine the role of BDNF in the modulation of metabolic markers in the hippocampus during exercise that have the capacity to influence learning and memory. In particular, we assessed various molecular systems involved with the monitoring, balance and transduction of cellular energy. AMP-activated protein kinase (AMPK) is a serine-threonine kinase, which is described as a ‘fuel gauge’ for cellular metabolism (Hardie, 2004) due to its ability to sense low energy levels and activate or inhibit the appropriate molecules to re-establish the proper energy balance of the cell. The ubiquitous mitochondrial creatine kinase (uMtCK) is involved in energy maintenance and transduction (Boero et al., 2003), and may function to modulate aspects of cognitive function possibly by interacting with the BDNF system. The mitochondrial uncoupling protein 2 (UCP-2) regulates energy metabolism via its ability to uncouple mitochondrial electron transport from ATP synthesis by permitting a proton leak across the mitochondrial membrane (Cheng et al., 2003; Kim-Han & Dugan, 2005). Ghrelin is secreted from an empty stomach and, when injected into the hippocampus, increases memory retention in rats (Carlini et al., 2002, 2004). Like BDNF, insulin-like growth factor I (IGF-I) plays a role in synaptic plasticity (Ramsey et al., 2005), neurotransmitter synthesis and release (Anlar et al., 1999), and can support cognitive function (Saatman et al., 1997; Carro et al., 2001). IGF-I also plays a major role in regulating the different aspects of general body metabolism, such as plasma lipid concentration (Zenobi et al., 1993) and insulin action (Cusi & DeFronzo, 2000).
Materials and methods
Exercise paradigm
Adult male Sprague–Dawley rats (n = 28, 3 months of age; Charles River, Wilmington, MA, USA) were individually housed in standard polyethylene cages in a 12/12 h light/dark cycle at 22–24°C, with food and water provided ad libitum. A voluntary exercise paradigm was chosen as it simulates aspects of human behavior by enabling animals to choose how much to run. Exercise rats were given access to a running wheel (diameter 31.8 cm, width 10 cm) that freely rotated against a resistance of 100 g, whose revolutions were monitored hourly by an attached receiver (VitalViewer Data Acquisition System software, Mini Mitter Co., Inc., Sunriver, OR, USA). Exercise was provided for a 1-week period prior to Morris water maze (MWM) training, during which the animals were exposed to the respective drug treatments as successfully used in our previous work (Vaynman et al., 2004b). Animals continued in their respective experimental conditions for the duration of the study until killing. There were four animal groups: exercise with injection of the BDNF blocker TrkB-IgG (Ex/IgG; n = 8), sedentary with TrkB-IgG injection (sed/IgG; n = 8), sedentary controls with injection of cytochrome C (cytC) (Sed/Con; n = 6) and exercise with cytC control injection (Ex/Con; n = 6). All animals were killed by decapitation on the morning following their last treatment day and their hippocampi were rapidly dissected out, immediately placed on dry ice and stored at −70°C. These studies were performed in accordance with the guidelines of the United States National Institute of Health Guide for the Care and Use of Laboratory Animals, and were approved by UCLA Animal Research Committees.
BDNF inhibitor preparation
Recombinant human TrkB-IgG chimera (in powder; R&D Systems, Inc., Minneapolis, MN, USA) comprises the extracellular domains of human TrkB and the Fc domain of IgG, and has been shown to be a highly potent and specific inhibitor of BDNF action (Shelton et al., 1995). We used cytC (Sigma, St Louis, MO, USA) as the control drug based on the successful results of previous studies (Lom & Cohen-Cory, 1999; Vaynman et al., 2003). The cytC was dissolved in sterile distilled water, with a stock concentration of 100 ng/μL. Fluorescent latex microbeads (Lumafluor Corp., Naples, FL, USA) were used as the vehicle for drug insertion into the hippocampus. Sterile phosphate-buffered saline containing 0.1% bovine serum albumin was added to the TrkB-IgG vial to prepare the stock solution (100 μg/mL). Infusion of TrkB-IgG into the hippocampus was performed according to previously published methods (Riddle et al., 1997; Lom & Cohen-Cory, 1999; Vaynman et al., 2004a). Briefly, these comprised coating the microbeads with each drug via passive absorbency for a 15 h incubation period at 4°C with a 1 : 5 mix of microbeads to TrkB-IgG (5 μg/μL in phosphate-buffered saline with bovine serum albumin) and cytC (100 ng/μL in sterile water) (Lom & Cohen-Cory, 1999). The solution was centrifuged at 14 000 g for 30 min and the microbeads were resuspended in sterile water at a 10% concentration.
Injection of drugs into the hippocampus
Exercise and sedentary rats received TrkB-IgG or the standard control injection. We used a unilateral injection to the right hippocampal tissue to be consistent with previous blocking experiments (Vaynman et al., 2004a). We did not use the contralateral hippocampus as a control as a unilateral injection can influence the contralateral side due to connecting fibers (Amaral & Witter, 1989). Injections were given to all animals in early morning such that an ample recovery time permitted all animals to begin running that same evening. All animals were anesthetized with Isoflurane (2–2.5%), utilizing the Mobile Laboratory Animal Anesthesia System (Vetequip Inc., Pleasanton, CA), and positioned in a stereotaxic apparatus that was used to secure the animal and to measure the site of injection. TrkB-IgG or cytC embedded in microbeads was injected into the right hippocampus (3.8 mm posterior to Bregma, 1 mm from the midline and 3.7 mm vertically) using a Hamilton syringe in a volume of 2 μL over 15 min. The location of the microbead injection was verified by florescence microscopy and this showed that the fluorescent microbead carrier was located in the central region of the hippocampus around the area stratum lacunosum moleculare.
