

Abstract
Acyl Carrier Protein (ACP) has a single reactive sulfhydryl necessary for function in covalently binding nascent fatty acids during biosynthesis. In Plasmodium falciparum, the causative agent of the most lethal form of malaria, fatty acid biosynthesis occurs in the apicoplast organelle during the liver stage of the parasite life cycle. During the blood stage, fatty acid biosynthesis is inactive and the redox state of the apicoplast has not been determined. We solved the crystal structure of ACP from P. falciparum in reduced and disulfide-linked forms, and observe the surprising result that the disulfide in the PfACP cross-linked dimer is sequestered from bulk solvent in a tight molecular interface. We assessed solvent accessibility of the disulfide with small molecule reducing agents and found that the disulfide is protected from BME but less so for other common reducing agents. We examined cultured P. falciparum parasites to determine which form of PfACP is prevalent during the blood stages. We readily detected monomeric PfACP in parasite lysate, but do not observe the disulfide-linked form, even under conditions of oxidative stress. To demonstrate that PfACP contains a free sulfhydryl and is not acylated or in the apo state, we treated blood stage parasites with the disulfide forming reagent diamide. We found that the effects of diamide are reversed with reducing agent. Together, these results suggest that the apicoplast is a reducing compartment, as suggested by models of P. falciparum metabolism, and that PfACP is maintained in a reduced state during blood stage growth.
Intro
Plasmodium falciparum is a protist parasite that causes malaria and has killed 881,000 people for the most recently tabulated year of 2006.1 This loss of life occurs despite recent reductions in malaria prevalence by intervention with insecticide treated bed nets and artemisinin-based combination drug therapy.2 Unfortunately, artemisinin drug resistance has already been observed and threatens to reverse recent reductions in malaria prevalence.3,4 Continued emphasis on malaria drug development and concomitant characterization of parasite biology are necessary to pursue the ultimate goal of malaria eradication.
Discovery that P. falciparum contains a plastid housing many proteins of prokaryotic origin led to immediate speculation that the metabolic pathways resident in this plastid, termed the apicoplast, could be targeted for drug development without side effects to the human host.5 Fatty acid biosynthesis is one such pathway that has received considerable attention since many antibiotics already exist to target this pathway.6 Two types of fatty acid synthase (FAS) pathway are found in nature. A type I FAS pathway is composed of large multifunctional proteins which catalyze all of the reactions required for de novo fatty acid synthesis, and is typically found in the cytosol of eukaryotic cells. A type II FAS pathway uses independent polypeptides to perform each catalytic step, and is typically bacterial. P. falciparum is unusual for a eukaryote in that it relies upon a complete type II pathway which is located in the apicoplast.
Central to the type II FAS pathway is a protein called acyl carrier protein (ACP). ACP acts as a hub for the type II FAS, shuttling the nascent fatty acid between enzymes of the pathway.7 All ACPs contain a flexible phosphopantetheine prosthetic group derived from coenzyme A, which is attached to a conserved serine residue. The phosphopantetheine group is attached post-translationally by holo-ACP synthase, converting apo-ACP into holo-ACP, here referred to as ACP for simplicity. Acyl groups are covalently bound to ACP by forming a thioester bond with the terminal sulfhydryl of this prosthetic group. Within bacteria such as E. coli (Ec), EcACP is thought to be monomeric, with the phosphopantetheine sulfhydryls either reduced or acylated.8 The phosphopantetheine sulfhydryl can also be readily oxidized, forming disulfide-linked ACP dimers. ACP dimer was observed in early work in E. coli9 and more recently in a selection of ACPs expressed heterologously in E. coli10, although the dimeric species is believed to form during gel electrophoresis and not in vivo.
In P. falciparum, the most dangerous form of malaria, PfACP has been localized exclusively to the apicoplast.11 PfACP contains a phosphopantetheine prosthetic group12 and can serve as a cofactor for fatty acid biosynthesis enzymes when it is reduced in the monomeric state.13 While oxidized dimers of PfACP are observed in vitro,14 presence of the dimer has not been reported in vivo in any model system. In E. coli, EcACP is present at 0.1 mM,15 and glutathione is present at 0.1–10 mM.16 When EcACP is not acylated, glutathione prevents oxidized EcACP dimers by forming an EcACP-glutathione mixed disulfide, as observed in stationary phase E. coli.9 The apicoplast shares common ancestry with chloroplasts,17 which also contain ACP functioning in a type II FAS. In spinach chloroplasts, ACP-glutathione conjugates are not observed, and ACP with free sulfhydryl was the major species in seedlings and leaves under light conditions, and acyl-ACP dominates in leaves left in the dark.18
During the human blood stage of P. falciparum, it is not clear that oxidized PfACP dimer can be prevented by the same mechanisms used in either bacteria or chloroplasts. Inactivity of FAS during the blood stage19,20 means that acyl-PfACP species are probably not synthesized. Since glutathione biosynthesis occurs in the cytosol of P. falciparum,21 PfACP-glutathione mixed disulfides are probably not formed in the apicoplast of the parasite. One candidate for maintaining a pool of reduced PfACP in the apicoplast is ferredoxin. Ferredoxin has been localized to the apicoplast and has been proposed as a source of reducing equivalents for the organelle.22 While metabolic models predict that many enzymes located in the apicoplast require a reducing environment to function, explicit experimental data on the redox potential within the apicoplast have not yet been described.
To address the biochemical and structural implications of an oxidized PfACP dimer, here we report the crystal structures of PfACP in both the reduced and oxidized disulfide-linked states. Surprisingly, we find constituents of the disulfide-linked dimer to be in close molecular contact, raising questions about accessibility of the disulfide bond to cellular reducing agents. We examine solvent accessibility of the disulfide bond in vitro as a function of ionic strength and reducing agent, finding that the dimer interface protects the disulfide bond when probed by BME, but less so for other common reducing agents. Finally, we directly probe the oxidative state of PfACP in blood stage parasites under conditions of either normal growth or oxidative stress. Using hydrogen peroxide or the disulfide inducing compound diamide, our studies show that PfACP is maintained in the reduced state in vivo, thus suggesting that the apicoplast is a highly buffered reducing compartment.
Methods
Plasmid Construction and Protein Purification
Vector derived amino acids were removed from the PfACP construct encoded by pSTP113 using QuickChange mutagenesis (Stratagene) with primers (F) CTCGGGATCGAGGGAAGGAGCTCTTTAAAAAGTACTTTTG and (R) CAAAAGTACTTTTTAAAGAGCTCCTTCCCTCGATCCCGAG. The resulting plasmid, pSPr29, was subject to further mutagenesis to insert a tobacco etch virus (TEV) protease cleavage site (residues ENLYFQ) using primers (F) CTCGGGATCGAGGGAAGGGAAAACCTGTATTTTCAGAGCTCTTTAAAAAGTACTTTTGATGATAT and (R) ATATCATCAAAAGTACTTTTTAAAGAGCTCTGAAAATACAGGTTTTCCCTTCCCTCGATCCCGAG. The resulting plasmid, pJGa7, encodes a fusion protein of the maltose binding protein (MBP) with PfACP. Cleavage with the TEV protease liberates mature PfACP (residues S57-Q137) without any additional vector derived amino acids. Plasmid pJGa7 was used to transform BL21 Star(DE3) cells (Invitrogen) that had previously been transformed with pRIL plasmid isolated from BL21- CodonPlus(DE3) cells (Stratagene). The resulting cells were used for PfACP protein expression.
