

Summary
Endogenous mono-ADP-ribosylation in eukaryotes is involved in regulating protein synthesis, signal transduction, cytoskeletal integrity and cell proliferation, though few cellular ADP-ribosyltransferases have been identified. The sirtuins are a highly conserved family of protein deacetylases and several family members have also been reported to carry out protein ADP-ribosylation. We characterized the ADP-ribosylation reaction of the nuclear sirtuin homolog from Trypanosoma brucei, TbSIR2RP1, on both acetylated and unacetylated substrates. We demonstrate that an acetylated substrate is not required for ADP-ribosylation to occur, indicating that the reaction carried out by TbSIR2RP1 is a genuine enzymatic reaction and not a side reaction of deacetylation. Biochemical and mass spectrometry data show that arginine is the major ADP-ribose acceptor for unacetylated substrates, while arginine does not appear to be the major ADP-ribose acceptor in reactions with acetylated histone H1.1. We carried out combined ab initio QM/MM molecular dynamics simulations, which indicate that sirtuin ADP-ribosylation at arginine is energetically feasible, and employs a concerted mechanism with a highly dissociative transition state. In comparison with the corresponding nicotinamide cleavage in deacetylation reaction, the simulations suggest that sirtuin ADP-ribosylation would be several orders slower but less sensitive to nicotinamide inhibition, which is consistent with experimental results. These results suggest that TbSIR2RP1 can carry out ADP-ribosylation using two distinct mechanisms that depend upon whether or not the substrate is acetylated.
Introduction
Mono-ADP-ribosylation is a reversible, covalent modification involving the transfer of the ADP-ribose moiety from beta-NAD+ to protein and DNA substrates. Since its initial discovery, mono-ADP-ribosylation has been implicated in a number of physiological processes including transcriptional regulation [1], bacterial toxigenicity [2,3], protein synthesis [4], intracellular signaling [5-7], innate immunity [8,9], and apoptosis [10,11]. Formation of the covalent adduct is carried out by ADP-ribosyltransferases (ARTs) while the ADP-ribose is removed by ADP-ribosylhydrolases (ARHs). ADP-ribosyltransferases have been identified in prokaryotes, eukaryotes and even viruses. The enzymatic activities of the bacterial exotoxins have been the best-characterized; for example, cholera toxin and pertussis toxin are bacterial proteins that ADP-ribosylate the alpha-subunits of G proteins in the infected host, downregulating GTPase activity and perturbing signaling pathways [12]. The clostridial toxins, including botulinum C2 toxin and C. perfringens iota toxin, ADP-ribosylate monomeric actin and thereby block polymerization [13]. These bacterial toxins restrict mono-ADP-ribosylation to specific amino acid acceptors and can be classified based on the identity of the ADP-ribose acceptor. Six amino acid-specific classes of bacterial ADP-ribosyltransferases have been identified to date, including arginine, asparagine, diphthamide (a modified histidine), glutamate, aspartate and cysteine specific enzymes [2,13-22].
Side chain-specific ADP-ribosyltransferases are also found in eukaryotes, where they have been implicated in diverse eukaryotic pathways. The GPI-anchored and secretory mono-ADP-ribosyltransferases represent the only eukaryotic family of ARTs characterized biochemically, with most members exhibiting side chain specificity for arginine as the site of ADP-ribosylation [23]. In addition to arginine, cysteine, asparagine and glutamate have been identified as ADP-ribose acceptors in eukaryotes [24-29]. Although the eukaryotic ARTs that have been identified to date are ectoenzymes, mono-ADP-ribosylation has been observed in intracellular compartments including the nucleus, golgi, mitochondria and endoplasmic reticulum [30-33]. The enzymes that modify these proteins, however, are not known. One putative family of eukaryotic intracellular ARTs is the sirtuin family. The sirtuins are a family of NAD+-dependent enzymes that function in various biological pathways including transcriptional silencing, DNA recombination, fat mobilization and apoptosis [34-37]. Most sirtuins are NAD+-dependent deacetylases that consume one molecule of NAD+ for each deacetylated lysine, generating nicotinamide and the product, O-acetyl-ADP-ribose (OAADPR) [38].
In addition to their deacetylase activity, several sirtuin family members exhibit mono-ADP-ribosyltransferase activity, including yeast SIR2, hSIRT4, mSIRT6, PfSIR2 and LiSIR2RP1 [39-42]. Indeed, the earliest reports of sirtuin enzymatic activity suggested that these proteins were mono-ADP-ribosyltransferases: CobB, the sirtuin from Salmonella typhimurium [43], was shown to exhibit phosphoribosyltransferase activity in vitro. Even the founding member of the sirtuin family, yeast SIR2, was shown to ADP-ribosylate histones and bovine serum albumin [41]. However, the finding of a more robust deacetylation activity for ySIR2 [44] called into question the validity of sirtuin ART activity, and it was suggested that sirtuin ADP-ribosylation occurs a side reaction of the deacetylation mechanism or via nonenzymatic reaction with OAADPR [45-47]. A recent study indicates sirtuin ADP ribosyltransferase activity to be 3-5 orders of magnitude weaker than the deacetylase activity [48]. Nevertheless, there is evidence that at least some members of the sirtuin family possess only ADP-ribosyltransferase activity. Human SIRT4 has no detectable deacetylase activity [49] but instead can ADP-ribosylate glutamate dehydrogenase (GDH), which culminates in reduced insulin secretion [49,50]. This clearly indicates that SIRT4 ADP-ribosylation is not due to an uncoupling of the deacetylation mechanism and though the reaction is relatively weak in vitro [46], it elicits biological effects in vivo.
While several studies have reported ADP-ribosylation in vitro by sirtuins, none have identified the targeted side chains. TbSIR2 is a chromatin-associated sirtuin possessing deacetylase and ADP-ribosyltransferase activity and has been shown to function in telomeric silencing and DNA repair [48,51,52]. Garcia-Salcedo and co-workers indicate that histone ADP-ribosylation by TbSIR2 may function in DNA repair [51]. Determining the amino specificity of sirtuin mono-ADP-ribosylation will be important towards uncovering its role in biological pathways. If sirtuin ADP-ribosylation is truly as side reaction occurring during deacetylation as theorized, we would not expect the reaction to exhibit amino acid specificity. We have assessed the amino acid specificity of TbSIR2 ADP-ribosylation and probed its dependence on substrate acetylation. Using histone H1.1 as an in vitro substrate we find that TbSIR2 ADP-ribosylation does not require an acetylated substrate, indicating that the enzyme possesses intrinsic ADP-ribosylation activity. Consistent with previous reports [48,53], we find that acetylated substrate, H1.1, is more highly ADP-ribosylated that the unacetylated form. We present results from peptide microarrays and enzymatic assays demonstrating that ADP-ribosylation of the unacetylated substrate is specific for arginine side chains, and provide direct, mass spectrometry evidence for the presence of the modification. In characterizing the mechanism of this reaction, we used ab initio quantum mechanical/molecular mechanical (QM/MM) molecular dynamics simulations. We found that sirtuin ADP-ribosylation of arginine would be energetically feasible but several orders slower than the corresponding deacetylation reaction, which is consistent with available biochemical results and explains why the deacetylase activity is more robust. The simulation results indicate a concerted mechanism for the ADP-ribosylation reaction with a highly dissociative transition state. The resulting product is found to have a lower free energy than the enzyme reactant complex, indicating that the ADP-ribosylation at arginine is stable and that the reaction would have low sensitivity to exogenous nicotinamide. In contrast with our experiments on unacetylated substrates, we find that arginine is not the principal ADP-ribose acceptor in the reaction with acetylated H1.1, suggesting that this reaction proceeds by a mechanism distinct from the ADP ribosylation of arginine in unacetylated substrates. We discuss the possible differences in the mechanism of ADP-ribosylation of acetylated substrates.
Results
TbSIR2 has intrinsic ADP-ribosyltransferase activity
Previous studies had indicated that the Trypanosoma brucei sirtuin, TbSIR2RP1 (abbreviated hereafter as TbSIR2) ADP-ribosylates core histones [48,51]. We tested human histone H1.1 as a substrate for ADP-ribosylation. We first verified that recombinant TbSIR2 had deacetylation activity using an acetylated p53 peptide and chemically acetylated recombinant histone H1.1 as substrates (see Methods and Fig. S1). Deacetylation of the peptide was assessed by a fluorescent NAD+ consumption assay (Fig. S2A), with TbSIR2 displaying robust activity. As expected, mutation of the active site histidine (H142Y) to tyrosine significantly reduces deacetylation activity (Fig. S2A) [51]. Incubating chemically acetylated histone H1.1 with recombinant TbSIR2 generated the deacetylation reaction products, nicotinamide and O-acetyl-ADP-ribose, in addition to ADP-ribose (Fig. S2B and C), confirming that the TbSIR2 enzyme was active. To determine if TbSIR2 could ADP-ribosylate an unacetylated substrate, reactions were performed using radiolabeled NAD+ (32P-NAD+) and unacetylated recombinant H1.1 as a substrate. As shown in Figure 1A, TbSIR2 ADP-ribosylated unacetylated H1.1. Consistent with previous reports, mutation of the TbSIR2 active site histidine, H142, to tyrosine [41,51] caused a significant loss of ADP-ribosylation activity (Fig. 1A lanes 1, 3 and 4). Additionally, the reaction exhibits dependence on TbSIR2 concentration (Fig. S3). These results demonstrate that TbSIR2 possesses genuine ADP-ribosyltransferase activity. Incorporation measurements show that wild type TbSIR2 is at least three times as efficient as the catalytic mutant (Fig. 1B). ADP-ribosylation activity was inhibited by increasing nicotinamide concentrations (Fig. 1C), further indicating that the ADP-ribosylation activity is enzymatic. However, high nicotinamide concentrations, well in excess of NAD+, were required to appreciably inhibit the reaction. Treatment of labeled histone H1.1 with snake venom phosphodiesterase-I removes virtually all of the radioactivity (Fig. 1D), indicating the presence of a pyrophosphate bond in the label and suggesting that the labeling is due to the covalent attachment of ADP-ribose to the protein.
Figure 1.

TbSIR2 ADP-ribosylates unacetylated H1.1.