Learning and memory test
The effect of exercise and BDNF inhibition on memory functions was evaluated by the MWM test. Animals were tested for spatial memory acquisition and retention (Morris et al., 1982; Sutherland et al., 1982). As previously discussed in Molteni et al. (2002), the swimming pool (130 cm diameter, 50 cm height) was divided into four quadrants. The escape platform (12 cm diameter) was fixed in a permanent position 1 cm under the water surface and the quadrant housing the platform was defined as the target zone. The water, kept at a steady 22°C, was made opaque with white non-toxic biodegradable dye to prevent the rats from seeing the platform. We used a stringent two-trial-per-day, 5 day MWM training protocol, which we have identified as a good discriminative test for the effect of exercise on learning and memory (Vaynman et al., 2004b). The animals were placed into the tank facing the wall from one of the equally spaced start locations, which were randomly altered for every trial. Spatial reference cues around the pool were maintained constant throughout the duration of the MWM training and probe trials. Each trial lasted until the rat found the platform or for a maximum duration of 60 s. If the rat failed to find the platform, it was gently placed on it. At the end of each trial, the rat was allowed to rest on the platform for 10 s. The time to reach the platform (escape latency) was recorded for each animal. To assess spatial memory retention, a probe trial was performed 2 days after the last training trial, during which the platform was removed from the pool, whereas all other factors remained constant. As previously described (Molteni et al., 2004), rats were allowed to swim for 60 s, during which the percentage of time spent in each quadrant was calculated and their swim paths were semi-automatically recorded by a video tracking system (Spontaneous Motor Activity Recording and Tracking; no. 35E4F-FA9, Pan Laboratory s.I., Barcelona, Spain). The MWM was performed early in the morning during the light phase of the circadian cycle.
Isolation of total RNA and real-time quantitative RT-PCR
Total RNA was isolated using the RNA STAT-60 kit (Tel-Test, Inc., Friendswood, TX, USA) as per the manufacturer's protocol. Quantification was carried out by absorption at 260 nm. The mRNAs for AMPK, uMtCK, UCP-2, ghrelin and IGF-I were measured by real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR) using a sequence detection instrument (prism model 7700, Applied Biosystems), which directly detects the RT-PCR product without downstream processing. This is achieved by monitoring the increase in fluorescence of a dye-labeled DNA probe, one that is specific for the factor of interest plus another that is specific for the glyceraldyehyde-3-phosphate dehydrogenase gene, which has been previously used as a successful endogenous assay control (Griesbach et al., 2002). Total RNA (100 ng) was converted into cDNA using TaqMan EZ RT-PCR Core reagents (Applied Biosystems, Branchburg, NJ, USA). The sequences of probes, forward and reverse primers (Integrated DNA Technologies, Coralville, IA, USA) were: AMPK, 5′-CTCAACCGGCAGAAGATTCGAAGCC-3′; forward, 5′-GACTGGACATAAAGTTGCTGTGAAG-3′; reverse, 5′-GGATTTTCCCGACCACGTC-3′; uMtCK, 5′-CCAGATTTGCGCCAGATCCTGCTC-3′; forward, 5′-CACACCCAGAGCAATCAACAGT-3′; reverse, 5′-TGGGATGCGGCTACCG-3′; UCP-2, 5′-TATCTCCGACCACCGCCAGCCCG-3′; forward, 5′-ATGTGGTAAAGGTCCGCTTCC-3′; reverse, 5′-GTAGCCTTCGACAGTGCTCTG-3′; ghrelin, 5′-CAAGGCCATGGTGTCTTCAGCGACT-3′; forward, 5′-CCGCCAGCATGCCTTC-3′; reverse, 5′-GGATTTTCCCGACCACGTC-3′; IGF-I, 5′-CCGGACCAGAGACCCTTTGCGG-3′; forward, 5′-CTTTACCAGCTCGGCCACA-3′; reverse, 5′-TTGGTCCACACACGAACTGAAG-3′; BDNF, 5′-AGTCATTTGCGCACAACTTTAAAAGTCTGCATT-3′; forward, 5′-GGACATATCCATGACCAGAAAGAAA-3′; reverse, 5′-GCAACAAACCACAACATTATCGAG-3′. The endogenous control probe, specific for the glyceraldyehyde-3-phosphate dehydrogenase gene, served to standardize the amount of RNA sample and consisted of the following oligonucleotide sequence (5′-CCGACTCTTGCCCTTCGAAC-3′). The reverse transcription reaction steps consisted of an initial 2 min incubation step at 50°C to activate uracil glycosylase and were followed by 30 min of reverse transcription at 60°C. A completion step for uracil glycosylase deactivation was performed for 5 min at 95°C. The 40 cycles of two-step polymerase chain reaction consisted of a 20-s period at 94°C and a 1-min period at 62°C.
We used glyceraldyehyde-3-phosphate dehydrogenase for RT-PCR as an internal standard as described previously in our other study (Molteni et al., 2002). Quantification of the TaqMan RT-PCR results was performed by plotting fluorescent signal intensities against the number of polymerase chain reaction cycles on a semilogarithmic scale. A threshold cycle was designated as the amplification cycle at which the first significant increase in fluorescence occurred. The threshold cycle value of each sample was compared with that of the internal standard. These processes were fully automated and carried out using the ABI sequence detector software version 1.6.3 (PE Biosystems). Taqman EZ RT-PCR values for uMtCK, AMPK, ghrelin, UCP-2, IGF-I and BDNF were corrected by subtracting values for glyceraldyehyde-3-phosphate dehydrogenase as previously described (Griesbach et al., 2002; Molteni et al., 2002). These corrected values were used to make cross-group comparisons.
Statistical analyses
All statistical analyses were performed by commercial software spss 16.0. A level of 5% probability was considered significant. Data are shown as the mean ± SEM. MWM and mRNA data were analysed by two-way anova (behavior, sedentary vs. exercise; drug, cytC vs. TrkB-IgG). Interaction effects were further analysed by performing means comparisons and desired contrast weights were specified. Analysis of correlation (linear regression) was performed to evaluate the association between BDNF mRNA expression and the metabolic proteins from the sedentary and exercised group injected with the control drug. Post-hoc analyses were conducted using Bonferroni comparisons.
Results
Exercise increases the expression of metabolic proteins in the hippocampus
We examined the expression of molecules associated with energy management in the hippocampus in exercised rats. AMPK is activated in response to ATP depletion, which has led to the idea that AMPK provides a good estimate of cellular energy (Fryer et al., 2002). We found that exercise (Ex/Con) significantly increased the mRNA levels of AMPK in the hippocampus to 124% (P < 0.05) of sedentary controls (Sed/Con) (Fig. 1A). Recent findings indicate that the creatine kinase uMtCK is involved in energy transduction that may support higher-order brain functions such as learning and memory (Boero et al., 2003). The results indicate that exercise (Ex/Con) significantly increases the mRNA level of uMtCK to 121% (P < 0.05) of sedentary controls (Sed/Con) (Fig. 1B). Lastly, UCP-2 has been shown to play a role in energy metabolism and may interact with the substrates for hippocampal synaptic plasticity (Vaynman et al., 2006). We found that exercise (Ex/Con) increased the mRNA level of UCP-2 to 130% (P < 0.01) of sedentary controls (Sed/Con) (Fig. 1C).