PfACP expression cells were grown to an OD600 of 0.8 in LB medium shaking at 37 °C. Protein expression was induced with 0.4 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) as the cultures were transferred to a 20 °C shaker for 10 hr. Cells were harvested by centrifugation and stored at −20 °C. Protein purification from cell pellets began by resuspending cells in 20 mL of lysis buffer (30 mM Tris pH 6.5, 1 mg/mL lysozyme, 2.5 μg/mL DNAse I, 1 mM PMSF) per liter of cell culture. Resuspended cells were lysed by sonication and the lysate was clarified by centrifugation at 4 °C for 20 min at 27000 g. The resulting supernatant was loaded on an amylose column (New England Biolabs) equilibrated with 30 mM Tris pH 6.5. The column was washed with equilibration buffer for 2 column volumes before connecting a HiTrap Q FF anion exchange column (GE Healthcare) in tandem. Proteins were then eluted from the amylose column onto the anion exchange column with 30 mM Tris pH 6.5, 500 mM glucose, and then the amylose column was removed. MBP-PfACP fusion protein was eluted from the anion exchange column with a linear gradient of NaCl from 0 to 400 mM. Fractions that contained fusion protein were pooled, and subjected to proteolytic cleavage in 1mM dithiothreitol (DTT) and 20 μg/mL TEV protease for three days at 4 °C. Cleaved MBP and uncut fusion protein were then separated from PfACP by subtractive amylose column followed by size exclusion chromatography on a S100 column (GE Healthcare) equilibrated with 30 mM Tris pH 6.5, 100 mM NaCl. Fractions containing pure PfACP were pooled and dialyzed into 20 mM Tris pH 6.5, 20 mM NaCl using a Slide-A-Lyzer dialysis window (Pierce). Dialyzed protein was concentrated to 16.7 mg/mL with an Amicon concentrator (Millipore) and flash frozen for storage at −80 °C.
Pure PfACP was used to generate rabbit antiserum using the custom antibody service of Cocalico Biologicals Inc. Briefly, 250 μg of PfACP mixed with Complete Freund’s Adjuvant was used for the initial inoculation followed by boosts of 125 μg of antigen 2, 5, 7 and 10 weeks later. Final exsanguination was performed on day 87. Specific antibodies were purified from serum using a PfACP affinity column. A 1mL NHS-activated HP column (GE Healthcare) was activated with 1 mM HCl according to the manufacturer’s instructions. Immediately after activation, 7.8 mg of PfACP in 4 mL of reaction buffer (100 mM NaCO3H, 500 mM NaCl pH 8.3) was pumped through the column at a constant rate of 0.1 mL/min. PfACP was circulated through the column for 24 hr at 4 °C at which point the release of N-hydroxysuccinimide reaction product was quantified by absorbance at 260 nM (ε=8600 M−1cm−1). The column was subsequently washed and blocked according to the manufacturer’s directions. Rabbit antiserum was prepared for antibody purification by diluting 4 mL of serum into 16 mL of phosphate buffered saline (PBS). Diluted serum was circulated over the affinity column at 4 °C for 12 hr at 0.25 mL/min. Nonspecific proteins were washed from the column with 10 mL of PBS followed by elution in 4 mL of 50 mM glycine pH 2.9 and 4 mL of 50 mM glycine pH 1.9. A total of 1.44 mg of αPfACP IgG was concentrated to 1mg/mL and stored at −20 °C in storage buffer (PBS, 50% glycerol, 0.02% NaN3).
Crystallization and Structure Determination
PfACP crystals were grown at 23 °C by the hanging drop method combining 1 μL of well solution with 1 μL PfACP at 16.7 mg/mL. Crystals were first observed in saturated NaCitrate solution at pH 7. Changing to a well solution of NaMalonate allowed higher solute concentrations to be used and resulted in improved crystal morphology.23 Crystals of disulfide-linked dimeric PfACP were obtained by the hanging drop method at 23 °C using 1 μL PfACP solution supplemented with 2 mM β-mercaptoethanol (BME), and 1 μL well solution consisting of 3.06 M NaMalonate pH 7.5 and 1 mM CuCl2. Crystal growth was visible after 3–5 days, and crystals matured over 1–3 weeks into a rectangular or wedge shaped morphology (Fig. S1A). Diffraction data were collected by capillary mount at 20 °C on a Bruker X8 Proteum (Bruker AXS Inc.). Crystals of reduced PfACP were obtained by diluting the PfACP solution 50% with 100 mM BME for 30 min at 23 °C before setting hanging drops with 1 μL PfACP/BME solution and 1 μL well solution of 3.03 M NaMalonate pH 7.2 with 80 mM BME. Crystal growth was visible after 3–5 days, and matured over 1–3 weeks into a bipyramidal morphology (Fig. S1B). Crystals were flash frozen in the diffractometer cryostream followed by collection of diffraction data at 100 K on a Bruker X8 Proteum. PfACP crystallization in malonate conditions with intermediate concentrations of BME (5–20 mM) resulted in plate clusters and crystal twinning.
Diffraction data were integrated and scaled using Bruker software SAINT and SADABS. Disulfide-linked PfACP diffraction data phases were determined using Phaser24 with molecular replacement target 1T8K.25 Coot26 and CCP427 were used to build and refine the atomic coordinates. Reduced PfACP diffraction data phases were solved with molecular replacement using the disulfide-linked PfACP model and refined with the same software. Rfree was calculated by omitting from refinement 5% of the diffraction data which was randomly distributed by resolution shell. The structures of reduced and disulfide-linked PfACP were analyzed with PROCHECK.28 PfACP pore length was determined using CAVER,29 then manually truncating paths that protruded beyond the surface of the protein. The path end was selected as the most buried point flush with the surface of the protein on at least two sides. Solvent accessible surface area was calculated with MSMS.30 Protein structure figures were generated using Pymol 0.99rc6 (DeLano Scientific LLC).
In Vitro and In Vivo Redox Assays
The redox state of PfACP samples was determined by separating monomeric and dimeric species on non-reducing polyacrylamide gels. Crystals of PfACP were dissolved in 1X NuPage sample buffer (Invitrogen) and resolved on a 4–12% BisTris NuPage gel (Invitrogen) run for 30 min at 200 V. Protein bands were visualized with SimplyBlue stain (Invitrogen), or with silver stain (Bio-Rad). For reducing agent dose-response experiments, PfACP samples were first oxidized to the dimeric state by air oxidation for 10–20 days at 23 °C in a closed eppendorf tube (periodically opened to the atmosphere). Buffer and reducing agent were then added at reported concentrations and incubated at 23 °C for 30 min. Disulfide exchange was quenched with 180 mM iodoacetamide for 1hr at 23 °C to block free sulfhydryls, then NuPage gel loading buffer was added and the samples were separated by non-reducing polyacrylamide gel electrophoresis.