(A) Autoradiograph of the enzyme-dependent ADP-ribosylation of unacetylated H1.1. Reactions were carried out using wild type TbSIR2. The catalytic mutants were used as control reactions. (B) 32P-incorporation into unacetylated H1.1 was determined for wild type TbSIR2 and the catalytic mutant TbH142Y. (C) TbSIR2 ADP-ribosylation of unacetylated H1.1 displays concentration dependent inhibition with nicotinamide. (D) Phosphodiesterase treatment removes the radiolabel from unacetylated H1.1 treated with TbSIR2 and 32P-NAD+. (E) Autoradiograph of ADP-ribosylation reactions using acetylated and unacetylated histone H1.1 as substrates.
Previous reports indicated that core histones are preferentially ADP-ribosylated when acetylated [53]. We therefore compared the labeling of unacetylated histone H1.1 with chemically acetylated substrate. As shown in Figure 1E, we find that chemically acetylated histone H1.1 exhibits more labeling than the unacetylated protein.
Arginine is the major ADP-ribose acceptor in unacetylated substrates
Peptide microarray technology was employed to identify unacetylated peptides derived from histone H1.1 that are ADP-ribosylated by TbSIR2. The enzyme was incubated with 32P-NAD+ on a peptide microarray containing overlapping 13mer peptides that span the amino acid sequence of residues 1 – 104 of bovine histone H1.1 (Fig. 2A). Peptides with high signal intensity were considered as substrates for TbSIR2 (data not shown). Four of the peptides, P12, P39, P40 and P44, that were labeled in this assay were re-synthesized and used in a solution-based ADP-ribosylation assay using radiolabeled NAD+ (Fig. 2B). Peptide P19, which showed no apparent labeling on the microarray, was used as negative control.
Figure 2.
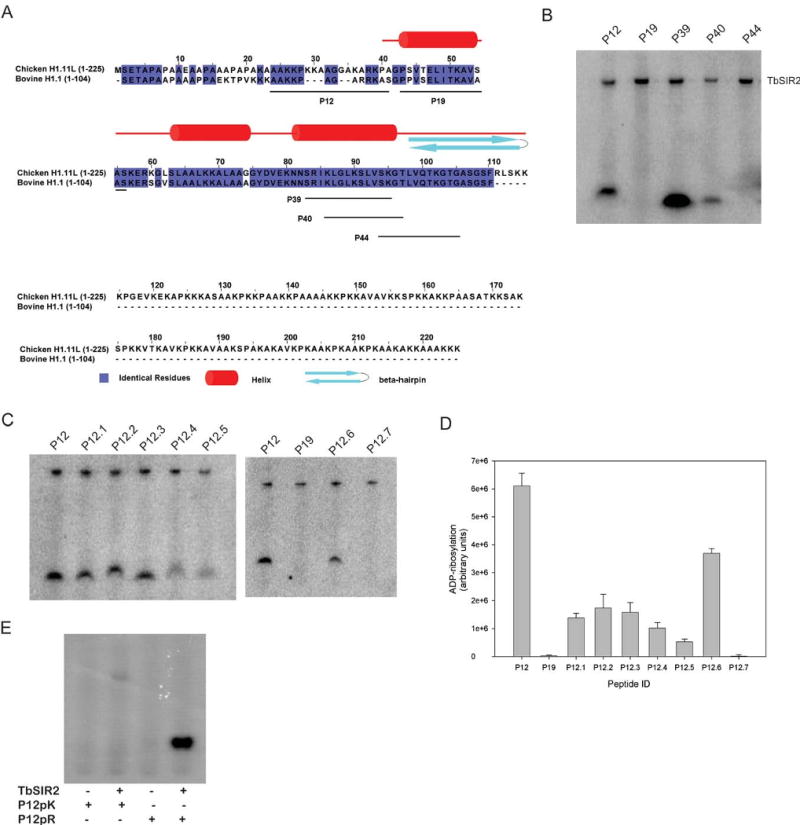
TbSIR2 preferentially labels arginine containing peptides.
(A) Sequence alignment of bovine histone H1.1 with chicken histone H1.11L. Chicken histone H1.11L is the closest homolog to bovine H1.1, whose structure has been partially determined. The identical residues are highlighted. The peptides used in ADP-ribosylation assays are underlined on the sequence of bovine H1.1. (B) Peptides which exhibited robust labeling from the microarray were used in solution-based assays. Peptide P19 was used as a negative control. (C) Amino acid substitutions of peptide P12 were used to determine the identity of the ADP-ribose acceptor. A double arginine variant of P12 was also tested. (D) ADP-ribosylation of the variant peptides was quantified using densitometry. (E) Autoradiograph of the ADP-ribosylation reactions of a poly-lysine (P12pK) and a poly-arginine (P12pR) peptide.
ADP ribosylation assays of peptides were carried out in solution and resolved by gel electrophoresis (Fig. 2B). Peptides P12 and P39 exhibit the highest levels of labeling, with P40 showing significantly lower levels and P44 showing no labeling. Interestingly, peptides P39 and P40 have a sequence overlap of 11 residues, suggesting that intense labeling of P39 may be due to the presence of the first two residues, serine and arginine, which are missing in peptide P40. Given that serine is common to all the peptides tested, including the negative control, we postulated that the increased labeling of P39 as compared with P40 might be due to the additional presence of arginine.
Since arginine is known to be modified by other classes of eukaryotic ADP-ribosyltransferases [23], we hypothesized that the robust labeling of peptides P12 and P39 might be due to the preferential ADP-ribosylation of arginine by TbSIR2. To test this, we synthesized peptides containing alanine point substitutions at each of the potential acceptor residues in peptide P12 (Table 2) and incubated the peptides with 32P-NAD+ and TbSIR2 (Fig. 2B). Significant peptide ADP-ribosylation occurs despite replacing serine (P12.6) or lysine residues (P12.1 – P12.3) with alanine (Fig. 2C and D). In contrast, peptides with substitutions at arginine residues (P12.4 and P12.5) show a dramatic reduction in labeling, suggesting that TbSIR2 ADP-ribosylates both arginines in the unaltered peptide. When both arginines are substituted with alanine (P12.7), we observe a complete lack of ADP-ribosylation, similarly implicating arginine as the acceptor residue.
Table 2.
Amino acid sequences of P12 variants used in identifying the ADP-ribose acceptor.
| Peptide Identification | Amino acid sequence |
|---|---|
| P12.1 | AAAKPAGARRKAS |
| P12.2 | AAKAPAGARRKAS |
| P12.3 | AAKKPAGARRAAS |
| P12.4 | AAKKPAGAARKAS |
| P12.5 | AAKKPAGARAKAS |
| P12.6 | AAKKPAGARRKAA |
| P12.7 | AAKKPAGAAAKAS |
| P12pR | AARRPAGARRRAR |
| P12pK | AAKKPAGAKKKAK |
Early investigations characterizing mono-ADP-ribosyltransferases used free amino acids or amino acid homopolymers as substrates in order to determine the amino acid specificity of individual proteins. Many of the arginine-specific enzymes, including the bacterial toxins and the membrane associated eukaryotic ADP-ribosyltransferases, were characterized using these strategies [3,7,54]. Since the amino acid homopolymers were amenable to mono-ADP-ribosyltransferase reactions, we tested the poly-lysine and poly-arginine peptides, P12pK and P12pR (Table 2), to assess the amino acid specificity of TbSIR2 ADP-ribosylation (Fig. 2E). The poly-arginine peptide exhibits robust, enzyme-dependent ADP-ribosylation as compared to the poly-lysine peptide, further supporting the arginine specificity of TbSIR2.
Mass spectrometry and enzymatic cleavage provides direct evidence for arginine ADP-ribosylation
Previous studies [7,55], as well as the experiments described above, relied on indirect measures of NAD+ incorporation to demonstrate ADP-ribosylation by sirtuins. We therefore employed MALDI mass spectrometry to confirm that the labeling was indeed due to the covalent attachment of ADP-ribose. Peptides P12 and P39 (Table 1) were incubated with cold NAD+ and TbSIR2 and subject to MALDI, as described Methods. As shown in Figure 3A, the peptides show mass changes of ∼540 Daltons, consistent with the addition of one or more ADP-ribose units to each peptide [9,56]. MALDI analysis of the peptide P12pR (Fig. 3B), similarly showed mass changes corresponding to the addition an ADP-ribose moiety. Multiple ADP-ribose moieties were incorporated into peptide P12pR (data not shown).
Table 1.
Amino acid sequences of peptides substrates identified using the microarray assay.
| Peptide Identification | Amino acid sequence |
|---|---|
| P12 | AAKKPAGARRKAS |
| P19 (negative control) | PPVSELITKAVAA |
| P39 | SRIKLGLKSLVSK |
| P40 | IKLGLKSLVSKGT |
| P44 | VSKGTLVQTKGTG |
Figure 3.
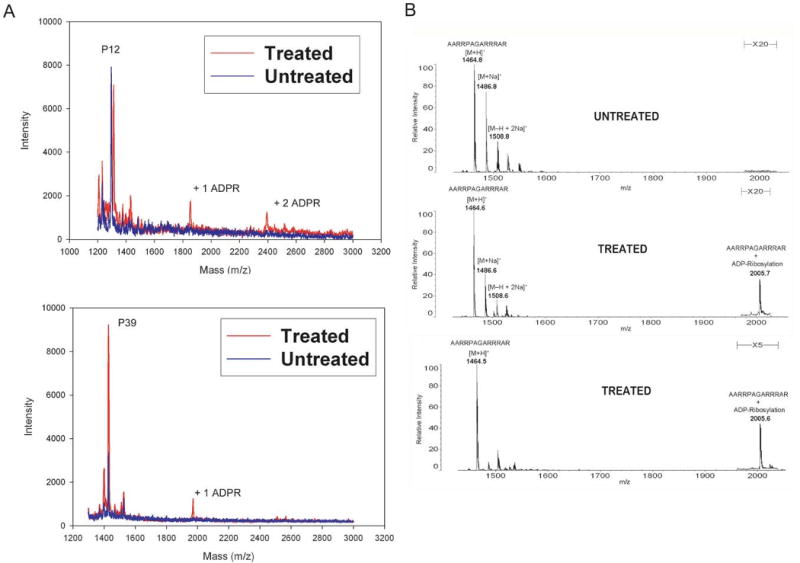
MALDI mass spectrometry confirms the incorporation of ADP-ribose in peptide substrates. (A) Peptides P12 and P39 were ADP-ribosylated in a non-radioactive assay. The treated peptides were analyzed by MALDI mass spectrometry. Samples treated with TbSIR2 show mass changes of 540 Daltons, consistent with the addition ADP-ribose moieties to the peptides. (B) MALDI mass spectrometry analysis of the ADP-ribosylation of peptide P12pR. The reactions were carried out using 20 μM peptide with 100 μM NAD+. Detection of the ADP-ribosylated P12pR was improved using higher concentrations of peptide and NAD+.