Fig. 1.

Effects of voluntary exercise on the mRNA levels of AMPK (A), uMtCK (B), UCP-2 (C), ghrelin (D) and IGF-I (E) in the hippocampus. Exercise (Ex/Con) elevated the expression of these metabolism-related molecules relative to sedentary animals (Sed/Con). The injection of the BDNF inhibitor TrkB-IgG into the hippocampus abolished these increases in exercise rats (Ex/IgG). Levels of AMPK, uMtCK and UCP-2 are expressed as a percent of sedentary controls (Sed/Con). Each value represents the mean ± SEM (two-way anova, *P < 0.05, **P < 0.01). Sed/IgG, sedentary with TrkB-IgG injection.
Ghrelin and IGF-I
Ghrelin is secreted by the stomach in response to energy depletion related to decreased food intake (Ariyasu et al., 2001; Tschop et al., 2001). Ghrelin has also been shown to affect cognitive function as injections of ghrelin into the hippocampus increase memory retention in rats (Carlini et al., 2002, 2004). Ghrelin is a hormone that is involved in energy metabolism and plays a role in cognitive function. Due to the dual role of ghrelin in energy metabolism and cognitive function, we examined the effects of exercise on ghrelin expression in the hippocampus. We found that exercise (Ex/Con) increased the mRNA level of ghrelin to 121% (P < 0.05) of sedentary controls (Sed/Con) (Fig. 1D). We also examined the trophic factor IGF-I, which has also been associated with energy metabolism and cognitive function under homeostatic and challenging conditions. In addition, IGF-I shares downstream pathways with BDNF (Yamada et al., 1997; Roudabush et al., 2000) and its receptors are abundantly expressed in the hippocampus (Bohannon et al., 1988; Araujo et al., 1989; Bondy et al., 1992). The results showed that exercise (Ex/Con) elevated IGF-I mRNA levels to 138% (P < 0.01) of sedentary controls (Sed/Con) (Fig. 1E) in the hippocampus.
Association between BDNF and AMPK, uMtCK, IGF-I and ghrelin
Exercise elevated BDNF mRNA levels to 140% (P < 0.05) of control values (Fig. 2A). We examined a possible association between BDNF and metabolic proteins by performing an analysis of correlation between the levels of BDNF and the levels of each of the proteins under study. The results showed that BDNF levels were positively correlated with the levels of AMPK (r = 0.779, P < 0.01; Fig. 2B) and uMtCK (r = 0.64, P < 0.05, Fig. 2C), and UCP-2 reached a value near significance (r = 0.515, P = 0.08). In addition, BDNF levels were positively correlated with the levels of ghrelin (r = 0.619, P < 0.05; Fig. 2D) and IGF-I (r = 0.669, P < 0.05; Fig. 2E).
Fig. 2.

Association between BDNF and metabolic molecular systems. (A) Exercise (Ex/Con) elevated the levels of BDNF mRNA relative to sedentary animals (Sed/Con) and this effect was counteracted by the hippocampal injection of the BDNF function blocker TrkB-IgG (Ex/IgG). The injection of TrkB-IgG also reduced BDNF mRNA levels in sedentary animals (Sed/IgG). Analysis of correlation showed an association between BDNF mRNA levels and mRNA levels of AMPK (B), uMtCK (C), ghrelin (D) and IGF-I (E) for sedentary and exercised animals. The injection of TrkB-IgG into the hippocampus of sedentary and exercised rats abolished the correlation (data not shown). Each value represents the mean ± SEM (two-way anova, *P < 0.05).
Blocking BDNF action abolishes the effects of exercise on proteins involved in energy metabolism
BDNF has been shown to play a crucial role in the ability of exercise to enhance learning and memory in the rat (Vaynman et al., 2004b). We wanted to determine how blocking BDNF action in the hippocampus during an exercise period that enhanced cognitive function would regulate key factors involved in energy metabolism. Hippocampal BDNF was inhibited with a specific immunoadhesin chimera (TrkB-IgG) that mimics the BDNF receptor TrkB to selectively sequester BDNF. Histological examination showed fluorescent microbeads distributed around the area of the lacunosum moleculare, whereas the transported drug was free to diffuse to adjacent hippocampal regions. A two-way anova analysis (behavior vs. drug) indicated the effects of behavior or drug on BDNF (behavior, F1,27 = 8.596, P < 0.007; drug, F1,27 = 32.828, P < 0.001). The same analysis showed the effects of behavior or drug on AMPK (behavior, F1,27 = 11.104, P < 0.003; drug, F1,27 = 5.050, P < 0.034) and an interaction of behavior × drug (uMtCK, F1,27 = 5.627, P < 0.01; UCP-2, F1,27 = 12.469, P < 0.01; ghrelin, F1,27 = 5.001, P < 0.05; IGF-I, F1,27 = 9.114, P < 0.01). Specifically, blocking the action of BDNF fully abolished the exercise-induced increase in the mRNA levels for AMPK (124 to 105%, P < 0.05), uMtCK (121 to 86%, P < 0.05), UCP-2 (130 to 95%, P < 0.01), ghrelin (121 to 101%, P < 0.01) and IGF-I (138 to 81%, P < 0.01; Fig. 1A–E). The BDNF blocker did not significantly alter the mRNA levels of AMPK, uMtCK, UCP-2 or ghrelin in sedentary animals (94, 89, 104 and 102%, Sed/IgG) compared with the control injection (Sed/Con). However, IGF-I mRNA levels were significantly (P < 0.05) decreased to 80% of Sed/Con. The BDNF blocking also disrupted the correlation between BDNF and metabolic markers (data not shown).