P. falciparum parasites, strain 3D7 or DD2attB,31 were maintained in human red blood cell culture at 1% hematocrit using the general method described by Trager and Jensen.32 Briefly, blood stage parasites were cultured in RPMI 1640 supplemented with 10% human serum, 28 mM NaCO3H, 25 mM HEPES, and 0.09 mM hypoxanthine. Cultures were gassed with 90% N2, 5% O2, 5% CO2 and incubated in sealed 75 cm2 flasks at 37 °C. Parasites were cultured to 15–30% parasitemia of late blood stage parasites before harvesting at 4 °C by centrifugation for 3 min at 500 g. Red blood cells were lysed with 0.02% saponin in PBS for 5 min at 4 °C, then excess PBS was added and the parasites were pelleted by centrifugation at 4 °C for 5 min at 5000 g. The parasite pellet was washed of hemoglobin by resuspension in cold PBS and centrifugation. Washes were repeated until the supernatant was clear.
For reduced samples, the parasite pellet was resuspended in 2X NuPage sample buffer with 50 mM DTT. Samples were vortexed at high speed for at least 3 min to break up DNA, and then briefly heated to 90 °C. Prepared samples were then run on a 4–12% BisTris NuPage gel for 30 min at 200 V, followed by transfer to 0.2μm pore nitrocellulose membrane (Invitrogen) by BioRad trans-blot semi-dry transfer using Towbin transfer buffer33 at 5 V for 2 hr. To prevent ACP from diffusing out of the membrane, the membrane was briefly rinsed in PBS, and then incubated in a cross-linking buffer of 4% formaldehyde and 0.1% glutaraldehyde in PBS for 1 hr at 23 °C. Then the membrane was blocked with 5% milk (Carnation non-fat dry milk) in PBS for 1 hr, washed three times with 1% milk in PBS, and probed with primary αPfACP antibodies (see above) at 1:5000 overnight at 4 °C. Subsequently, the blot was washed three times with 1% milk in PBS, and probed with secondary αrabbit HRP conjugate (GE Healthcare) at 1:3500 for 1 hr. The blot was then washed three times and visualized with Pierce ECL western substrate and exposed to film sequentially for 30 sec, 3 min, and overnight.
For non-reduced samples, a lysis buffer of 140 μL was prepared and mixed with a parasite pellet volume of approximately 60 μL. In 200 μL, the concentrations of the lysis buffer components were: 1X NuPage LDS gel loading buffer, 25 mM EDTA pH 8.0, 0.02 mg/mL pepstatin, 1X Complete Protease Inhibitor (Roche), 0.1% Triton X-100, 150 mM iodoacetamide. Following saponin lysis and washing of the parasite pellet, the lysis buffer was mixed with the parasite pellet to simultaneously lyse cells and block all free sulfhydryls, thereby quenching disulfide exchange. The lysate was vortexted and then incubated at 23 °C for 3 hr. The long incubation time ensures complete reaction of iodoacetamide with free sulfhydryls, since the reaction should be complete well before then.8 As previously shown, iodoacetamide does not attack disulfides, allowing them to be preserved. Samples were then visualized by gel electrophoresis and transferred to membrane exactly as in reduced sample preparation. After transfer to membrane and cross-linking, one additional step was added to reduce the background of heme derived species reacting with the Pierce ECL western substrate. The membrane was incubated in a minimum volume composed of 10 mM bathophenanthrolinedisulfonic acid disodium salt and 250 mM ascorbic acid for 1 hr at 37 °C. The membrane was then blocked, probed with antibody, and visualized as described for the reduced samples.
Diamide or hydrogen peroxide treated P. falciparum parasites were first liberated from host red blood cells as previously described, then incubated in 1X PBS with either diamide or hydrogen peroxide while maintained on ice. Lysis was performed without iodoacetamide or reducing agent, and then Western analysis was performed as described for non-reduced samples.
Results
Disulfide-linked PfACP Crystal Structure
Disulfide-linked dimer crystals of PfACP diffracted to 2.55 Å at 23 °C. Despite efforts to identify cryogenic data collection conditions that preserved diffraction quality, none were found. While it was possible to grow crystals starting with air oxidized PfACP, crystal twinning was problematic. The best diffracting crystals began with protein pre-treated with equimolar amounts of reducing agent, then the crystallization condition was augmented with Cu(II) as a catalyst for disulfide bond formation. The diffraction pattern from these crystals was determined to be space group I41, yielding one molecule in the asymmetric unit. Initial phases were determined by molecular replacement using EcACP, and the model was refined to an R value of 24.2% and Rfree of 26.7% (Table I). The final model resolves all amino acids of the PfACP protein construct plus the 4′-phosphopantetheine prosthetic group.
TABLE 1.
PfACP Structure Statistics.
| Disulfide-linked PfACP | Reduced PfACP | |
|---|---|---|
| PDB Code | 3GZL | 3GZM |
| Number of Crystals | 1 | 4 |
| Refinement Resolution | 2.55 – 43.56Å | 1.80 – 38.32Å |
| Data Collection Temp. | 293K | 100K |
| Redundancy | 5.8X (5.1X) | 60.1X (38.3X) |
| Completeness | 99.7% (100%) | 100% (100%) |
| Rsym/Rmerge | 6.8% (29.4%) | 19.2% (60.0%) |
| No. of unique reflections | 3550 (441) | 14747 (2143) |
| Total No. of atoms | 664 | 1551 |
| No of solvent atoms | 0 | 175 |
| R-factor | 24.180 % | 20.562 % |
| R-free | 26.703 % | 27.607 % |
| RMS deviations | ||
| Bonds | 0.006 A | 0.020 A |
| Angles | 1.005 degrees | 2.112 degrees |
| Ramachandran Plot | ||
| Most Favored | 93.6 % | 94.1% |
| Additionally Allowed | 6.4 % | 5.9% |
| Generously Allowed | 0 % | 0% |
| Disallowed | 0 % | 0% |
| Average B Factor | 70.027 Å2 | 22.915 Å2 |
Values in parenthesis are for the highest resolution shell. For more complete diffraction data statistics, see supplementary Fig. S5 and Fig. S6.
The overall fold of PfACP is closely related to the family of known ACP structures, as described previously.34 The core of the protein consists of a four helix bundle which tethers the N and C termini to the bottom of the structure (Fig. 1A). An extended ordered loop region connects helices 1 and 2 and packs over the top of the four helix bundle. The attachment site of the phosphopantetheine prosthetic group at Serine 39 is at the beginning of helix 2. A brief run of amino acids in extended conformation connect the bottom of helix 2 to helices 3 and 4, which pack up and down the back of the helix bundle to form a pocket that can open to allow fatty acids to bind when attached to the phosphopantetheine group.
FIGURE 1.

Crystal Structure of disulfide-linked PfACP dimer. (A) Four helix bundle topology of ACP. The phosphopantetheine attaches at the beginning of helix 2 and helices 2–4 form the fatty acid binding pore. (B) Side view of PfACP dimer showing the disulfide bond buried within the dimer interface and defining orientation for (C) Top view of PfACP dimer with 2Fo-Fc electron density contoured at 1 sigma. The phosphate (orange) can be seen connecting the pantetheine prosthetic group to serine 39. The isopentyl moiety of the pantetheine group blocks solvent access to the disulfide bond. (D) Bottom view of the dimer interface highlighting reciprocal polar interactions with 2Fo-Fc electron density contoured at 1.5 sigma.