Since the peptide ADP-ribosylation by TbSIR2 pointed towards arginine as the acceptor site, we used recombinant human arginine-specific ADP-ribosylhydrolase (hADPRH), to detect the presence of ADP-ribosylarginine in protein acceptors. The hADPRH enzyme specifically cleaves ADP-ribose from arginine side chains [57]. We first confirmed the specificity of recombinant GST-hADPRH for ADP-ribosylated arginine residues. Chemically ADP-ribosylated poly-arginine (P12pR) and poly-lysine (P12pK) peptides were generated by incubating peptides with 10 mM NAD+ at high temperature, which promotes the breakdown of NAD+ and nonenzymatic ADP-ribosylation of peptides (Fig. 4A). The modified peptides were then treated with either GST-hADPRH or phosphodiesterase I and analyzed by MALDI mass spectrometry. As predicted, treatment with GST-hADPRH removes the ADP-ribosyl group from the poly-arginine peptide, but fails to remove ADP-ribose from poly-lysine (Fig. 4B). By contrast, phosphodiesterase I treatment hydrolyzes the pyrophosphate bond for ADP-ribosylarginine and ADP-ribosyllysine alike, as shown by the loss of the mono-ADP-ribosylation peaks (Fig. 4B). This assay therefore confirmed that the recombinant GST-hADPRH enzyme retains the specificity for ADP-ribosylated arginine.
Figure 4.
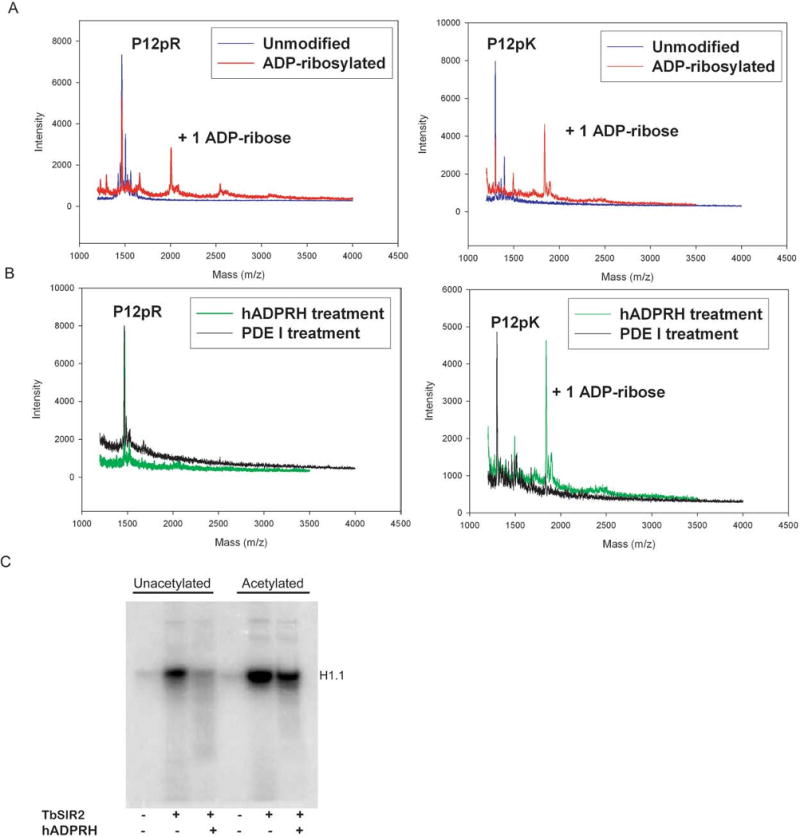
Arginine is the major ADP-ribose acceptor for unacetylated histone H1.1, but is not the major ADP-ribose acceptor for acetylated histone H1.1. (A) Nonenzymatic ADP-ribosylation of peptides P12pR and P12pK, generating an ADP-ribosylarginine substrate and an ADP-ribosyllysine substrate. (B) Enzymatic detection of ADP-ribosylarginine. Human ADP-ribosylarginine hydrolase (hADPRH) exhibits specificity for ADP-ribosylarginine, cleaving ADP-ribose from arginine side chains but not from lysine side chains. Phosphodiesterase I hydrolyzes the pyrophosphate bond for ADP-ribosylarginine and ADP-ribosyllysine. (C) Unacetylated and acetylated histone H1.1 were modified in ADP-ribosylation reactions then treated with hADPRH.
GST-tagged hADPRH was used to determine whether TbSIR2 ADP-ribosylates arginine residues in unacetylated histone H1.1 (Fig. 4C). Recombinant histone H1.1 was first incubated with TbSIR2 and radiolabeled NAD+, followed by treatment with ADPRH. As shown in Figure 4C, addition of hADPRH reduces the signal due to 32P-NAD+ to background levels, consistent with the specific cleavage of ADP-ribose from arginine side chains.
Since acetylated histone H1.1 exhibits enhanced labeling when incubated with TbSIR2 and 32P-NAD+ (Fig. 4C), we next asked whether TbSIR2-dependent ADP-ribosylation of acetylated histone H1.1 also targets arginine residues. We therefore examined whether the radioactive label could be removed by treatment with hADPRH. In contrast with the virtually complete removal of ADP-ribose from unacetylated H1.1, the radioactive label associated with acetylated H1.1 is only somewhat reduced after treatment with GST-hADPRH (Fig. 4C). We did not observe further loss of the radioactive label with longer incubation times (data not shown). This suggests that additional residues other than arginine are likely ADP-ribosylated when the H1.1 substrate is acetylated, as compared with the unacetylated form. We were unable to identify the additional ADP-ribose acceptors by mass spectrometry.
ADP-ribosylation simulations predict a highly dissociative transition state with a large activation barrier
Our results indicate that TbSIR2 can ADP-ribosylate arginine residues in unacetylated substrates, independently of the deacetylation reaction. We characterized the reaction mechanism using ab initio QM/MM molecular dynamics simulations. The Sir2Tm ternary complex structure, which contains the enzyme bound to both acetylated peptide and NAD+ [58] was used to model the reactant complex for our calculations.
We modeled the ADP-ribosylation of arginine by replacing the acetyl lysine with deprotonated arginine in the Sir2Tm ternary complex (Fig. 5A). No other alterations were made to the rest of the peptide or to the NAD+ molecule. Prior to the QM/MM calculations, the whole system was subject to a series of minimizations and equilibrations. The trajectory of this complex was quite stable over a 10 ns MD simulation.
Figure 5.
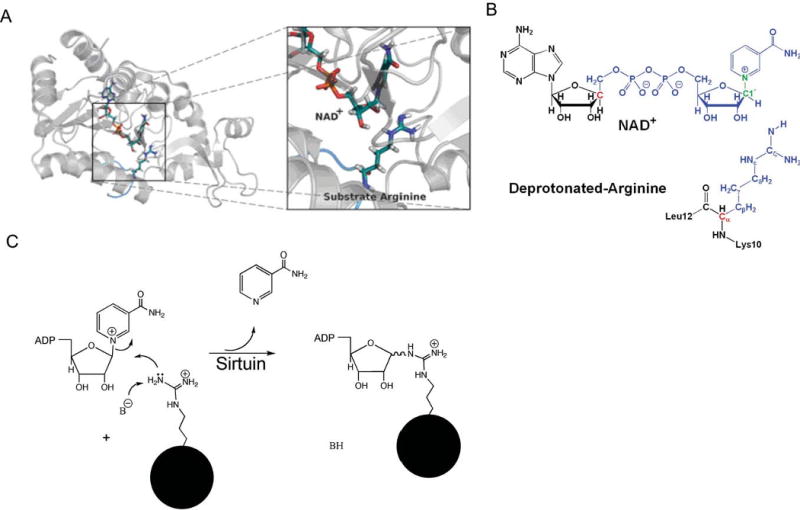
Reactant complex model for QM/MM calculations. (A) Substrate arginine binding in Sir2Tm enzyme. The overall substrate peptide binding pattern is very similar to that with acetyl-lysine substrate. (B) Illustration of the QM/MM partition in Sir2Tm ribosylation. Color notations: blue, QM subsystem; red, QM/MM boundary atoms described by improved pseudobond parameters; green, C1′-N1 bond; black, MM subsystem. (C) Proposed arginine ADP-ribosylation mechanism.
We hypothesized that the ADP-ribosylation of arginine involves breaking the C1′-N1 bond to nicotinamide and forming a Nζ–C1′ bond with arginine (Fig. 5B and C). The two-dimensional potential energy surface associated with the C1′–N1 and Nζ–C1′ bonds (Fig. 6A) indicates that this mechanism is energetically feasible and it is appropriate to choose dC1′–N1 − dNζ–C1′ as the reaction coordinate. The presence of a single transition state near the center of the map suggests that the reaction follows a concerted mechanism. It is unlikely that a stable oxacarbenium intermediate would be formed in the reaction.
Figure 6.