Exercise enhances learning and memory under the action of BDNF
The paradigm used to determine the efficacy of exercise on BDNF-mediated hippocampal energy metabolic markers has a demonstrated ability in enhancing learning and memory on the MWM (Vaynman et al., 2004b). Accordingly, we aimed to assess the association between exercise-enhanced spatial learning acquisition, BDNF and energy metabolism. We used a challenging two-trial-per-day, 5 day MWM paradigm. The results showed that exercise decreased (P < 0.05) the latency to locate the platform on days 3–5 of MWM training as compared with sedentary controls (Fig. 3A). Exercise also increased the learning speed (the slope of the escape latency across the 5 days of learning; Fig. 3B). Blocking the action of BDNF in exercising animals abolished the exercise-induced enhancement of learning acquisition as demonstrated by an increase in the escape latency and a reduction in the learning speed. Although the BDNF blocker was not sufficient to alter the escape latencies in the sedentary condition (Fig. 3A), it showed a decreasing trend to reduce learning speed in the MWM (Fig. 3B). To evaluate a possible association between BDNF-mediated energy metabolic markers and behavior, we assessed the correlation between learning speed in the MWM with levels of the metabolic markers (uMtCK, AMPK and UCP-2) and levels of IGF-I and ghrelin (Fig. 3B). The results showed an association between the learning speed and the levels of AMPK (r = 0.66, P = 0.02; Fig. 3C), uMtCK (r = 0.59, P = 0.04; Fig. 3D), IGF-I (r = 0.59, P = 0.04; Fig. 3E) and ghrelin (r = 0.59, P = 0.047; Fig. 3F) for rats maintained under sedentary and exercise conditions. This association was not observed in the group of rats injected with the BDNF inhibitor (data not shown).
Fig. 3.

(A) Exercise (Ex/Con) decreased the latency to locate the platform on days 3–5 in the MWM as compared with sedentary controls (Sed/Con). Blocking BDNF action with TrkB-IgG abolished the exercise-induced enhancement of learning acquisition (Ex/TrkB-IgG) but did not alter the escape latencies in the sedentary condition (Sed/IgG). (B) These effects were reflected in changes in the slope of the learning curve for the exercise condition. TrkB-IgG showed a decreasing trend in the sedentary group. A correlation analysis showed an association between learning speed and mRNA levels for AMPK (C), uMtCK (D), IGF-I (E) and ghrelin (F) for the total pool of exercise and sedentary rats. The association between learning speed and levels of mRNAs was disrupted by the injection of the BDNF blocker TrkB-IgG (data not shown). Each value represents the mean ± SEM (two-way anova, *P < 0.05, #P < 0.01).
To evaluate memory retention, we performed a probe trial, at 2 days after the last MWM training day, in which rats had to swim for 60 s in the pool in which they received their training but with the escape platform removed. In agreement with our previous study (Vaynman et al., 2004b), the Ex/Con group spent a greater (P < 0.05) percentage of time in quadrant P (148% compared with Sed/Con, Fig. 4A). Blocking the action of BDNF prevented the exercise-induced preference for the target quadrant, such that there was no difference between the amount of time spent in quadrant P by Ex/IgG (104%) and Sed/Con (100%) rats (Fig. 4A). We performed a correlation analysis to evaluate a possible association between energy molecules and memory retention. We found a positive correlation between the IGF-I mRNA level and the memory retention ability (r = 0.68, P = 0.015; Fig. 4B) in the Sed/Con and Ex/Con rats. This association was disrupted in the group of animals injected with the BDNF blocker (data not shown).
Fig. 4.

(A) The probe trial showed a preference of exercised rats (Ex/Con) for the quadrant in which they had to swim for 60 s in the pool in which they received their training but with the escape platform removed. Blocking BDNF action prevented the exercise-induced preference for the target quadrant (Ex/IgG). Blocking the action of BDNF was selective for exercise as it did not affect the learning acquisition or recall abilities of sedentary animals (Sed/IgG). Each value represents the mean ± SEM (anova, *P < 0.05). (B) We performed a correlation analysis to evaluate a possible association between energy molecules and memory retention. We found a positive correlation between IGF-I mRNA levels and the memory retention ability (r = 0.68, P = 0.01) in the sedentary control (Sed/Con) and Ex/Con rats for the total pool of exercise and sedentary rats. The association between retention and levels of mRNAs was disrupted by the injection of the BDNF blocker TrkB-IgG (data not shown).
Discussion
The capacity of exercise to engage energy metabolism and enhance cognitive function makes exercise an excellent model to study the association between metabolic energy and cognitive function (Vaynman et al., 2004b). Studies have been centered on the possibility that BDNF can mediate the action of voluntary exercise on energy metabolism and cognitive function. The results showed that exposure to voluntary exercise for 1 week elevates the hippocampal mRNA levels of the energy regulators AMPK, uMtCK and UCP-2, and the plasticity effectors BDNF, IGF-I and ghrelin. We also show that the hippocampal levels of most of the molecular systems under study varied proportionally to the levels of BDNF mRNA. Blocking the action of BDNF during exercise reduced the expression of all systems measured, in conjunction with counteracting the enhanced ability of exercised animals to learn the location of the platform. The association between learning speed and the levels of AMPK, uMtCK, IGF-I and ghrelin was disrupted after BDNF blocking. These findings suggest that the ability of exercise to enhance cognitive function involves the action of BDNF on metabolic processes, such that BDNF may function as a metabotrophin in the hippocampus.
Involvement of AMPK and UCP-2 in exercise and cognition
AMPK
Voluntary exercise increased the hippocampal expression of AMPK, a molecule emerging as a central regulator of energy balance (Yamauchi et al., 2002; Minokoshi et al., 2004). AMPK has been shown to increase the cellular energy supply by switching on catabolic pathways that generate ATP and shutting off processes that consume it (Hardie & Carling, 1997; Hardie et al., 1998; Kemp et al., 1999; Carling, 2004). The rise in AMPK levels observed in the present study suggests that exercise may activate similar mechanisms to conserve ATP levels in the hippocampus, with subsequent effects on the modulation of intracellular processes. In addition to the AMPK control of energy through phosphorylation of metabolic enzymes, AMPK activation regulates numerous transcription factors (Habinowski & Witters, 2001; Leclerc et al., 2001; Yang et al., 2001; Zhou et al., 2001; Bronner et al., 2004; Hardie, 2004) that may link intracellular energy levels with protein synthesis (Jones et al., 2005). Our results, showing an association between AMPK and BDNF mRNA levels, and AMPK and learning ability, suggest that AMPK may serve to support the ability of exercise to influence cognitive function. It has been shown that, in conditions of mild energy restriction, AMPK activity in the hippocampus works to improve cognitive function (Dagon et al., 2005). Accordingly, it is possible that a mild energy depletion state associated with our moderate exercise regimen may engage AMPK to enhance learning and memory performance.