The terminal sulfur atoms of the 4′-phosphopantetheine groups from symmetry related PfACP molecules were found to sterically clash, suggesting that they were covalently bonded. A disulfide bond restrain was added to refinement, setting the S-S bond length to 2.0 Å. In addition to this covalent bond, the dimer is stabilized by a 1494 Å2 buried surface area including 4 symmetric intermolecular hydrogen bonds between N30 – N66, T32 – L63, and an ionic interaction between K33 – D70 (Fig. 1D). Intermolecular hydrophobic contacts are formed between L42 – L63, as well as L63 and the isopentyl group of the phosphopantetheine arm. Only a single conformation of the phosphopantetheine arm is consistent with the electron density, due to constraints imposed by the intermolecular interface (Fig. 1B). The disulfide bond is sequestered from solvent at the dimer interface, with the isopentyl groups of both phosphopantetheine arms blocking the shortest route to bulk solvent (Fig. 1C). Crystals were confirmed to contain covalent dimer by non-reducing, SDS polyacrylamide gel electrophoresis (PAGE). The observed molecular weight was twice the expected size for monomer. PfACP solution made from crystals was found to be readily reduced by stoichiometric amounts of DTT (Fig. 2A). Residual dimer in the reduced crystals was likely due to nonspecific chemical cross-linking after several weeks crystallized at 23 °C. Indeed, small populations of higher-order multimers can be seen in the non-reduced crystals as well. PfACP dimer solution was made from air oxidation of monomer, and was found to be equally susceptible to DTT reduction as the crystals of PfACP dimer (Fig. 2A).
FIGURE 2.

Reduction of PfACP dimer in vitro. (A) Crystals of disulfide-linked PfACP and air oxidized PfACP solution were titrated with DTT at 0, 0.1, 1, 10, and 50 mM for 30 min in LDS, and then visualized with silver stain. Bands corresponding to PfACP monomer and the disulfide-linked dimer are clearly resolved. Higher order multimers exist which were likely formed through chemical cross-linking due to the high concentration in the crystalline state. They are only visible by silver stain, and as was particularly evident in the protein crystals, not susceptible to reducing agent. (B–F) Air oxidized PfACP dimer (0.36 mM) treated with reducing agent at 0, 0.5, 1.5, 4.5, and 13.5 mM for 30min in varying buffer conditions. Samples were visualized with SimplyBlue stain (Invitrogen). PfACP dimer is the upper band and monomer is the lower band. (B) Dimer treated with DTT under native conditions (50 mM Tris) compared to denaturing conditions (LDS). (C) Dimer treated with Glutathione under native conditions (200 mM Tris) compared to denaturing conditions (LDS). (D) Dimer treated with TCEP under native conditions (200 mM Tris) compared to denaturing conditions (LDS). (E) Dimer treated with BME under native conditions compared to denaturing conditions (LDS). (F) Dimer treated with BME in native conditions compared to high ionic strength conditions (2 M NaMalonate).
In Vitro Reduction Assays
The disulfide linkage observed in the dimeric PfACP structure may be protected from solvent by the protein conformation, so the ability of native structure to block disulfide bond reduction was assayed. While the actual kinetics of disulfide bond reduction involve the formation of several intermediates capable of competing with the direct reaction of ACP dimer with reducing agent, we have employed a simple electrophoresis assay to measure net reduction of disulfide-linked dimer in a fixed time interval. Tris buffer with a pH of 7.5 was chosen to represent native conformation, as PfACP is well folded in this simple buffer. Lithium dodecyl sulfate (LDS) gel loading buffer is a strong denaturant that was used to disrupt PfACP structure. 1.8 mM PfACP was air oxidized at 23 °C, then 0.36 mM PfACP was titrated with 0, 0.5, 1.5, 4.5, and 13.5 mM reducing agent for 60 min under varying buffer conditions. Differences between native and denatured PfACP dimer were insignificant when treated with DTT, suggesting that the native conformation does not protect the disulfide from DTT reduction (Fig. 2B). Quantified differences measured by densitometry show a maximal deviation of 5% between comparable reducing agent concentrations. Repeating the assay with glutathione as the reducing agent (Fig. 2C) shows similar results to DTT, although at 1.5 and 4.5 mM glutathione, the difference between native and denatured conditions is measured as 9% and 6% protection, respectively. The non-thiol based reducing agent TCEP readily reduces PfACP and no differences are observable between native and denatured samples (Fig. 2D). However, when the same assay was performed using BME, reduction of the native PfACP dimer consistently required more BME than the denatured sample (Fig. 2E). Quantified differences determined by densitometry show that native PfACP was protected from reduction by 0%, 18%, 27%, 16%, and 3% across the five BME concentrations, respectively. To determine if the high ionic strength crystallographic conditions confer additional protection, native conditions were compared to 2 M NaMalonate pH 7.5 using BME as a reducing agent. Surprisingly, no further stabilization of the interface is observed (Fig. 2F), and quantification by densitometry shows a maximal deviation of 6% between comparable reducing agent concentrations.
Reduced PfACP Crystal Structure
Reduced PfACP crystals that were grown in 80 mM BME diffracted to 1.8Å under cryogenic conditions, and were found to belong to space group P41212. Crystallization conditions with lower BME concentrations produced a range of results including needle clusters, plates, and rampant crystal twinning. The structure was solved with molecular replacement using the disulfide-linked PfACP structure and was refined to an R value of 20.6% and Rfree of 27.6% (Table I). Two PfACP molecules were found in the asymmetric unit and the packing of these two molecules is similar to that of the disulfide-linked structure (Fig. 3A). The difference in packing between these two molecules in the disulfide-linked and reduced structures can be summarized by a translation of 3.5 Å and rotation of 14 ° (Fig. 3B). In comparison to the disulfide-linked PfACP intermolecular interface, the reduced PfACP interface is smaller, burying 1110 Å 2 of solvent accessible surface area.
FIGURE 3.

Crystal structure of the reduced PfACP monomer with 2 molecules in the asymmetric unit. (A) After aligning reduced ACP:A and the disulfide-linked structure (both in green), reduced ACP:B (dark blue) closely matches the symmetry related molecule of the disulfide-linked structure (cyan). (B) View of the reduced PfACP crystal contact interface from the side and (C) top with 2Fo-Fc electron density contoured at 1 sigma, showing that the phosphopantetheine groups do not form a disulfide. High electron density is observed for the phosphate (orange), but the density is weaker further along the phosphopantetheine side chain. (D) View of the reduced PfACP crystal contact interface from the bottom with 2Fo-Fc electron density contoured at 1.5 sigma, showing polar interactions similar to those observed the disulfide-linked structure.
In the reduced state, the phosphopantetheine group shows evidence of several conformations. The conformation which best fits the crystallographic data extends upwards away from PfACP into a solvent pocket within the crystal lattice (Fig. 3C). This conformation renders the phosphopantetheine group completely solvent exposed, and juxtaposes two phosphopantetheines from the asymmetric unit with two more from symmetry related molecules. The high density of phosphopantetheine side chains in this small solvent cavity may explain the requirement for unusually concentrated reducing agent needed to prevent crystal twinning. Although this solvent exposed conformation best fits the electron density, it is likely that the phosphopantetheine groups are statically disordered. Two other conformations gave plausible results for the beta-mercaptoethylamine moieties of phosphopantetheines, but less satisfactory results for the carbonyls of the hydrophilic portion. The first alternative conformation reaches back down the side of PfACP towards its own fatty acid binding pore, and the second alternative conformation extends towards the entrance of the fatty acid binding pore of a symmetry related molecule.