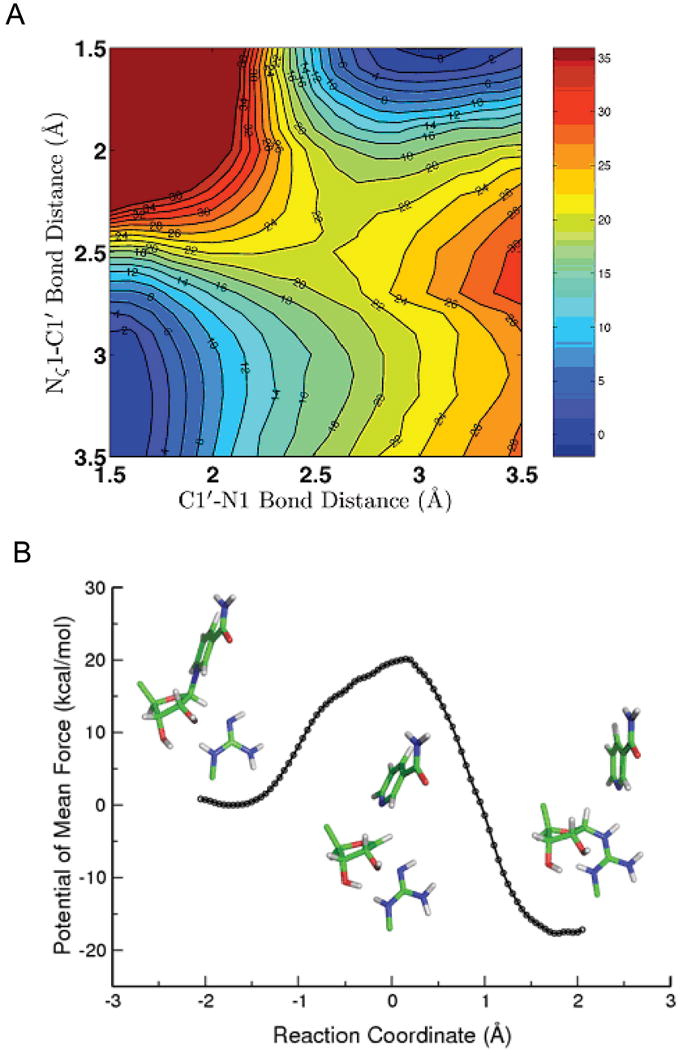
(A) Two-dimensional minimum energy surface along C1′–N1 and Nζ–C1′ bonds. The reactant is on the lower left corner, the product is on the upper right corner, and the transition state is well located in the middle. (B) Potential of mean force (PMF) for the Sir2Tm catalyzed ribosylation determined with B3LYP/6-31G(d) QM/MM MD simulations. The active site geometries at reactant, transition state, and product and some key bond lengths are also shown.
The calculated free energy of activation for Sir2Tm ribosylation is 20.1 kcal mol-1 (Fig. 6B), which is about 4.5 kcal mol-1 higher than the nicotinamide cleavage step catalyzed by Sir2Tm in the deacetylation reaction [59]. This suggests that the ADP-ribosylation at arginine catalyzed by Sir2 would be about 3-4 orders slower than the corresponding deacetylation reaction, consistent with experimental results. The transition state is mainly dissociative with calculated bond lengths of C1′-N1 and Nζ-C1′ are 2.67 ± 0.18 and 2.54 ± 0.18 Å, respectively. The is more dissociative than the transition state for the nicotinamide cleavage step of Sir2Tm deacetylation [59] with C1′-N1 and O-C1′ bond lengths of 2.60 ± 0.10 Å and 2.35 ± 0.10 Å. Interestingly, the enzyme contribution to transition state stabilization with the arginine substrate is comparable to that with the acetyl-lysine substrate (Fig. S4). In addition, arginine exhibits limited charge migration in progressing from reactant to transition state (Fig. 7), indicating that arginine has less of a role in forming the transition state. Meanwhile, the difference of nicotinamide charge between the transition state and the product is very small, less than 0.06 e. This indicates that the glycosidic bond is almost broken at the transition state, which is consistent with a highly dissociative transition state. This more highly dissociative transition state may explain why the activation energy barrier for the ADP-ribosylation is higher.
Figure 7.
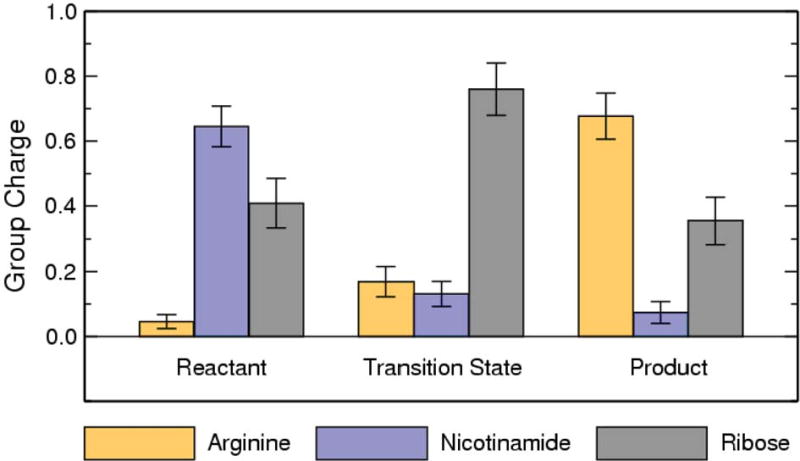
Computed group partial charges of arginine, nicotinamide, and ribose at reactant, transition state, and product. The di-phosphate portion has almost the constant partial charge of -2.10e during the reaction.
Discussion
The mechanistic basis for the ADP-ribosylation activity of sirtuins, the substrate specificity of the reaction, and the relevance of this activity in vivo has remained an ongoing subject of investigation. In this study, we provide the most direct evidence to date for enzymatic mono-ADP-ribosylation by a sirtuin and show that the trypanosomal sirtuin, TbSIR2, ADP-ribosylates arginine side chains in unacetylated substrates. The reaction is inhibited by exogenous nicotinamide, consistent with reports of other mono-ADP-ribosyltransferases [60]. While several studies have reported ADP-ribosylation in vitro by sirtuins, none have shown specificity for amino acid context. Amino acid specificity is a common trait of the mono-ADP-ribosyltransferases and uncovering the linkage specificity of sirtuin mono-ADP-ribosylation will be important in studying the potential biological role of this modification.
Biochemical data and QM/MM calculations support a mechanism of direct ADP-ribosyltransfer to an arginine acceptor residue (Fig. 9A). This reaction can be understood in the context of the NAD+-dependent deacetylation reaction [38]. During deacetylation, the acetyl-lysine promotes binding of the nicotinamide of NAD+ in the adjacent C pocket [58]. Interactions with the enzyme destabilize the glycosidic bond and promote formation of an oxacarbenium-like transition state, which is attacked by the acetyl oxygen at the C1′ of NAD+ to form a short-lived 1′-O-alkylamidate intermediate [61,62] (Fig. 9B), while nicotinamide is released. Following this step, the 2′-hydroxyl of the ribose attacks the O-alkylamidate intermediate to yield the deacetylated lysine and a 1′,2′-bicyclic intermediate, which is then hydrolyzed to yield 2′-O-acetyl-ADP-ribose (OAADPR). Our calculations support a model in which the arginine side chain replaces the acetyl lysine in the active site, and that the arginine attacks the developing oxacarbenium-like ion to form a stable ADP-ribosylated arginine. By analogy with the bacterial exotoxins, the transition state of this reaction is likely to be highly dissociative [4,63-65]. Structural studies of arginine-specific ADP-ribosyltransferases suggest that the arginine is deprotonated by the enzyme during catalysis [66]. This would enhance the nucleophilicity of arginine and facilitate the attack at the C1′ of the ribose. Further work is needed to determine whether arginine is deprotonated during TbSIR2 catalyzed ADP-ribosylation. Our computational studies suggest that Sir2Tm ADP-ribosylation of arginine follows a concerted but highly dissociative mechanism in which the deprotonated arginine has limited participation in formation of the transition state. Our calculations also suggest that the stability of ADP-ribosylation product makes the reverse reaction unfavorable. This is consistent with the absence of nicotinamide exchange activity for TbSIR2 ADP-ribosylation (Fig. S6).
The enhanced ADP-ribosylation observed with acetylated histone H1.1 can also be understood in the context of the NAD+-dependent deacetylation reaction [38]. The 1′-O-alkylamidate intermediate of the deacetylation reaction is short-lived and sensitive to attack by nicotinamide re-binding to the C pocket, regenerating NAD+ in a reaction known as base exchange [67]. The 1′-O-alkylamidate intermediate is also susceptible to methanolysis, yielding 1′-O-methyl-ADP-ribose [68,69]. This raises the possibility that side chain nucleophiles could also react with the deacetylation intermediate to give an ADP-ribosylated product (Fig. 9C), as has been previously suggested [47,62]. The enhanced labeling of acetylated histone H1.1 may therefore be due to the attack of neighboring H1.1 side chains on the O-alkylamidate intermediate. Since only a modest proportion of the ADP-ribose was removed from acetylated H1.1 by hADPRH, an arginine-specific ADP ribose hydrolase, side chains other than arginine are likely to have been modified by this alternate ADP ribosylation mechanism. [70,71]. Based on the amino acid sequence of histone H1.1 several nucleophilic side chains could possibly react with the O-alkylamidate, including lysine, serine, asparagine, and glutamate. We note the high concentrations of 2 putative acceptors: over 25% of the histone H1.1 sequence consists of lysines; serine makes up over 10% of the amino acid content. We attempted to determine the identities of the major ADP-ribose acceptors in acetylated H1.1 by analyzing tryptic peptides with mass spectrometric methods. We did not identify the acceptor residues, possibly due to the low abundance of the modification on individual peptides and the loss of ADP-ribose under our experimental conditions.
We cannot rule out the possibility of additional nonenzymatic labeling of side chains by O-acetyl-ADP-ribose that is generated by the NAD+-dependent deacetylation reaction catalyzed by TbSIR2. This form of ADP ribosylation would only occur in the case of labeling of acetylated substrates. Nonenzymatic labeling by OAADPR has previously been reported [48], although the side chain specificity is not known. Further studies will be needed to identify the side chains that are modified in the deacetylation-dependent reaction, along with the spatial relationship between those side chains and acetyl lysines. However, sirtuins such as hSIRT4 [49] that do not have detectable deacetylation activity, but carry out protein ADP-ribosylation are unlikely to utilize the deacetylation-dependent mechanism to facilitate ADP-ribosyl transfer. Assuming that they do not generate the 1′-O-alkylamidate intermediate, these sirtuins would ADP-ribosylate proteins only through a direct mechanism.
Arginine-specific ADP-ribosylation has been shown to regulate protein function in many pathways. In nitrogen-fixing bacteria ADP-ribosylation of a specific arginine in dinitrogen reductase inhibits the enzyme's ability to reduce nitrogen to ammonia. The enzyme is fully reactivated by an ADP-ribosylarginine hydrolase known as dinitrogenase reductase activating glycohydrolase [72]. Desmin, the intermediate filament protein in eukaryotes, is endogenously ADP-ribosylated at arginine [73-75]. Mono-ADP-ribosylation prevents desmin from assembling into intermediate filaments and causes existing filaments to disassemble. Arginine ADP-ribosylation has also been detected in the nucleus [76]. Arginine-linked ADP-ribose was detected in histone H3 and H4 [53] and arginine 33 was identified as the ADP-ribosylation site for histone H1.3 [26]. Our studies demonstrate that TbSIR2, a nuclear sirtuin, has arginine-specificity in the ADP-ribosyltransferase reaction. Assuming this specificity is conserved among eukaryotes, other nuclear sirtuins may be responsible for some arginine-specific mono-ADP-ribosylation in the nucleus.