uMtCK and UCP-2
We found that exercise increases the hippocampal expression of the creatine kinase uMtCK involved in energy maintenance and transduction (Fig. 1B). The transfer of high-energy phosphates between sites of energy production and consumption by the creatine kinase/phosphocreatine system is an essential mechanism by which the brain maintains its high-energy requirements. The isoform known as uMtCK is brain specific and located in the mitochondrial intermembrane space (Kottke et al., 1991), where it functions to synthesize phosphocreatine, which is then exported to the cytosol (Jacobus & Lehninger, 1973; Jacobus, 1985; Rojo et al., 1991). uMtCK is highly expressed in hippocampal granule and pyramidal cells (Eppenberger et al., 1967), and its regulation by neural activity may provide a mechanism to protect neurons during periods of increased energy demand (Boero et al., 2003). Accordingly, phosphocreatine can buffer the elevated ATP consumption resulting from heightened neuronal activity by donating its phosphate group to ADP. Recent findings that mice lacking uMtCK show slower spatial learning acquisition, exploration and habituation emphasize the possibility that learning and memory dysfunction may be associated with energy mismanagement (Streijger et al., 2004). The present results show that a potential action of uMtCK in cognition may be associated with the function of BDNF by integrating mechanisms of energy homeostasis and synaptic plasticity. The results also show that exercise increases the levels of UCP-2 and that BDNF seems to mediate these effects. High levels of UCP-2 expression have been shown to protect neonatal neurons from excitotoxic cell death by inhibiting reactive oxygen species production and preventing mitochondrial dysfunction (Sullivan et al., 2003).
Involvement of IGF-I in exercise and cognition
Our results demonstrate that exercise regulates two molecular systems by which the body signals to the brain, i.e. ghrelin and IGF-I, about aspects of energy homeostasis that are crucial for the regulation of cognitive function. The results also show that BDNF blocking abolishes the effects of exercise on IGF-I mRNA. It appears that both BDNF and IGF-I are mutually regulated in response to exercise, with resulting implications for the management of energy homeostasis. BDNF and IGF-I have been shown to protect cultured hippocampal neurons from serum deprivation-induced cell death using similar downstream pathways (Zheng & Quirion, 2004). Our results showing that the BDNF blocker abolished the association between IGF-I and learning and memory retention suggest that IGF-I can work in conjunction with BDNF to modulate the action of exercise on cognitive function. Although IGF-I receptor is abundantly expressed in the hippocampus (Bohannon et al., 1988; Araujo et al., 1989; Bondy et al., 1992), IGF-I can also be produced in peripheral tissue, such as skeletal muscle and liver in response to exercise (Carro et al., 2000). Several actions of IGF-I have been described in the central nervous system, such as supporting regeneration during development (Anlar et al., 1999), synaptic plasticity in adulthood (Ramsey et al., 2005) and cognition after brain trauma (Saatman et al., 1997; Carro et al., 2001). It has also been proposed that IGF-I administration may help to reduce age-related cognitive deficits (Markowska et al., 1998; Sonntag et al., 2000). Accordingly, it is possible that the association of IGF-I with elements of the energy metabolic machinery may play a crucial role in the effects of exercise enhancing cognitive abilities throughout life.
Involvement of ghrelin in exercise and cognition
Ghrelin is generally secreted from the oxycintic gland of the stomach (Kojima et al., 1999) in response to energy restriction and/or depletion (Ariyasu et al., 2001). The present results show that exercise elevates hippocampal ghrelin mRNA and that these changes may be associated with the influence of exercise on cognitive function. Ghrelin can bind hippocampal receptors (Guan et al., 1997) with profound effects on hippocampal synaptic plasticity, altering long-term potentiation and hippocampal-dependent learning and memory (Diano et al., 2006). The present results, showing that BDNF blocking reduced the effect of exercise on ghrelin mRNA, provide new information to ponder how metabolic signals from food and exercise can integrate to regulate cognitive function. Exercise, similar to dietary restriction, produces a negative energy balance that would explain the increase in ghrelin expression. Intravenous injections of ghrelin given to human subjects increase appetite and energy intake (Wren et al., 2001), and it has been shown that central and peripheral ghrelin production contributes to modulating the energy balance (Cowley et al., 2003; van der Lely et al., 2004). Ghrelin may represent a class of factors that work to maintain homeostasis by engaging aspects of cognitive function to facilitate food procurement for survival, particularly during times of limited energy availability.
Exercise-mediated hippocampal expression of metabolic proteins is dependent on BDNF
Our results reaffirm a central role of BDNF in coordinating the metabolic and cognitive effects of physical activity (Fig. 5). Exercise increases the mRNA levels of the energy mediators AMPK, uMtCK and UCP-2, and the plasticity effectors IGF-I and ghrelin upon dependence on the activity of BDNF. TrkB receptors are present in hippocampal neurons as well as in brain mitochondria (Wiedemann et al., 2006), which may account for the observed reliance of uMtCK, AMPK and UCP-2 expression on BDNF. In addition to the effects of BDNF on energy metabolites, the opposite situation may also be possible, as it has recently been shown that AMPK actively modulates BDNF expression in cultured C6 glioma and neuroblastoma cells (Yoon et al., 2008). This mutual interaction between BDNF and energy metabolism can have crucial effects on the modulation of neuronal plasticity in response to exercise and other stimuli associated with energy metabolism. The function of BDNF in energy metabolism can be perceived in disorders of energy balance, where reduced BDNF levels can result in obese and hyperglycemic mice (Kernie et al., 2000).
Fig. 5.
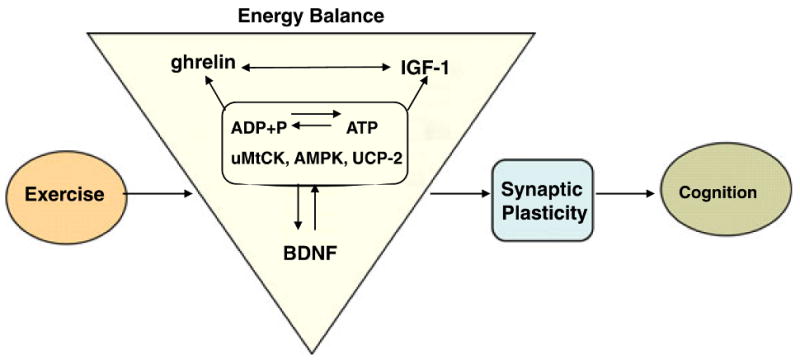
Proposed mechanism by which exercise enhances cognitive function by engaging aspects of cellular energy metabolism. There is a crucial association between metabolic energy and synaptic plasticity, in which BDNF plays a crucial role. The effects of exercise on hippocampal BDNF would activate several molecular systems involved in the metabolism of energy, thereby modulating the capacity of the synapse to process information relevant to cognitive function. In particular, molecular systems, such as uMtCK, AMPK and UCP-2, may work at the interface between energy and synaptic plasticity. IGF-I, ghrelin and energy-related molecules can interact with BDNF to modulate synaptic plasticity and cognitive function. Therefore, BDNF appears to be a central integrator for the effects of exercise on synaptic markers and energy metabolic processes to affect cognitive function.