Ordered solvent molecules are found spanning the interface between the two PfACP molecules and in the fatty acid binding pockets. Two well defined malonate molecules lie near the loop connecting helices 1 and 2 in both monomers of PfACP. A third well defined malonate sits over the fatty acid binding cleft in PfACP monomer B. A BME molecule occupies the entrance to the fatty acid binding cleft of PfACP monomer A. The sulfur atom of this BME occupies an interesting position. The sulfurs from the phosphopantetheine in acyl-ACP structures heptanoyl-ACP (2FAD:B) and decanoyl-ACP (2FAE:A)35 lie in this same position, as does the sulfur atom of an ordered detergent molecule in a cytochrome P450-ACP complex.36 This particular location on ACP seems to favor interaction with sulfur. Remaining solvent molecules in the reduced PfACP structure were modeled as malonate, however, alternative models can be made with other solvent molecules (beta-mercaptoethanol, 2-hydroxyethyl disulfide, and tris(hydroxymethyl)aminomethane) resulting in only slightly poorer crystallographic statistics. Overall the model occupies 84.5% of the unit cell.
Comparison of Structures
ACP proteins are known to be dynamic molecules capable of conformational change in response to bound fatty acid. To compare ACP structures, we focused first on the conformation of the protein backbone, and second on the phosphopantetheine prosthetic group. We were surprised to find Cα RMSD values between PfACP and a wide variety of different ACP crystal structures to be within 0.5–0.8 Å (Fig. 4). RMSD values between the disulfide-linked and the two reduced PfACP monomers reported here are of the same magnitude as RMSD values between our unliganded PfACP structures and fatty acid bound crystal structures. Hexanoyl-EcACP and decanoyl-EcACP show high similarity to PfACP by Cα RMSD. The crystal structure of unliganded ACP from the related apicomplexan parasite Toxoplasma gondii shows highest similarity to PfACP as measured by Cα RMSD. Differences between PfACP crystal structures and reported NMR structures are larger, even when excluding the earliest ACP NMR structures solved without the benefit of modern NMR methodology. Notably included in this set are the PfACP NMR structures, 2FQ0, and 2FQ2.34 RMSD values between crystal structures reported here and previously reported NMR structures range from 1.5–3 Å with most differences found in the loops, but differences in helix packing angles were observed as well. It is worth noting that this observation is not limited to PfACP or comparisons across species. When comparing recent EcACP NMR structures37 to an EcACP crystal structure representative of apo-ACP, such as 1T8K25, or fatty acid derivitized ACP, such as 1L0I38, all Cα RMSD values range between 1.5–1.9 Å. The largest Cα RMSD we observed between PfACP and any ACP crystal structure was 1.2 Å, with all the rest being less than 1.0 Å (Fig. S2). Differences between ACP structures have often been attributed to the presence or absence of fatty acid derivatives, or to the phosphopantetheine group itself. Our classification of the differences in main chain conformation of the PfACP crystal structures compared to other known ACP structures is further described in the discussion.
FIGURE 4.

Cα RMSD values between representative ACP structures. Disulfide-linked PfACP is labeled “oxACP”, and reduced PfACP is labeled “redACP.” PfACP crystal structures have RMSD values of 0.5–0.6 Å when compared to each other, and slightly higher values (0.6–0.8 Å) when compared to other parasite and bacterial crystal structures of both holo and acylated forms. NMR structures of either holo or acylated ACP have significantly larger RMSD values when compared to the PfACP crystal structures. See supplementary Fig. S2 for a more complete listing of ACP structures.
The conformation of the phosphopantetheine prosthetic group differs widely between reduced and oxidized PfACP crystal structures, and both differ from the previously reported PfACP NMR structure.34 In the oxidized PfACP crystal structure, the phosphopantetheine group extends down helix 2 before turning towards the fatty acid binding pore where it terminates with a disulfide link to an adjacent PfACP molecule. The packing between the two disulfide-linked PfACP molecules occludes the position of the prosthetic group previously reported in the NMR structure. In the reduced PfACP crystal structure, the prosthetic group points away from the protein core and orthogonal to helix 2. This orientation is opposite that reported in the PfACP NMR structure, where it points back down the ordered coil structure preceding helix 2. Closely examining the prosthetic group refinement constraints reported for the PfACP NMR structure indicates a single long range NOE (|i – j| > 4) connecting Valine 41, which is one helical turn down helix 2, and the isopentyl moiety of the prosthetic group. Yet the prosthetic group points away from Valine 41 in the NMR structure, separated by a distance of 7.3 Å. Had the modeled conformation of the reported NMR structure pointed instead down helix 2 towards residue Valine 41 (Valine 43 in our crystal structures), it would have been similar to the oxidized PfACP conformer, as well as the phosphopantetheine conformer previously reported for ACP from Bacillus subtilis in complex with holo-ACP synthase.39 In the oxidized PfACP crystal structure, the distance between atoms described by the NOE is 5.2 Å. In the reduced structure, the isopentyl group is even closer to helix 2 while the rest of the prosthetic group extends away from the protein. The same distances spanned by the NOE when measured in the two reduced conformers are 2.9 Å and 2.3 Å. While the conformation of the phosphopantetheine group in our crystal structures appears to differ widely from the previously reported NMR structure, we find that our model is actually well supported by the NMR restraints.
While the phosphopantetheine group in the oxidized crystal structure is sterically constrained, the reduced structure is not, and exhibits weaker electron density, particularly beyond the iosopentyl group. This suggests that the conformation is heterogeneous for an un-tethered prosthetic group. Lack of other long range NOEs in the NMR structure also implies that the phosphopantetheine has substantial flexibility. The phosphopantetheine prosthetic group has 10 rotatable bonds and is hydrophilic, allowing for great conformational heterogeneity when not constrained, as seen in both the reduced PfACP crystal structure, or implied by the previously reported NMR structure.
PfACP Redox State In Vivo
Uncertainty about the molecular reducing agents that might be active in the apicoplast or even the metabolic state of the apicoplast during blood stage growth motivated us to probe the redox state of PfACP in vivo. Cultured P. falciparum parasites were lysed in the presence of iodoacetamide to block all free sulfhydryls while preserving native disulfide bonds. Iodoacetamide-treated samples were separated by SDS PAGE without reducing agent, and analyzed by western blot with αPfACP antibodies. The primary species of PfACP had the same apparent molecular weight as reduced monomeric PfACP (Fig. 5). A small population of unprocessed PfACP precursor that had not yet undergone apicoplast import is also clearly visible, which provides a good measure of sensitivity. The PfACP dimer was not detected in iodoacetamide-treated samples. This indicates that PfACP does not form the covalent dimer in vivo. This implies that either the apicoplast is a reducing environment, or that the exposed phosphopantetheine sulfhydryl is blocked by a covalent modification. It is also possible that holo-ACP synthase does not attach the phosphopantetheine group during the blood stages. To distinguish between these possibilities we attempted to determine whether PfACP disulfides can be induced under oxidizing conditions.
FIGURE 5.