Methods
Plasmid constructs
Trypanosoma brucei genomic DNA was used as a template for PCR. TbSIR2RP1 was amplified using primers 5′-GAAAACCTGTATTTTCAGGGCATGACAGAA CCGAAGTTAGC-3′ and 5′-GTGATGGTGATGGTGGTGACCCTCAACGACTTTTTCC-3′ to incorporate an N-terminal TEV cleavage site and a C-terminal hexahistidine tag. In a subsequent PCR, the gene was amplified with primers 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCT CCGAAAACCTGTATTTTCAGGGC-3′ and 5′- GGGGACCACTTTGTACAAGAAAGCTGG GTATCAG TGATGGTGATGGTGGTG-3′ to introduce attB recombination sites to facilitate recombination cloning. Using the Gateway system (Invitrogen), recombination reactions were used to insert the full length TbSIR2RP1 sequence into a pMAL c2g vector (NEB) that was modified for recombination cloning. The final construct encoded full length TbSIR2 with a TEV-cleavable N-terminal maltose binding protein tag and a C-terminal hexahistidine tag.
TbSIR2 mutants were generated using the Change-IT Multiple mutation site-directed mutagenesis kit (USB) as described by the manufacturer. The active site histidine was changed to tyrosine, producing the point mutant TbH142Y. A double mutant, named TbDm, consisting of the catalytic histidine mutant (H142Y) and mutation of aspartate 125 to asparagine (D125N), was also generated.
The human H1.1 construct, in pET11a, was a gift from Dr. Katherine Wilson (Johns Hopkins University, School of Medicine). The Human ADP-ribosylarginine hydrolase construct, in pGEX-2T, was provided by Dr. Joel Moss (National Heart, Lung and Blood Institute, National Institute of Health).
Protein Expression and Purification
Plasmids encoding wild type and mutant TbSIR2 were transformed into E. coli BL21 (DE3) RIL cells and grown in M9ZB media containing 100 μg/ml of carbenicillin and 25 μg/ml chloramphenicol at 37°C. After reaching and OD600 of about 0.9, cultures were induced with 0.5 mM isopropyl-beta-D-thiogalactopyranoside (IPTG) for 3 hours at 20°C. Cells were harvested by centrifugation and stored at -80°C prior to lysis. Thawed cells were lysed in 50 mM Tris pH 8.0, 500 mM NaCl, 20 mM imidazole, 10 mM 2-mercaptoethanol and 0.2 mM phenylmethylsulfonyl fluoride (PMSF) using a microfluidizer. The TbSIR2 constructs were purified by affinity chromatography using HisTrap columns (GE Healthcare) and dialyzed in a low salt buffer at pH 8.0. TbSIR2 was purified by anion exchange chromatography with a MonoQ column (GE Healthcare). Purified protein was dialyzed and stored in 50 mM Tris pH 8.0, 50 mM NaCl, 10 mM 2-mercaptoethanol and 15% glycerol.
Histone H1.1 was expressed in E. coli Rosetta cells. Cultures were grown in M9ZB media with 100 μg/ml of carbenicillin and 25 μg/ml chloramphenicol at 37°C and induced at an OD600 of about 0.6. Cultures were induced with 0.5 mM IPTG for 2 hours, harvested and stored at -80°C until lysis. Cells were resuspended in phosphate-buffered saline (PBS) with 0.2 mM PMSF and lysed. Perchloric acid was added to the lysate at a final concentration of 5% (v/v) and the mixture was incubated on ice for 30 minutes. After centrifugation for 20 minutes at 15,000 rpm, the supernatant was collected and the pellet was subjected to a second round of perchloric acid extraction. Trichloroacetic acid was added to the pooled supernatant to a final concentration of 20% (v/v) for 1 hour on ice. The mixture was then centrifuged for 20 minutes at 15,000 rpm and the precipitated protein was washed with cold acetone. The pellet was solubilized in 10 mM HCl, then neutralized with Tris buffer. The soluble protein was further purified by cation exchange chromatography with a SP sepharose Fast Flow column. Protein was stored in 50 mM Hepes pH 8.2, 100 mM NaCl.
GST-hADPRH was expressed in Rosetta cells (Novagen). Cultures were grown in M9ZB media with 100 μg/ml of carbenicillin and 25 μg/ml chloramphenicol at 37°C and induced at an OD600 of about 0.6. Cultures were induced with 0.5 mM IPTG for 2 hours, harvested and stored at -80°C. Thawed cells were lysed in 1 × PBS with 0.2 mM PMSF and purified using a GSTPrep Fast Flow column (GE Healthcare) as described by the manufacturer. The protein was exchanged into 50 mM Hepes pH 7.5, 200 mM NaCl, 0.2 mM PMSF buffer and further purified by size exclusion chromatography using a Superdex 200 10/300 GL column (GE Healthcare). Protein aliquots were stored at -80°C in 50 mM Hepes pH 7.5, 200 mM NaCl, 0.2 mM PMSF and 15% glycerol.
Histone H1.1 acetylation
Histone H1.1 in 50 mM Hepes pH 8.2, 100 mM NaCl was chemically acetylated using 100 mM acetic anhydride for 30 minutes at 4°C. The reaction was quenched with excess Tris pH 8.2 then dialyzed overnight in 50 mM Tris pH 8.2 with 100 mM NaCl. Acetylation was confirmed using MALDI mass spectrometry.
Peptide Synthesis
Custom made peptides were synthesized at the JHU School of Medicine Synthesis and Sequencing Facility and Pi Proteomics / Global Peptide Services, LLC.
Deacetylation Assays
A fluorescent NAD+ consumption assay was used to measure deacetylase activity [77]. NAD+ standard solutions ranging from 0 to 100 μM were used to produce a calibration curve as follows: fifty microliters of the NAD+ solutions were added in triplicate to a 96-well flat bottomed fluorescence plate and 20 μL of 2M KOH solution followed by 20 μL of 20% acetophenone (in ethanol). The plate was incubated in an oven at 110°C for 5 minutes, cooled and read on a Synergy™ HT Multi-Mode Microplate Reader with an excitation wavelength of 360 nm and an emission of 460 nm. The deacetylation reactions were performed in 50 μL assay buffer containing 50 mM Tris pH 8.0 and 100 μM NAD+. The reactions were incubated with 0.25 μM recombinant enzyme, 100 μM NAD+ and increasing concentrations of acetylated p53 peptide (0 – 1000 μM) for 10 minutes at 37°C.
Deacetylation reactions with chemically acetylated H1.1 were analyzed using an HPLC assay [67]. Reactions were carried out in 250 μL containing 50 mM Hepes pH 7.5 and 400 μM NAD+. The reactions were incubated with 0.3 μM recombinant SIR2, and 4.5 μM acetylated H1.1 at 37°C for 30 minutes. Samples were quenched by flash freezing in liquid nitrogen. Prior to analysis by HPLC each sample was thawed and combined with 250 μL of 20 mM ammonium acetate pH 7.5. The samples were assayed for deacetylation products by HPLC using a Waters Nova-Pak®C18 column with an isocratic gradient of 20 mM ammonium acetate pH 7.5. Chromatograms were collected at a wavelength of 254 nm with the Waters 2996 Photodiode array.
ADP-ribosylation Assays
The radioactive reactions contained 0.3 μM recombinant SIR2 in 50 mM Tris pH 8.0, 100 mM NaCl, 10 mM DTT, 20 μM NAD+ and 5 μCi of 32P-NAD+. Unless specified, reactions contained 5 μg unacetylated or acetylated histone H1.1. Peptide substrates were maintained at 300 μM. Reactions with histone H1.1 were incubated for 2 hours at 37°C, while the reactions with peptide substrates were incubated for 4 hours. The samples were quenched by acetone precipitation followed by 3 acetone washes. For SDS-PAGE analysis, the reactions were mixed in sample buffer, boiled for 1 minute and resolved on 4-12% Bis-Tris NuPAGE gels (Invitrogen). Gels were stained with Coomassie blue, dried and exposed to a phosphor screen. Histone H1.1 ADP-ribosylation was quantitated by densitometry using ImageQuant software, version 5.2 (Molecular Dynamics) with background correction. A 32P-NAD+ standard curve and was used to determine the amount of 32P incorporation. Quantification of peptide ADP-ribosylation was determined using ImageQuant software, version 5.2 (Molecular Dynamics) and the results expressed in terms of arbitrary units. The reactions were carried out in triplicate.
ADP-ribosylation of bovine histone H1.1 peptide microarrays (Jerini Peptide Technologies) were performed in assay buffer containing 50 mM Tris pH 8.0, 100 mM NaCl, 10 mM DTT, 330 μCi of 32P-NAD+ and 5 μM TbSIR2. The peptide arrays were sealed with Gene-Frame™ incubation chambers (Abgene), and the chambers were filled with 330 μl of assay buffer. The peptide arrays were incubated at 37°C for 2 – 2.5 hours. The microarrays were washed once with 0.1 M phosphoric acid for 3 minutes followed by duplicate washes with 0.1% Sodium Dodecyl Sulfate (SDS), de-ionized water and methanol. The glass slide was then dried at room temperature. Incorporated radioactivity was detected by exposing the microarrays to a phosphorscreen overnight at -80°C, followed by readout with a Phosphor Imager.
Non-radioactive ADP-ribosylation of peptides was performed in 50 mM Hepes pH 7.5, 10 mM DTT with 0.5 – 1 mM NAD+, 250 μM peptide and 2 μM TbSIR2. The reactions were incubated at 37°C for 15 – 30 minutes and quenched by acetone precipitation followed by 3 acetone washes. Samples were dried, resuspended in 50% acetonitrile and submitted for Matrix Assisted Laser Desorption/Ionization (MALDI) mass spectrometry.
Nonenzymatic ADP-ribosylation of a poly-lysine peptide and a poly-arginine peptide was performed in 50 mM Hepes pH 7.5 with 1 mM NAD+ and 250 μM peptide. The reactions were incubated at 80°C for 20 minutes, cooled and subjected to acetone precipitation followed by 3 acetone washes. The samples were dried for use in de-ribosylation assays.