Our results also demonstrate that blocking the action of BDNF abolished the exercise-related enhancement in cognitive performance and disrupted the association between learning speed and the expression of key factors involved in energy metabolism. Our findings that the TrkB-IgG injection did not affect learning parameters in sedentary animals do not imply that BDNF is not important for learning and memory under normal conditions. Indeed, abundant evidence indicates that the action of BDNF is important for hippocampal learning (Kesslak et al., 1998; Linnarsson et al., 1997; Minichiello et al., 1999; Broad et al., 2002; Heldt et al., 2007). Our experimental design was directed to evaluate whether BDNF inhibition could block the effects of exercise. Accordingly, the BDNF inhibitor was injected on the first day of exercise, with expected maximal effects during the 7 day exercise period. Previous studies reported that neurotrophin-embedded microbeads can deliver neurotrophins for 4 days (Riddle et al., 1997), whereas TrkB-IgG-embedded microbeads can be active for 7 days in the hippocampus (Vaynman et al., 2004b). The proper management of cellular energy is a vital requirement for the synaptic function underlying cognition. Previous findings show that BDNF and IGF-I share a common signaling cascade that activates CAM-KII, Akt and CREB, which in turn can affect learning and memory (Fig. 5).
In conclusion, taken together, these findings suggest that BDNF is part of a central mechanism through which physical activity integrates with elements of energy metabolism to impact aspects of hippocampal function. These findings support the evolutionary contention that learning ability is intimately related to energy balance, an attribute that may have developed to maximize motor operations that increased the chances of obtaining food and the probability of survival.
Acknowledgments
This work was supported by National Institutes of Health Grant NS50465. We thank Dr Soo Kim for helpful discussions.
Abbreviations
- AMPK
AMP-activated protein kinase
- BDNF
brain-derived neurotrophic factor
- cytC
cytochrome C
- IGF-I
insulin-like growth factor I
- MWM
Morris water maze
- RT-PCR
reverse transcription-polymerase chain reaction
- UCP-2
uncoupling protein 2
- uMtCK
ubiquitous mitochondrial creatine kinase
References
- Alsina B, Vu T, Cohen-Cory S. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci. 2001;4:1093–1101. doi: 10.1038/nn735. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Anlar B, Sullivan KA, Feldman EL. Insulin-like growth factor-I and central nervous system development. Horm Metab Res. 1999;31:120–125. doi: 10.1055/s-2007-978708. [DOI] [PubMed] [Google Scholar]
- Araujo DM, Lapchak PA, Collier B, Chabot JG, Quirion R. Insulin-like growth factor-1 (somatomedin-C) receptors in the rat brain: distribution and interaction with the hippocampal cholinergic system. Brain Res. 1989;484:130–138. doi: 10.1016/0006-8993(89)90355-7. [DOI] [PubMed] [Google Scholar]
- Ariyasu H, Takaya K, Tagami T, Ogawa Y, Hosoda K, Akamizu T, Suda M, Koh T, Natsui K, Toyooka S, Shirakami G, Usui T, Shimatsu A, Doi K, Hosoda H, Kojima M, Kangawa K, Nakao K. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J Clin Endocrinol Metab. 2001;86:4753–4758. doi: 10.1210/jcem.86.10.7885. [DOI] [PubMed] [Google Scholar]
- Boero J, Qin W, Cheng J, Woolsey TA, Strauss AW, Khuchua Z. Restricted neuronal expression of ubiquitous mitochondrial creatine kinase: changing patterns in development and with increased activity. Mol Cell Biochem. 2003;244:69–76. [PubMed] [Google Scholar]
- Bohannon NJ, Corp ES, Wilcox BJ, Figlewicz DP, Dorsa DM, Baskin DG. Localization of binding sites for insulin-like growth factor-I (IGF-I) in the rat brain by quantitative autoradiography. Brain Res. 1988;444:205–213. doi: 10.1016/0006-8993(88)90931-6. [DOI] [PubMed] [Google Scholar]
- Bondy C, Werner H, Roberts CT, Jr, LeRoith D. Cellular pattern of type-I insulin-like growth factor receptor gene expression during maturation of the rat brain: comparison with insulin-like growth factors I and II. Neuroscience. 1992;46:909–923. doi: 10.1016/0306-4522(92)90193-6. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Poo MM. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci. 1999;2:346–351. doi: 10.1038/7258. [DOI] [PubMed] [Google Scholar]
- Broad KD, Mimmack ML, Keverne EB, Kendrick KM. Increased BDNF and trk-B mRNA expression in cortical and limbic regions following formation of a social recognition memory. Eur J Neurosci. 2002;16:2166–2174. doi: 10.1046/j.1460-9568.2002.02311.x. [DOI] [PubMed] [Google Scholar]
- Bronner M, Hertz R, Bar-Tana J. Kinase-independent transcriptional co-activation of peroxisome proliferator-activated receptor alpha by AMP-activated protein kinase. Biochem J. 2004;384:295–305. doi: 10.1042/BJ20040955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade – a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Carlini VP, Monzon ME, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR. Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem Biophys Res Commun. 2002;299:739–743. doi: 10.1016/s0006-291x(02)02740-7. [DOI] [PubMed] [Google Scholar]
- Carlini VP, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR. Differential role of the hippocampus, amygdala, and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem Biophys Res Commun. 2004;313:635–641. doi: 10.1016/j.bbrc.2003.11.150. [DOI] [PubMed] [Google Scholar]
- Carro E, Nunez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci. 2000;20:2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor I mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J Neurosci. 2001;21:5678–5684. doi: 10.1523/JNEUROSCI.21-15-05678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G, Polito CC, Haines JK, Shafizadeh SF, Fiorini RN, Zhou X, Schmidt MG, Chavin KD. Decrease of intracellular ATP content downregulated UCP2 expression in mouse hepatocytes. Biochem Biophys Res Commun. 2003;308:573–580. doi: 10.1016/s0006-291x(03)01409-8. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, Horvath TL. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661. doi: 10.1016/s0896-6273(03)00063-1. [DOI] [PubMed] [Google Scholar]