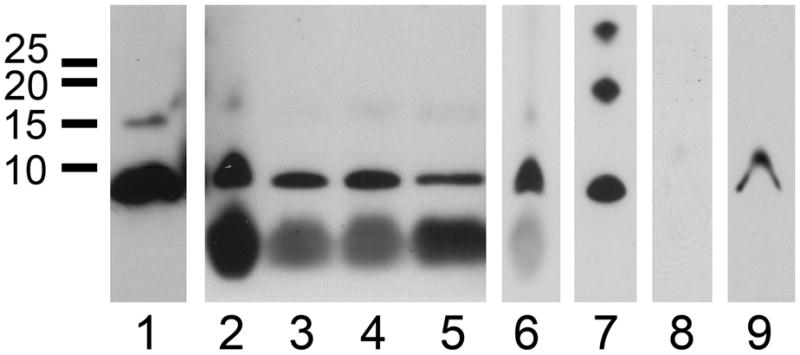
Western blots of PfACP from P. falciparum cell culture under normal and oxidizing conditions. (1) Reduced PfACP runs just under 10 kDa, and PfACP with its apicoplast transit peptide can be seen as a minor band at 15 kDa. (2) Parasites in culture medium were treated with H2O2 at 0 mM, (3) 10 mM, and (4) 100 mM. The large low molecular weight band is residual heme reacting directly with the ECL substrate. (5) Parasites liberated from their red blood cells were treated with H2O2 at 10 mM and (6) 20 mM. (7) Air oxidized recombinant PfACP provides a marker for monomer, dimer, and trimer. (8) P. falciparum parasites were treated with 20 mM diamide for 30 min at 4 °C, then lysed without reducing agent or iodoacetamide, and analyzed by Western. (9) The same sample used in lane 8, except 300 mM DTT was added before electrophoresis. Lanes 6 and 7 are from the same blot, and lanes 8 and 9 are from the same blot. Exposure settings are similar across gels, and identical within them.
Oxidative stress could affect the redox conditions in the parasite, leading to formation of the PfACP dimer. Blood stage P. falciparum parasites are believed to be under elevated oxidative stress associated with the accumulation of heme in the parasite during the degradation of host cell hemoglobin. To assess if PfACP dimer can be formed under conditions of increased oxidative stress, we challenged P. falciparum parasites with hydrogen peroxide and observed the resulting state of PfACP by Western blot. Parasites cultured in standard culture media spiked with varying amounts of hydrogen peroxide for 30 minutes were analyzed and found to contain only PfACP monomer (Fig. 5). These experiments were potentially complicated by the presence of reduced glutathione, which is a standard component of the culture medium. This complication was addressed by isolating parasites from the culture system before treatment with hydrogen peroxide. Cultured parasites were liberated from host red blood cells with saponin and directly treated with 10 mM hydrogen peroxide in PBS for 20 minutes on ice. Surprisingly, PfACP monomer was still observed. Repeating the experiment with 20 mM hydrogen peroxide for 1 hour produced the same result. No dimeric PfACP was detected by Western analysis (Fig. 5). In similar experiments, the antimalarial drug chloroquine was used to inhibit the detoxification of heme and increase oxidative stress in the parasite. When cultured 3D7 parasites were treated with 20 nM chloroquine for 3 hours followed by Western blot analysis, PfACP monomer was the only species observed (Fig. 5). From these results we conclude that either PfACP is in a state which is unaffected by oxidative stress, or that the apicoplast is able to maintain a reducing environment in spite of formidable levels of hydrogen peroxide.
To determine if PfACP dimer can be formed in any conditions, we made use of the compound diamide, which is known to directly, rapidly, and reversibly induce disulfide bond formation, and has proven effective in cell culture.40,41 Cultured P. falciparum parasites were liberated from their red blood cells and treated with 20 mM diamide in PBS for 30 min on ice. Lysis was performed without iodoacetamide or reducing agent, and then visualized by Western blot as previously described. Interestingly, the PfACP monomer is only faintly visible after diamide treatment of intact parasites. Addition of 300 mM DTT to the same cell lysate before electrophoresis and analyzed in the same Western blot shows recovery of the signal for PfACP monomer. We interpret the faint band in diamide treated lysate to mean the PfACP has been dispersed throughout the sample via disulfide bonds to molecules of diverse size. Treatment with DTT breaks these bonds and PfACP monomer is observed again. Susceptibility to diamide strongly suggests that the majority of PfACP contains a free sulfhydryl during blood stage growth, thus requiring the apicoplast to be a reducing cellular compartment.
Discussion
PfACP Susceptibility to Reduction
While PfACP disulfide-linked dimers have long been thought to behave as two beads on a string, pushed apart by electrostatic repulsion, our structure of disulfide-linked PfACP caused us to question this assumption. Particularly if the disulfide bond is sequestered from bulk solvent as observed in our crystal structure, the kinetics of disulfide reduction by cellular reducing agents could be significantly slowed. PfACP’s susceptibility to reduction in vitro shows an interesting dependence on the choice of reducing agent. While DTT was equally effective in reducing native or denatured PfACP, BME failed to reduce native PfACP as readily as it reduced denatured PfACP. Glutathione was nearly as effective as DTT at reducing PfACP disulfide-linked dimer, although a small difference was observed between native and denatured protein. The non-thiol based reducing agent TCEP has a much faster intrinsic reaction rate42 and is therefore able to more effectively reduce PfACP even at low concentration.
It is somewhat remarkable that any differences at all can be observed between native and denatured conditions of PfACP after 1 hr incubation with reducing agent. Both DTT and glutathione show reasonable activity against the dimer, albeit slower than predicted from calculated rate constants43 under the assumption of irreversible reduction during the experiment. But BME appears anomalously poor at reducing native PfACP disulfide-linked dimer. Perhaps BME is not as capable of interacting with the dimer interface, being slightly more hydrophobic than DTT. High ionic strength conditions are expected to further stabilize the dimer interface because the electrostatic repulsion between negatively charged monomers would be mitigated. Ionic strength is proportional to the square of the charge, and malonate is divalent, making it four times as potent as sodium chloride. The highest concentration feasible for in vitro reduction assays was 2 M malonate, which was expected to promote self association of the dimer interface, and protect the disulfide bond. But this was not observed, either since the ionic strength was still not as high as crystallographic conditions, or perhaps because BME had a role in stabilizing the dimer interface and no additional effect remained.
Disulfide cross-linking of PfACP molecules results in a dramatic increase in the effective concentration of the monomers with respect to each other. This can be estimated geometrically by modeling a disulfide-linked PfACP dimer with fully extended phosphopantetheine groups. In this configuration, protein centroids are 71 Å apart, allowing the concentration to be estimated as 1 molecule in 713 Å3 or 5 mM. The effective concentration of PfACP dimer interface is estimated to be even higher. A distance of 39 Å separates the Cα of each Serine 39, which equates to a local concentration of 27 mM. The bottom of the interface may be further apart in an extended conformation. Distance between Cα atoms of N30 and N66 represent the bottom of the interface, and are 62 Å apart, or 7 mM. At these concentrations, only 2.5–3.5 kcal/mol of binding energy is needed to populate 50% of the bound conformation. Estimated local concentrations of PfACP approach those found in our crystals, where proteins are packed at a concentration of 57 mM in the disulfide-linked state, and 89 mM in the reduced state.