Nicotinamide Inhibition Assay
Nicotinamide sensitivity of the ADP-ribosylation reaction was assessed using the aforementioned radioactive assay. Reactions were performed using 20 μg unacetylated histone H1.1 and peptide P39 with or without nicotinamide at the concentrations indicated. The samples were quenched by acetone precipitation, washed with acetone, dried and analyzed by SDS-PAGE. Gels were stained with Coomassie blue, dried and exposed to a phosphor screen to obtain the autoradiograph.
De-ribosylation assays
The phosphodiesterase reaction was performed in 50 mM Hepes pH 7.5 and 10 mM MgCl2 with 1.5 μM snake venom phosphodiesterase I. Reactions with human ADP-ribosylarginine hydrolase were carried out in 50 mM Hepes pH 7.5 and 10 mM MgCl2 using an enzyme concentration of 2 μM.
Mass Spectrometry
Matrix Assisted Laser Desorption/Ionization (MALDI) mass spectrometry was performed at the Middle-Atlantic Mass Spectrometry Laboratory. One microliter of the peptide sample was applied to the MALDI sample plate and allowed to air dry. Upon air drying, 1.0 μL of alpha-cyano-4-hydroxycinnamic acid (10 mg/mL) in 70% acetonitrile and 0.1% trifluoroacetic acid (TFA) was added and allowed to dry. All samples were analyzed using a Shimadzu Corporation (Manchester, UK) Axima TOF2 mass spectrometer. MALDI TOF mass spectra were acquired in linear mode with a 337 nm N2 laser. Each spectrum was acquired as average of 150 profiles (10 shots per profile). The instrument was externally calibrated using a four-point calibration with Bradykinin ([M + H]+mono = 757.39), Angiotensin II ([M + H]+mono = 1046.54), P14R ([M + H]+mono = 1533.85), and ACTH fragment 18-39 ([M + H]+mono = 2465.19). Samples were also analyzed by MALDI TOF Mass spectrometry at the JHU Mass Spectrometry and Proteomics Facility. Peptides samples were prepared as described above. Protein samples were applied to the MALDI sample plate and allowed to air dry. One microliter of sinapinic acid (10 mg/mL) in 50% acetonitrile in 0.05% TFA was applied to the sample spots and allowed to air dry. Samples were analyzed using a Voyager DE-STR MALDI TOF MS (Applied Biosystems, Framingham, MA); external calibration was conducted using standard protein mixtures.
Theoretical Calculations
Combined ab initio quantum mechanical/molecular mechanical (QM/MM) molecular dynamics simulations were employed to study SIR2 ADP-ribosylation of arginine. As structures of TbSIR2RP1 were not available, the Sir2Tm ternary complex, based on the crystal structure 2H4F [58], was used for our calculations. The acetyl-lysine residue was replaced with deprotonated arginine, while the rest of the substrate remained unchanged. The protonation states of charged residues were determined with pdb2pqr [78] at pH 7. The reactant complex was solvated into a 60×60×84 Å3 solvent box with 10 Å buffer distance. Seven sodium ions were added to neutralize the net charge and the resulting system had 30935 atoms. The entire system was subject to a series of minimizations and equilibrations with periodic boundary conditions using the AMBER10 molecular dynamics package [79] with Amber99SB force field [80-82] and TIP3P model [83]. The partial charges of nonstandard residue deprotonated arginine were fitted with HF/6-31G(d) calculations and RESP module in AMBER package. The MD trajectory was very stable after 2 ns, and a snapshot at 6 ns has been employed for the subsequent QM/MM studies.
In the QM/MM model, the enzyme complex and the solvents within 27 Å of reaction center (C1′ of NAD+) were retained. The resulting system had 9096 atoms. The nicotinamide, puckered ribose and di-phosphate portions of NAD+ as well as the side chain of deprotonated arginine were treated by B3LYP functional with 6-31G(d) basis set. The QM/MM interface was described by the pseudobond approach [84,85] with the improved parameters [86]. The Amber99SB molecular mechanical force field and TIP3P water model were employed for the rest of enzyme environment and solvents, respectively. The prepared system was first minimized by QM/MM calculations, and then a two-dimensional potential energy surface associated with C1′–N1 and Nζ–C1′ bonds was mapped out. The results suggest a linear combination of these two bond lengths as the reaction coordinate: RC = dC1′–N1 − dNζ–C1′. Then for each of 24 structures along the diagonal, it was first minimized with the restraint on the corresponding reaction coordinate with ab initio QM/MM calculations, and then further equilibrated by carrying out 500 ps MD simulations with MM force field while fixing the QM region. Finally, the resulting snapshot was used as the starting structure for ab initio QM/MM MD simulation with umbrella sampling. The simulation windows covered the reaction coordinate from -2.0 to 2.0 Å, and the total potential energy was biased with a harmonic constraint of 40 ∼ 60 kcal mol-1 Å-2, centered on the successive values of the reaction coordinate. Each simulation window had been sampled for 30 ps, first 10 of which were for equilibration and the last 20 ps were used for data analysis. All the QM/MM MD simulations were performed at B3LYP/6-31G(d) QM/MM level. Finally, the probability distributions along the reaction coordinate were determined for each window and pieced together with weighted histogram analysis method (WHAM) [87-89] to obtain the potential of mean force (PMF).
All our ab initio QM/MM calculations were preformed with modified Q-Chem [90] and Tinker [91] programs. A time step of 1 fs was used, and the Newton equations of motion were integrated with Beeman algorithm [92]. In the QM/MM simulations, the spherical boundary condition had been applied over the atoms beyond 20 Å from the C1′ atom of NAD+. The cutoffs of 18 and 12 Å were employed for electrostatic and van der Waals interactions, respectively. Note that there was no electrostatic cutoff between QM and MM subsystems. The Berendsen thermostat method [93] was used to maintain the system temperature at 300 K.
Supplementary Material
Supplementary Figure S1. MALDI mass spectrometry confirms the chemical acetylation of histone H1.1.
Supplementary Figure S2. TbSIR2 can deacetylate chemically acetylated histone H1.1.
Supplementary Figure S3. ADP-ribosylation of unacetylated histone H1.1 is dependent on the concentration of TbSIR2.
Supplementary Figure S4. Calculated enzyme contributions to QM subsystem at reactant (shown in black) and transition state (shown in red) of the nicotinamide cleavage step for deacetylation and ADP-ribosylation.
Figure 8.

Proposed mechanisms for deacetylation and ADP-ribosylation catalyzed by TbSIR2. (A) Deacetylation-independent ADP-ribosylation involving the direct transfer of the ADP-ribose moiety to arginine. (B) Proposed mechanism of the NAD+-dependent deacetylation reaction. (C) Deacetylation-dependent ADP-ribosylation in which the O-alkylamidate intermediate reacts with a neighboring protein nucleophile.
Acknowledgments
We would like to thank Rocio Montes de Oca for technical assistance with histone H1.1 purification. This work was supported by the National Science Foundation (MCB-0615815) and supported in part by the National Institutes of Health Roadmap grant U54 RR020839. The computational part of work was supported by NIH (R01-GM-079223) and NSF (CHE-CAREER-0448156) to YZ, and we thank NYU-ITS and NCSA for providing computational resources.
References
- 1.Sommer N, Salniene V, Gineikiene E, Nivinskas R, Ruger W. T4 early promoter strength probed in vivo with unribosylated and ADP-ribosylated Escherichia coli RNA polymerase: a mutation analysis. Microbiology. 2000;146:2643–2653. doi: 10.1099/00221287-146-10-2643. [DOI] [PubMed] [Google Scholar]
- 2.Sekine A, Fujiwara M, Narumiya S. Asparagine residue in the rho gene product is the modification site for botulinum ADP-ribosyltransferase. The Journal of Biological Chemistry. 1989;264:8602–8605. [PubMed] [Google Scholar]
- 3.Simpson LL, Stiles BG, Zepeda HH, Wilkins TD. Molecular Basis for the Pathological Actions of Clostridium perfringens Iota Toxin. Infection and Immunity. 1987;55:118–122. doi: 10.1128/iai.55.1.118-122.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parikh SL, Schramm VL. Transition State Structure for ADP-Ribosylation of Eukaryotic Elongation Factor 2 Catalyzed by Diphtheria Toxin. Biochemistry. 2004;43:1204–1212. doi: 10.1021/bi035907z. [DOI] [PubMed] [Google Scholar]
- 5.Vecchio MD, Balducci E. Mono ADP-ribosylation inhibitors prevent inflammatory cytokine release in alveolar epithelial cells. Molecular and Cellular Biochemistry. 2008;310:77–83. doi: 10.1007/s11010-007-9667-3. [DOI] [PubMed] [Google Scholar]
- 6.Lupi R, Corda D, Di Girolamo M. Endogenous ADP-ribosylation of the G Protein b Subunit Prevents the Inhibition of Type 1 Adenylyl Cyclase. The Journal of Biological Chemistry. 2000;275:9418–9424. doi: 10.1074/jbc.275.13.9418. [DOI] [PubMed] [Google Scholar]
- 7.Inageda K, Nishina H, Tanuma Si. Mono-ADP-ribosylation of Gs by an Eukaryotic Arginine-specific ADP-ribosyltransferase Stimulates the Adenylate Cyclase System. Biochemical and Biophysical Research Communications. 1991;176:1014–1019. doi: 10.1016/0006-291x(91)90383-i. [DOI] [PubMed] [Google Scholar]
- 8.Fu ZQ, Guo M, Jeong Br, Tian F, Elthon TE, Cerny RL, Staiger D, Alfano JR. A type III effector ADP-ribosylates RNA-binding proteins and quells plant immunity. Nature. 2007;447:284–288. doi: 10.1038/nature05737. [DOI] [PubMed] [Google Scholar]