- Cusi K, DeFronzo R. Recombinant human insulin-like growth factor I treatment for 1 week improves metabolic control in type 2 diabetes by ameliorating hepatic and muscle insulin resistance. J Clin Endocrinol Metab. 2000;85:3077–3084. doi: 10.1210/jcem.85.9.6827. [DOI] [PubMed] [Google Scholar]
- Dagon Y, Avraham Y, Magen I, Gertler A, Ben-Hur T, Berry EM. Nutritional status, cognition, and survival: a new role for leptin and AMP kinase. J Biol Chem. 2005;280:42142–42148. doi: 10.1074/jbc.M507607200. [DOI] [PubMed] [Google Scholar]
- Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA, Morley JE, Pinto S, Sherwin RS, Xu L, Yamada KA, Sleeman MW, Tschop MH, Horvath TL. Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- Eppenberger HM, Dawson DM, Kaplan NO. The comparative enzymology of creatine kinases. I Isolation and characterization from chicken and rabbit tissues J Biol Chem. 1967;242:204–209. [PubMed] [Google Scholar]
- Fordyce DE, Wehner JM. Physical activity enhances spatial learning performance with an associated alteration in hippocampal protein kinase C activity in C57BL/6 and DBA/2 mice. Brain Res. 1993;619:111–119. doi: 10.1016/0006-8993(93)91602-o. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- Grealy MA, Johnson DA, Rushton SK. Improving cognitive function after brain injury: the use of exercise and virtual reality. Arch Phys Med Rehabil. 1999;80:661–667. doi: 10.1016/s0003-9993(99)90169-7. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Molteni R, Gomez-Pinilla F. Alterations in BDNF and synapsin I within the occipital cortex and hippocampus after mild traumatic brain injury in the developing rat: Reflections of injury-induced neuroplasticity. J Neurotrauma. 2002;19:803–814. doi: 10.1089/08977150260190401. [DOI] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F. Voluntary exercise following traumatic brain injury: brain-derived neurotrophic factor upregulation and recovery of function. Neuroscience. 2004;125:129–139. doi: 10.1016/j.neuroscience.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, Smith RG, Van der Ploeg LH, Howard AD. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997;48:23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- Habinowski SA, Witters LA. The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem Biophys Res Commun. 2001;286:852–856. doi: 10.1006/bbrc.2001.5484. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Med Sci Sports Exerc. 2004;36:28–34. doi: 10.1249/01.MSS.0000106171.38299.64. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D. The AMP-activated protein kinase–fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobus WE. Respiratory control and the integration of heart high-energy phosphate metabolism by mitochondrial creatine kinase. Annu Rev Physiol. 1985;47:707–725. doi: 10.1146/annurev.ph.47.030185.003423. [DOI] [PubMed] [Google Scholar]
- Jacobus WE, Lehninger AL. Creatine kinase of rat heart mitochondria. Coupling of creatine phosphorylation to electron transport. J Biol Chem. 1973;248:4803–4810. [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through trkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kemp BE, Mitchelhill KI, Stapleton D, Michell BJ, Chen ZP, Witters LA. Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem Sci. 1999;24:22–25. doi: 10.1016/s0968-0004(98)01340-1. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesslak JP, So V, Choi J, Cotman CW, Gomez-Pinilla F. Learning upregulates brain-derived neurotrophic factor messenger ribo-nucleic acid: a mechanism to facilitate encoding and circuit maintenance? Behav Neurosci. 1998;112:1012–1019. doi: 10.1037//0735-7044.112.4.1012. [DOI] [PubMed] [Google Scholar]
- Kim-Han JS, Dugan LL. Mitochondrial uncoupling proteins in the central nervous system. Antioxid Redox Signal. 2005;7:1173–1181. doi: 10.1089/ars.2005.7.1173. [DOI] [PubMed] [Google Scholar]
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- Kottke M, Adams V, Wallimann T, Nalam VK, Brdiczka D. Location and regulation of octameric mitochondrial creatine kinase in the contact sites. Biochim Biophys Acta. 1991;1061:215–225. doi: 10.1016/0005-2736(91)90287-i. [DOI] [PubMed] [Google Scholar]
- Kramer AF, Hahn S, Cohen NJ, Banich MT, McAuley E, Harrison CR, Chason J, Vakil E, Bardell L, Boileau RA, Colcombe A. Ageing, fitness and neurocognitive function. Nature. 1999;400:418–419. doi: 10.1038/22682. [DOI] [PubMed] [Google Scholar]
- Laurin D, Verreault R, Lindsay J, MacPherson K, Rockwood K. Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch Neurol. 2001;58:498–504. doi: 10.1001/archneur.58.3.498. [DOI] [PubMed] [Google Scholar]
- Leclerc I, Lenzner C, Gourdon L, Vaulont S, Kahn A, Viollet B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes. 2001;50:1515–1521. doi: 10.2337/diabetes.50.7.1515. [DOI] [PubMed] [Google Scholar]
- van der Lely AJ, Tschop M, Heiman ML, Ghigo E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004;25:426–457. doi: 10.1210/er.2002-0029. [DOI] [PubMed] [Google Scholar]
- Linnarsson S, Björklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S. Brain-derived neurotrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization in vivo. J Neurosci. 1999;19:9928–9938. doi: 10.1523/JNEUROSCI.19-22-09928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowska AL, Mooney M, Sonntag WE. Insulin-like growth factor-1 ameliorates age-related behavioral deficits. Neuroscience. 1998;87:559–569. doi: 10.1016/s0306-4522(98)00143-2. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- Molteni R, Barnard JR, Ying Z, Roberts CK, Gomez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–814. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- Molteni R, Wu A, Vaynman S, Ying Z, Barnard RJ, Gomez-Pinilla F. Exercise reverses the effects of consumption of a high-fat diet on synaptic and behavioral plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience. 2004;123:429–440. doi: 10.1016/j.neuroscience.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Morris RGM, Garrud P, Rawlins JNP, O'Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297:681–683. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Neeper SA, Gómez-Pinilla F, Choi J, Cotman CW. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 1996;726:49–56. [PubMed] [Google Scholar]