While crystal contacts alone are by no means indicative of a functional biological interface, we note the coincidence that the same amino acids were found to participate in the disulfide-linked dimer interface and the interface packing two reduced monomers. The bottom of the interface shares the same hydrogen bond network, and the top of the interface juxtaposes phosphopantetheine groups in both disulfide-linked and reduced states. The difference between the interfaces is limited to a slight change in the packing angle between monomers. This suggests that if PfACP is forced to interact with itself, whether in the reduced or disulfide-linked state, it does so via the helix 3 and the loop between helix 1 and 2, juxtaposing prosthetic groups and fatty acid binding pores. It is worth noting that when the apicoplast protein Ferredoxin-NADP+ Reductase was crystallized, it too was found to be a disulfide-linked dimer, which trapped the catalytically inactive state.44,49
The active reducing agent in the apicoplast is not known. Evidence for glutathione in the apicoplast is presently indirect, leaving ferredoxin,45 thioredoxin,46 or even lipoic acid47 as the reducing agent responsible for reducing disulfide-linked PfACP. The redox potential of cysteine is −0.22 mV.48 If the phosphopantetheine sulfhydryl has a similar potential, then it could be readily reduced by apicoplast resident protein ferredoxin, experimentally determined to have a redox potential of −0.266 mV.45 Under normal growth conditions these agents could be responsible for maintaining PfACP as a reduced monomer.
The Apicoplast Is a Reducing Environment
Treatment of cells with hydrogen peroxide is a common tool for probing redox responsive elements in vivo. Doing so can demonstrate which components respond first to oxidative stress, and reveal the threshold of hydrogen peroxide that the cell is capable of enduring without showing ill effects. We examined the oxidative state of PfACP in the apicoplast and found it to be reduced under normal growth conditions. Yet when the cells were probed for an oxidative stress phenotype (visualized as PfACP oxidation), surprisingly harsh conditions could be endured. Previous work on murine malarias established that hydrogen peroxide was effective at killing malaria, and at concentrations of 10 mM, definite growth phenotypes were observed.42 While it is difficult to identify proteins first responding to oxidative stress, it has been done in E. coli successfully using 4 mM hydrogen peroxide treatment for 2–30 min.8 These experiments broadly define 1–10 mM hydrogen peroxide as stressful for cells, and higher levels are expected to be destructive. Our data show that treatment with 10–20 mM hydrogen peroxide does not affect PfACP, indicating a high level of protection from the effects of oxidative stress. Only when treating cultured parasites with 20 mM diamide can we observed an effect on PfACP, which is reversible with the addition of DTT. Interestingly, diamide treatment causes PfACP to disperse rather than dimerize, suggesting that the concentration of PfACP in the apicoplast is much lower than the concentration of other available disulfide bonding partners. Taken together, these results suggest that the observed PfACP monomers have free sulfhydryls, and that the apicoplast is able to maintain the reduced state of PfACP despite significant oxidative stress.
Assignment of Open or Closed ACP Conformation
ACP is known to change conformation when bound to its acyl chain ligand.51 Two lines of evidence suggest that our PfACP backbone conformation may be skewed toward a fatty acid bound state even though the structure contains no fatty acid. First, ordered solvent molecules occupy the top of the fatty acid binding pore in the reduced PfACP structure. Second, the high ionic strength of the crystallization conditions may effect structure compaction. Previous work has shown that mutating acidic residues to neutral ones stabilized the compact conformation in Vibrio harveyi ACP.52 Because a variety of ACP structures are available from different organisms and in different liganded states, we wanted to define a reaction coordinate related to fatty acid binding and then assign our PfACP structure along this coordinate.
Several models were considered as a reaction coordinate for conformational change upon ligand binding. Visual inspection of previous ACP structures suggests that helix 4 may angle outward in an iris-like motion, allowing a pore to open up between helices 2, 3, and 4. This model will be referred to as the “helix angle model.” If the helix bundle opens without angular motion of the helices, a mean displacement between helices would result, and this will be called the “helix distance model.” Wu et al.37 found by NMR of EcACP that the distance between T39 at the beginning of helix 2 and D56 at the beginning of helix 3 increases between apo- and holo-EcACP, and increases again in butyryl-EcACP. Their model will be referred to here as the “helix tip distance.” These models were compared to calculated pore depths as characterized with the program CAVER.
Support for the models was found by comparing specific liganded and unliganded structures. The helix angle model was evaluated by the crossing angle of helices 2 and 4, and was best supported by comparing the liganded NMR structure 2FVA with the unliganded crystal structure 1T8K. The helix distance model reports on bulk motion of the backbone, and was evaluated by the distance between centers of helices 2 and 3. This was best supported by recent NMR work comparing liganded and unliganded EcACP in otherwise identical conditions.37 The helix tip distance model was well supported by the NMR structures that Wu et al. used to define it, and according to these structures the helix tip distance was predicted to track the helix distance closely, expanding slightly to accommodate a larger pore size.
We were surprised to find that helix angle (Fig. S3) and helix distance (Fig. S4) appeared uncorrelated with pore depth, and are uncorrelated with each other (Fig. S5). Only when using the more directed metric of comparing the helix tip distance as in Wu et al., and then considering only x-ray structures, do we see a trend which can be fit linearly with an R2 of 0.68 (Fig. 6). This analysis includes acyl-EcACP 2FAE:B, which contains bound fatty acid but no electron density was observed for the ligand. By excluding this structure from the analysis, R2 improves to 0.79. Instead of expanding to allow the acyl chain to fill the protein interior, the trend shows slight compaction. This is in agreement with EcACP’s behavior on conformationally sensitive urea gels.51 When plotted on the same axes, our PfACP structure lies midway between E. coli unbound and fatty acid bound structures. Unlike EcACP, PfACP does not show a significant mobility shift by conformationally sensitive PAGE when bound to different fatty acids.53 Our structure suggests that this may be due to PfACP already assuming a more compact conformation in the unbound state. Interestingly, 2QNW from related parasite Toxoplasma gondii shows the same property even though it was crystallized in 30% PEG 1500, which is not a high ionic strength condition. Still, the compaction is slight, and when NMR structures are included in the analysis, the trend disappears (Fig. S6). This analysis suggests that the majority of the rearrangements for fatty acid binding involve side chain repacking instead of backbone motion. Since the conclusions differ between xray (Fig. 6) and NMR (Fig. S6) methodologies, it is possible that fatty acid binding by ACP is primarily manifest not by a mean structural displacement upon binding but by a change in protein dynamics.54
FIGURE 6.

Various metrics of ACP conformation compared to ACP pore depth as measured by CAVER. (A–C) Visual representation of the metrics for: (A) helix crossing angle between helices 2 and 4, (B) Helix distance between helices 2 and 3, and (C) Helix tip distance as defined by Wu et al. measuring the top of helix 2 to the tip of helix 3. (D–F) Correlation plots of various metrics of ACP conformation with pore depth as measured by CAVER. Solid shapes represent structures with bound ligand, and open shapes represent structures without ligand. Triangles represent crystal structures and circles represent NMR structures. Asterisks denote the PfACP crystal structures reported here. (D) Helix crossing angle is not predictive of pore depth in crystal or NMR structures. A crossing angle of 30 ° appears favored irrespective of the method or liganded state. (E) Helix distance between helices 2 and 3 is not predictive of pore depth despite the fact that the acyl chain binds between these two helices. (F) Helix tip distance does show correlation with pore depth, but only when considering crystal structures. Crystal structures with deeper pores tend to show a reduced helix tip distance. For the numerical values used in these correlation plots, see the supplementary information.