- 9.Paone G, Wada A, Stevens LA, Matin A, Hirayama T, Levine RL, Moss J. ADP ribosylation of human neutrophil peptide-1 regulates its biological properties. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8231–8235. doi: 10.1073/pnas.122238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adriouch S, Bannas P, Schwarz N, Fliegert R, Guse AH, Seman M, Haag F, Koch-Nolte F. ADP-ribosylation at R125 gates the P2X7 ion channel by presenting a covalent ligand to its nucleotide binding site. The FASEB Journal. 2008;22:861–869. doi: 10.1096/fj.07-9294com. [DOI] [PubMed] [Google Scholar]
- 11.Kawamura H, et al. P2X7 Receptor-Dependent and -Independent T Cell Death Is Induced by Nicotinamide Adenine Dinucleotide. The Journal of Immunology. 2005;174:1971–1979. doi: 10.4049/jimmunol.174.4.1971. [DOI] [PubMed] [Google Scholar]
- 12.Reisine T, Zhang YL, Sekura R. Pertussis toxin treatment blocks the inhibition of somatostatin and increases the stimulation by forskolin of cyclic AMP accumulation and adrenocorticotropin secretion from mouse anterior pituitary tumor cells. The Journal of Pharmacology and Experimental Therapeutics. 1985;232:275–282. [PubMed] [Google Scholar]
- 13.Aktories K, Wegner A. ADP-ribosylation of Actin by Clostridial Toxins. The Journal of Cell Biology. 1989;109:1385–1387. doi: 10.1083/jcb.109.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serres MH, Ensign JC. Endogenous ADP-ribosylation of proteins in Mycobacterium smegmatis. Journal of Bacteriology. 1996;178:6074–6077. doi: 10.1128/jb.178.20.6074-6077.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry. 1998;37:5296–5304. doi: 10.1021/bi972592c. [DOI] [PubMed] [Google Scholar]
- 16.Pope MR, Saari LL, Ludden PW. N-Glycohydrolysis of Adenosine Diphosphoribosyl Arginine Linkages by Dinitrogenase Reductase Activating Glycohydrolase (Activating Enzyme) from Rhodospirillum rubrum. The Journal of Biological Chemistry. 1986;261:10104–10111. [PubMed] [Google Scholar]
- 17.Oppenheimer NJ. Structural Determination and Stereospecificity of the Cholaregen catalyzed Reaction of NAD+ with Guanidines. The Journal of Biological Chemistry. 1978;253:4907–4910. [PubMed] [Google Scholar]
- 18.Moss J, Vaughn M. Mechanism of Action of Cholaragen. Evidence for ADP-ribosyltransferase activity with arginine as an acceptor. The Journal of Biological Chemistry. 1977;252:2455–2457. [PubMed] [Google Scholar]
- 19.Margarit SM, Davidson W, Frego L, Stebbins CE. A Steric Antagonism of Actin Polymerization by a Salmonella Virulence Protein. Structure. 2006;14:1219–1229. doi: 10.1016/j.str.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 20.von Olleschik-Elbheim Lars, eB A, S MA. In: Methods in Molecular Biology. Holst O, editor. Vol. 145. Humana Press Inc.; Totowa, NJ: 2000. [Google Scholar]
- 21.Hsia JA, Tsai SC, Adamik R, Yost DA, Hewlett EL, Moss J. Amino acid-specific ADP-ribosylation. Sensitivity to hydroxylamine of [cysteine(ADP-ribose)]protein and [arginine(ADP-ribose)]protein linkages. The Journal of Biological Chemistry. 1985;260:16187–16191. [PubMed] [Google Scholar]
- 22.Hochmann H, Pust S, von Figura G, Aktories K, Barth H. Salmonella enterica SpvB ADP-Ribosylates Actin at the Position Arginine-177--Characterization of the Catalytic Domain within the SpvB Protein and a Comparison to Binary Clostridial Actin-ADP-Ribosylating Toxins. Biochemistry. 2006;45:1271–1277. doi: 10.1021/bi051810w. [DOI] [PubMed] [Google Scholar]
- 23.Okazaki IJ, Zolkiewska A, Takada T, Moss J. Characterization of mammalian ADP-ribosylafion cycles. Biochimie. 1995;77:319–325. doi: 10.1016/0300-9084(96)88141-7. [DOI] [PubMed] [Google Scholar]
- 24.Ord MG, Stocken LA. Adenosine Diphosphate Ribosylated Histones. Biochemical Journal. 1977;161:583–592. doi: 10.1042/bj1610583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanigawa Y, Tsuchiya M, Imai Y, Shimoyama M. ADP-ribosyltransferase from Hen Liver Nuclei. Purification and Characterization. The Journal of Biological Chemistry. 1984;259:2022–2029. [PubMed] [Google Scholar]
- 26.Ushiroyama T, Tanigawa Y, Tsuchiya M, Matsuura R, Ueki M, Sugimoto O, Shimoyama M. Amino acid sequence of histone H1 at the ADP-ribose-accepting site and ADP-ribose*histone-H1 adduct as an inhibitor of cyclic-AMP-dependent phosphorylation. European Journal of Biochemistry. 1985;151:173–177. doi: 10.1111/j.1432-1033.1985.tb09082.x. [DOI] [PubMed] [Google Scholar]
- 27.Moss J, Stanley SJ. Amino Acid-specific ADP-ribosylation. Identification of an arginine-dependent ADP-ribosyltransferase in rat liver. The Journal of Biological Chemistry. 1981;256:7830–7833. [PubMed] [Google Scholar]
- 28.Smith JA, Stocken LA. Chemical and Metabolic Properties of Adenosine Diphosphate Ribose Derivatives of Nuclear Proteins. Biochemical Journal. 1975;147:523–529. doi: 10.1042/bj1470523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, Shilton BH, Luscher B. Substrate-Assisted Catalysis by PARP10 Limits Its Activity to Mono-ADP-Ribosylation. Molecular Cell. 2008;32:57–69. doi: 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 30.Herrero-Yraola A, Bakhit SMA, Franke P, Weise C, Schweiger M, Jorcke D, Ziegler M. Regulation of glutamate dehydrogenase by reversible ADP-ribosylation in mitochondria. The EMBO Journal. 2001;20:2404–2412. doi: 10.1093/emboj/20.10.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogata N, Ueda K, Kagamiyama H, Hayaishi O. ADP-ribosylation of Histone H1. The Journal of Biological Chemistry. 1980;255:7616–7620. [PubMed] [Google Scholar]
- 32.Leno GH, Ledford BE. Reversible ADP-ribosylation of the 78 kDa glucose-regulated protein. FEBS Letters. 1990;276:29–33. doi: 10.1016/0014-5793(90)80499-9. [DOI] [PubMed] [Google Scholar]
- 33.Silletta MG, et al. Role of brefeldin A-dependent ADP-ribosylation in the control of intracellular membrane transport. Molecular and Cellular Biochemistry. 1999;193:43–51. [PubMed] [Google Scholar]
- 34.Kobayashi T, Horiuchi T, Tongaonkar P, Vu L, Nomura M. SIR2 Regulates Recombination between Different rDNA Repeats, but Not Recombination within Individual rRNA Genes in Yeast. Cell. 2004;117:441–453. doi: 10.1016/s0092-8674(04)00414-3. [DOI] [PubMed] [Google Scholar]
- 35.Li C, Mueller JE, Bryk M. Sir2 Represses Endogenous Polymerase II Transcription Units in the Ribosomal DNA Nontranscribed Spacer. Molecular Biology of the Cell. 2006;17:3848–3859. doi: 10.1091/mbc.E06-03-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griswold AJ, Chang KT, Runko AP, Knight MA, Min KT. Sir2 mediates apoptosis through JNK-dependent pathways in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8673–8678. doi: 10.1073/pnas.0803837105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Picard F, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The Biochemistry of Sirtuins. Annual Review of Biochemistry. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 39.Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 Homolog SIRT6 Is a Nuclear ADP-ribosyltransferase. The Journal of Biological Chemistry. 2005;280:21313–21320. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- 40.Merrick CJ, Duraisingh MT. Plasmodium falciparum Sir2: an Unusual Sirtuin with Dual Histone Deacetylase and ADP-Ribosyltransferase Activity. Eukaryotic Cell. 2007;6:2081–2091. doi: 10.1128/EC.00114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An Enzymatic Activity in the Yeast Sir2 Protein that Is Essential for Gene Silencing. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 42.Tavares J, Ouaissi A, Santarem N, Sereno D, Vergnes B, Sampaio P, Cordeiro-Da-Silva A. The Leishmania infantum cytosolic SIR2-related protein 1 (LiSIR2RP1) is an NAD+-dependent deacetylase and ADP-ribosyltransferase. The Biochemical Journal. 2008;415:377–386. doi: 10.1042/BJ20080666. [DOI] [PubMed] [Google Scholar]
- 43.Tsang AW, Escalante-Semerena JC. CobB, a New Member of the SIR2 Family of Eucaryotic Regulatory Proteins, Is Required to Compensate for the Lack of Nicotinate Mononucleotide:5,6-Dimethylbenzimidazole Phosphoryltransferase Activity in cobT Mutants during Cobalamin Biosynthesis in Salmonella typhimurium LT2. The Journal of Biological Chemistry. 1998;273:31788–31794. doi: 10.1074/jbc.273.48.31788. [DOI] [PubMed] [Google Scholar]
- 44.Imai Si, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 45.Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du J, Jiang H, Lin H. Investigating the ADP-ribosyltransferase Activity of Sirtuins with NAD Analogues and 32P-NAD. Biochemistry. 2009;48:2878–2890. doi: 10.1021/bi802093g. [DOI] [PubMed] [Google Scholar]
- 47.Tanny JC, Moazed D. Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: Evidence for acetyl transfer from substrate to an NAD breakdown product. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:415–420. doi: 10.1073/pnas.031563798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kowieski TM, Lee S, Denu JM. Acetylation-dependent ADP-ribosylation by Trypanosoma brucei Sir2. The Journal of Biological Chemistry. 2008;283:5317–5326. doi: 10.1074/jbc.M707613200. [DOI] [PubMed] [Google Scholar]
- 49.Haigis MC, et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic beta Cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- 50.Ahuja N, Schwer B, Carobbio S, Waltregny D, North BJ, Castronovo V, Maechler P, Verdin E. Regulation of Insulin Secretion by SIRT4, a Mitochondrial ADP-ribosyltransferase. The Journal of Biological Chemistry. 2007;282:33583–33592. doi: 10.1074/jbc.M705488200. [DOI] [PubMed] [Google Scholar]