- Ramsey MM, Adams MM, Ariwodola OJ, Sonntag WE, Weiner JL. Functional characterization of des-IGF-1 action at excitatory synapses in the CA1 region of rat hippocampus. J Neurophysiol. 2005;94:247–254. doi: 10.1152/jn.00768.2004. [DOI] [PubMed] [Google Scholar]
- Riddle DR, Katz LC, Lo DC. Focal delivery of neurotrophins into the central nervous system using fluorescent latex microspheres. Biotechniques. 1997;23:928–934. 936–937. doi: 10.2144/97235rr02. [DOI] [PubMed] [Google Scholar]
- Rojo M, Hovius R, Demel R, Wallimann T, Eppenberger HM, Nicolay K. Interaction of mitochondrial creatine kinase with model membranes. A monolayer study FEBS Lett. 1991;281:123–129. doi: 10.1016/0014-5793(91)80374-c. [DOI] [PubMed] [Google Scholar]
- Roudabush FL, Pierce KL, Maudsley S, Khan KD, Luttrell LM. Transactivation of the EGF receptor mediates IGF-1-stimulated shc phosphorylation and ERK1/2 activation in COS-7 cells. J Biol Chem. 2000;275:22583–22589. doi: 10.1074/jbc.M002915200. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Contreras PC, Smith DH, Raghupathi R, McDermott KL, Fernandez SC, Sanderson KL, Voddi M, McIntosh TK. Insulin-like growth factor-1 (IGF-1) improves both neurological motor and cognitive outcome following experimental brain injury. Exp Neurol. 1997;147:418–427. doi: 10.1006/exnr.1997.6629. [DOI] [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Mason CA, Morrison ME. TrkB signaling modulates spine density and morphology independent of dendrite structure in cultured neonatal Purkinje cells. J Neurosci. 1998;18:8559–8570. doi: 10.1523/JNEUROSCI.18-21-08559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag WE, Lynch C, Thornton P, Khan A, Bennett S, Ingram R. The effects of growth hormone and IGF-1 deficiency on cerebrovascular and brain ageing. J Anat. 2000;197:575–585. doi: 10.1046/j.1469-7580.2000.19740575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streijger F, Jost CR, Oerlemans F, Ellenbroek BA, Cools AR, Wieringa B, Van der Zee CE. Mice lacking the UbCKmit isoform of creatine kinase reveal slower spatial learning acquisition, diminished exploration and habituation, and reduced acoustic startle reflex responses. Mol Cell Biochem. 2004;256–257:305–318. doi: 10.1023/b:mcbi.0000009877.90129.e3. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol. 2003;53:711–717. doi: 10.1002/ana.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RJ, Kolb B, Whishaw IQ. Spatial mapping: definitive disruption by hippocampal or medial frontal cortical damage in the rat. Neurosci Lett. 1982;31:271–276. doi: 10.1016/0304-3940(82)90032-5. [DOI] [PubMed] [Google Scholar]
- Tonra JR. Classical and novel directions in neurotrophin transport and research: anterograde transport of brain-derived neurotrophic factor by sensory neurons. Microsc Res Tech. 1999;45:225–232. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<225::AID-JEMT6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Tschop M, Wawarta R, Riepl RL, Friedrich S, Bidlingmaier M, Landgraf R, Folwaczny C. Post-prandial decrease of circulating human ghrelin levels. J Endocrinol Invest. 2001;24:RC19–RC21. doi: 10.1007/BF03351037. [DOI] [PubMed] [Google Scholar]
- Tsuchida A, Nonomura T, Ono-Kishino M, Nakagawa T, Taiji M, Noguchi H. Acute effects of brain-derived neurotrophic factor on energy expenditure in obese diabetic mice. Int J Obes Relat Metab Disord. 2001;25:1286–1293. doi: 10.1038/sj.ijo.0801678. [DOI] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. Interplay between BDNF and signal transduction modulators in the regulation of the effects of exercise on synaptic-plasticity. Neuroscience. 2003;122:647–657. doi: 10.1016/j.neuroscience.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. Exercise induces BDNF and synapsin I to specific hippocampal subfields. J Neurosci Res. 2004a;76:356–362. doi: 10.1002/jnr.20077. [DOI] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Gomez-Pinilla F. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci. 2004b;20:2580–2590. doi: 10.1111/j.1460-9568.2004.03720.x. [DOI] [PubMed] [Google Scholar]
- Vaynman S, Ying Z, Wu A, Gomez-Pinilla F. Coupling energy metabolism with a mechanism to support brain-derived neurotrophic factor-mediated synaptic plasticity. Neuroscience. 2006;139:1221–1234. doi: 10.1016/j.neuroscience.2006.01.062. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Siemen D, Mawrin C, Horn TF, Dietzmann K. The neurotrophin receptor TrkB is colocalized to mitochondrial membranes. Int J Biochem Cell Biol. 2006;38:610–620. doi: 10.1016/j.biocel.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992. doi: 10.1210/jcem.86.12.8111. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gómez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur J Neurosci. 2004;19:1699–1707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- Yacoubian TA, Lo DC. Truncated and full-length TrkB receptors regulate distinct modes of dendritic growth. Nat Neurosci. 2000;3:342–349. doi: 10.1038/73911. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ohnishi H, Sano S, Nakatani A, Ikeuchi T, Hatanaka H. Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J Biol Chem. 1997;272:30334–30339. doi: 10.1074/jbc.272.48.30334. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276:38341–38344. doi: 10.1074/jbc.C100316200. [DOI] [PubMed] [Google Scholar]
- Yoon H, Oh YT, Lee JY, Choi JH, Lee JH, Baik HH, Kim SS, Choe W, Yoon KS, Ha J, Kang I. Activation of AMP-activated protein kinase by kainic acid mediates brain-derived neurotrophic factor expression through a NF-kappaB dependent mechanism in C6 glioma cells. Biochem Biophys Res Commun. 2008;371:495–500. doi: 10.1016/j.bbrc.2008.04.102. [DOI] [PubMed] [Google Scholar]
- Zenobi PD, Holzmann P, Glatz Y, Riesen WF, Froesch ER. Improvement of lipid profile in type 2 (non-insulin-dependent) diabetes mellitus by insulin-like growth factor I. Diabetologia. 1993;36:465–469. doi: 10.1007/BF00402285. [DOI] [PubMed] [Google Scholar]
- Zheng WH, Quirion R. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J Neurochem. 2004;89:844–852. doi: 10.1111/j.1471-4159.2004.02350.x. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]