- 51.Garcia-Salcedo JA, Purificacion Gijon, Nolan DP, Tebabi P, Pays E. A chromosomal SIR2 homologue with both histone NAD-dependent ADP-ribosyltransferase and deacetylase activities is involved in DNA repair in Trypanosoma brucei. The EMBO Journal. 2003;22:5851–5862. doi: 10.1093/emboj/cdg553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alsford S, Kawahara T, Isamah C, Horn D. A sirtuin in the African trypanosome is involved in both DNA repair and telomeric gene silencing but is not required for antigenic variation. Molecular Microbiology. 2007;63:724–736. doi: 10.1111/j.1365-2958.2006.05553.x. [DOI] [PubMed] [Google Scholar]
- 53.Golderer G, Grobner P. ADP-ribosylation of core histone and their acetylated subspecies. The Biochemical Journal. 1991;277:607–610. doi: 10.1042/bj2770607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moss J, Garrison S, Oppenheimer NJ, Richardson SH. NAD-dependent ADP-ribosylation of Arginine and Proteins by Escherichia coli Heat-labile Enterotoxin. The Journal of Biological Chemistry. 1979;254:6270–6272. [PubMed] [Google Scholar]
- 55.Van Dop C, Yamanaka G, Steinberg F, Sekura RD, Manclark CR, Stryer L, Bourne HR. ADP-ribosylation of Transducin by Pertussis Toxin Blocks the Light-stimulated Hydrolysis of GTP and cGMP in Retinal Photoreceptors. The Journal of Biological Chemistry. 1984;259:23–26. [PubMed] [Google Scholar]
- 56.Ponnuraj RK, Rubio LM, Grunwald SK, Ludden PW. NAD-, NMN-, and NADP-dependent modification of dinitrogenase reductases from Rhodospirillum rubrum and Azotobacter vinelandii. FEBS Letters. 2005;579:5751–5758. doi: 10.1016/j.febslet.2005.09.057. [DOI] [PubMed] [Google Scholar]
- 57.Takada T, Iida K, Moss J. Cloning and SIte-directed Mutagenesis of Human ADP-ribosylarginine Hydrolase. The Journal of Biological Chemistry. 1993;268:17837–17843. [PubMed] [Google Scholar]
- 58.Hoff KG, Avalos JL, Sens K, Wolberger C. Insights into the Sirtuin Mechanism from Ternary Complexes Containing NAD+ and Acetylated Peptide. Structure. 2006;14:1231–1240. doi: 10.1016/j.str.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 59.Hu P, Wang S, Zhang Y. Highly Dissociative and Concerted Mechanism for the Nicotinamide Cleavage Reaction in Sir2Tm Enzyme Suggested by Ab Initio QM/MM Molecular Dynamics Simulations. Journal of the American Chemical Society. 2008;130:16721–16728. doi: 10.1021/ja807269j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rankin PW, Jacobson EL, Benjamin RC, Moss J, Jacobson MK. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. The Journal of Biological Chemistry. 1989;264:4312–4317. [PubMed] [Google Scholar]
- 61.Sauve AA, Celic I, Avalos J, Deng H, Boeke JD, Schramm VL. Chemistry of Gene Silencing: The Mechanism of NAD+-Dependent Deacetylation Reactions. Biochemistry. 2001;40:15456–15463. doi: 10.1021/bi011858j. [DOI] [PubMed] [Google Scholar]
- 62.Denu JM. The Sir2 family of protein deacetylases. Current Opinion in Chemical Biology. 2005;9:431–440. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 63.Berti PJ, Blanke SR, Schramm VL. Transition State Structure for the Hydrolysis of NAD Catalyzed by Diphtheria Toxin. Journal of the American Chemical Society. 1997;119:12079–12088. doi: 10.1021/ja971317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou GC, Parikh SL, Tyler PC, Evans GB, Furneaux RH, Zubkova OV, Benjes PA, Schramm VL. Inhibitors of ADP-ribosylating bacterial toxins based on oxacarbenium ion character at their transition states. Journal of the American Chemical Society. 2004;126:5690–5698. doi: 10.1021/ja038159+. [DOI] [PubMed] [Google Scholar]
- 65.Rising KA, Schramm VL. Transition State Analysis of NAD+ Hydrolysis by the Cholera Toxin Catalytic Subunit. Journal of the American Chemical Society. 1997;119:27–37. [Google Scholar]
- 66.Han S, Craig JA, Putnam CD, Carozzi NB, Tainer JA. Evolution and mechanism from structures of an ADP-ribosylating toxin and NAD complex. Nature Structural Biology. 1999;6:932–936. doi: 10.1038/13300. [DOI] [PubMed] [Google Scholar]
- 67.Sauve AA, Schramm VL. Sir2 Regulation by Nicotinamide Results from Switching between Base Exchange and Deacetylation Chemistry. Biochemistry. 2003;42:9246–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]
- 68.Smith BC, Denu JM. Sir2 Protein Deacetylases: Evidence for Chemical Intermediates and Functions of a Conserved Histidine. Biochemistry. 2006;45:272–282. doi: 10.1021/bi052014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.French JB, Cen Y, Sauve AA. Plasmodium falciparum Sir2 is an NAD+-Dependent Deacetylase and an Acetyllysine-Dependent and Acetyllysine-Independent NAD+ Glycohydrolase. Biochemistry. 2008;47:10227–10239. doi: 10.1021/bi800767t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cervantes-Laurean D, Minter DE, Jacobson EL, Jacobson MK. Protein Glycation by ADP-Ribose: Studies of Model Conjugates. Biochemistry. 1993;32:1528–1534. doi: 10.1021/bi00057a017. [DOI] [PubMed] [Google Scholar]
- 71.Cervantes-Laurean D, Jacobson EL, Jacobson MK. Glycation and Glycoxidation of Histones by ADP-ribose. The Journal of Biological Chemistry. 1996;271:10461–10469. doi: 10.1074/jbc.271.18.10461. [DOI] [PubMed] [Google Scholar]
- 72.Saari LL, Pope MR, Murrell SA, Ludden PW. Studies on the activating enzyme for iron protein of nitrogenase from Rhodospirillum rubrum. The Journal of Biological Chemistry. 1986;261:4973–4977. [PubMed] [Google Scholar]
- 73.Huang HY, Graves DJ, Robson RM, Huiatt TW. ADP-Ribosylation of the intermediate filament desmin and inhibition of desmin assembly in vitro by muscle ADP-ribosyltransferase. Biochemical and Biophysical Research Communications. 1993;197:570–577. doi: 10.1006/bbrc.1993.2517. [DOI] [PubMed] [Google Scholar]
- 74.Yuan J, Huiatt TW, Liao CX, Robson RM, Graves DJ. The Effects of Mono-ADP-Ribosylation on Desmin Assembly-Disassembly. Archives of Biochemistry and Biophysics. 1999;363:314–322. doi: 10.1006/abbi.1998.1096. [DOI] [PubMed] [Google Scholar]
- 75.Huang HY, Zhou H, Huiatt TW, Graves DJ. Target Proteins for Arginine-Specific Mono(ADP-Ribosyl) Transferase in Membrane Fractions from Chick Skeletal Muscle Cells. Experimental Cell Research. 1996;226:147–153. doi: 10.1006/excr.1996.0213. [DOI] [PubMed] [Google Scholar]
- 76.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-Ribosylation Reactions in Mammalian Cells: Where Are We Today and Where Are We Going? Microbiology and Molecular Biology Reviews. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Putt KS, Hergenrother PJ. An enzymatic assay for poly(ADP-ribose) polymerase-1 (PARP-1) via the chemical quantitation of NAD+: application to the high-throughput screening of small molecules as potential inhibitors. Analytical Biochemistry. 2004;326:78–86. doi: 10.1016/j.ab.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 78.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Research. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Case DA, et al. AMBER 10. University of California; San Francisco: 2008. [Google Scholar]
- 80.Cornell WD, et al. A 2nd Generation Force-Field for the Simulation of Proteins, Nucleic-Acids, and Organic-Molecules. Journal of the American Chemical Society. 1995;117:5179–5197. [Google Scholar]
- 81.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry. 2000;21:1049–1074. [Google Scholar]
- 82.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins-Structure Function and Bioinformatics. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 84.Zhang Y, Lee TS, Yang W. A pseudobond approach to combining quantum mechanical and molecular mechanical methods. Journal of Chemical Physics. 1999;110:46–54. [Google Scholar]
- 85.Zhang Y. Pseudobond ab initio QM/MM approach and its applications to enzyme reactions. Theoretical Chemistry Accounts. 2006;116:43–50. [Google Scholar]
- 86.Zhang Y. Improved pseudobonds for combined ab initio quantum mechanical/molecular mechanical methods. Journal of Chemical Physics. 2005;122:024114. doi: 10.1063/1.1834899. [DOI] [PubMed] [Google Scholar]
- 87.Souaille M, Roux B. Extension to the weighted histogram analysis method combining umbrella sampling with free energy calculations. Computer Physics Communicaations. 2001;135:40–57. [Google Scholar]
- 88.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules .1. the Method. Journal of Computational Chemistry. 1992;13:1011–1021. [Google Scholar]
- 89.Ferrenberg AM, Swendsen RH. New Monte-Carlo Technique for Studying Phase-Transitions. Physical Review Letters. 1988 December;61 doi: 10.1103/PhysRevLett.61.2635. [DOI] [PubMed] [Google Scholar]
- 90.Shao Y, et al. Advances in methods and algorithms in a modern quantum chemistry program package. Physical Chemistry Chemical Physics. 2006;8:3172–3191. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- 91.Ponder JW. TINKER, Software Tools for Molecular Design, Version 4.2. 2004 doi: 10.1021/acs.jctc.8b00529. The most updated version for the TINKER program can be obtained from J. W. Ponder's World Wide Web site at http://dasher.wustl.edu/tinker. [DOI] [PMC free article] [PubMed]
- 92.Beeman D. Some Multistep Methods for Use in Molecular-Dynamics Calculations. Journal of Computational Physics. 1976;20:130–139. [Google Scholar]
- 93.Berendsen HJC, Postma JPM, Vangunsteren WF, Haak JR. Molecular-Dynamics with Coupling to an External Bath. Journal of Chemical Physics. 1984;81:3684–3690. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. MALDI mass spectrometry confirms the chemical acetylation of histone H1.1.
Supplementary Figure S2. TbSIR2 can deacetylate chemically acetylated histone H1.1.
Supplementary Figure S3. ADP-ribosylation of unacetylated histone H1.1 is dependent on the concentration of TbSIR2.
Supplementary Figure S4. Calculated enzyme contributions to QM subsystem at reactant (shown in black) and transition state (shown in red) of the nicotinamide cleavage step for deacetylation and ADP-ribosylation.