Conclusions
We have shown that it is possible for disulfide-linked PfACP dimers to form a closely associated complex which sequesters the disulfide bond within the dimer interface. In vitro reduction of PfACP dimers shows that the dimer interface can partially shield the disulfide bond from reduction by BME. The redox state of PfACP was observed in P. falciparum cell culture to evaluate the ability of organellar reducing agents to maintain the reduced state of PfACP. In vivo results show PfACP to be reduced during normal blood stage growth under all but the harshest oxidizing conditions. This result provides experimental evidence that the apicoplast is a reducing cellular compartment and suggests that it is highly redox buffered.
Supplementary Material
Acknowledgments
The disulfide-linked and reduced structures of PfACP have been deposited in the Protein Data Bank under accession codes 3GZL and 3GZM, respectively. This work was supported by the National Institutes of Health (R01 AI065853), and the Johns Hopkins Bloomberg School of Public Health Faculty Innovation Fund.
References
- 1.WHO. World Malaria Report 2008. Geneva, Switzerland: World Health Organization; 2008. Report nr. [Google Scholar]
- 2.Bhattarai A, Ali AS, Kachur SP, Martensson A, Abbas AK, Khatib R, Al-Mafazy AW, Ramsan M, Rotllant G, Gerstenmaier JF, Molteni F, Abdulla S, Montgomery SM, Kaneko A, Bjorkman A. Impact of artemisinin-based combination therapy and insecticide-treated nets on malaria burden in Zanzibar. PLoS Med. 2007;4(11):e309. doi: 10.1371/journal.pmed.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Afonso A, Hunt P, Cheesman S, Alves AC, Cunha CV, do Rosario V, Cravo P. Malaria parasites can develop stable resistance to artemisinin but lack mutations in candidate genes atp6 (encoding the sarcoplasmic and endoplasmic reticulum Ca2+ ATPase), tctp, mdr1, and cg10. Antimicrob Agents Chemother. 2006;50(2):480–489. doi: 10.1128/AAC.50.2.480-489.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359(24):2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 5.Ralph SA, D’Ombrain MC, McFadden GI. The apicoplast as an antimalarial drug target. Drug Resist Updat. 2001;4(3):145–151. doi: 10.1054/drup.2001.0205. [DOI] [PubMed] [Google Scholar]
- 6.Gornicki P. Apicoplast fatty acid biosynthesis as a target for medical intervention in apicomplexan parasites. Int J Parasitol. 2003;33(9):885–896. doi: 10.1016/s0020-7519(03)00133-4. [DOI] [PubMed] [Google Scholar]
- 7.Rock CO, Cronan JE. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim Biophys Acta. 1996;1302(1):1–16. doi: 10.1016/0005-2760(96)00056-2. [DOI] [PubMed] [Google Scholar]
- 8.Leichert LI, Jakob U. Protein thiol modifications visualized in vivo. PLoS Biol. 2004;2(11):e333. doi: 10.1371/journal.pbio.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rock CO, Cronan JE, Jr, Armitage IM. Molecular properties of acyl carrier protein derivatives. J Biol Chem. 1981;256(6):2669–2674. [PubMed] [Google Scholar]
- 10.De Lay NR, Cronan JE. In vivo functional analyses of the type II acyl carrier proteins of fatty acid biosynthesis. J Biol Chem. 2007;282(28):20319–20328. doi: 10.1074/jbc.M703789200. [DOI] [PubMed] [Google Scholar]
- 11.Waller RF, Reed MB, Cowman AF, McFadden GI. Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. Embo J. 2000;19(8):1794–1802. doi: 10.1093/emboj/19.8.1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waters NC, Kopydlowski KM, Guszczynski T, Wei L, Sellers P, Ferlan JT, Lee PJ, Li Z, Woodard CL, Shallom S, Gardner MJ, Prigge ST. Functional characterization of the acyl carrier protein (PfACP) and beta-ketoacyl ACP synthase III (PfKASIII) from Plasmodium falciparum. Mol Biochem Parasitol. 2002;123(2):85–94. doi: 10.1016/s0166-6851(02)00140-8. [DOI] [PubMed] [Google Scholar]
- 13.Prigge ST, He X, Gerena L, Waters NC, Reynolds KA. The initiating steps of a type II fatty acid synthase in Plasmodium falciparum are catalyzed by pfACP, pfMCAT, and pfKASIII. Biochemistry. 2003;42(4):1160–1169. doi: 10.1021/bi026847k. [DOI] [PubMed] [Google Scholar]
- 14.Sharma SK, Modak R, Sharma S, Sharma AK, Sarma SP, Surolia A, Surolia N. A novel approach for over-expression, characterization, and isotopic enrichment of a homogeneous species of acyl carrier protein from Plasmodium falciparum. Biochem Biophys Res Commun. 2005;330(4):1019–1026. doi: 10.1016/j.bbrc.2005.03.094. [DOI] [PubMed] [Google Scholar]
- 15.Magnuson K, Jackowski S, Rock CO, Cronan JE., Jr Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol Rev. 1993;57(3):522–542. doi: 10.1128/mr.57.3.522-542.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masip L, Veeravalli K, Georgiou G. The many faces of glutathione in bacteria. Antioxid Redox Signal. 2006;8(5–6):753–762. doi: 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- 17.Gould SB, Waller RF, McFadden GI. Plastid evolution. Annu Rev Plant Biol. 2008;59:491–517. doi: 10.1146/annurev.arplant.59.032607.092915. [DOI] [PubMed] [Google Scholar]
- 18.Post-Beittenmiller D, Jaworski JG, Ohlrogge JB. In vivo pools of free and acylated acyl carrier proteins in spinach. Evidence for sites of regulation of fatty acid biosynthesis. J Biol Chem. 1991;266(3):1858–1865. [PubMed] [Google Scholar]
- 19.Yu M, Kumar TR, Nkrumah LJ, Coppi A, Retzlaff S, Li CD, Kelly BJ, Moura PA, Lakshmanan V, Freundlich JS, Valderramos JC, Vilcheze C, Siedner M, Tsai JH, Falkard B, Sidhu AB, Purcell LA, Gratraud P, Kremer L, Waters AP, Schiehser G, Jacobus DP, Janse CJ, Ager A, Jacobs WR, Jr, Sacchettini JC, Heussler V, Sinnis P, Fidock DA. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe. 2008;4(6):567–578. doi: 10.1016/j.chom.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaughan AM, O’Neill MT, Tarun AS, Camargo N, Phuong TM, Aly AS, Cowman AF, Kappe SH. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol. 2008 doi: 10.1111/j.1462-5822.2008.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller S. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol Microbiol. 2004;53(5):1291–1305. doi: 10.1111/j.1365-2958.2004.04257.x. [DOI] [PubMed] [Google Scholar]
- 22.Kimata-Ariga Y, Saitoh T, Ikegami T, Horii T, Hase T. Molecular interaction of ferredoxin and ferredoxin-NADP+ reductase from human malaria parasite. J Biochem. 2007;142(6):715–720. doi: 10.1093/jb/mvm184. [DOI] [PubMed] [Google Scholar]
- 23.Holyoak T, Fenn TD, Wilson MA, Moulin AG, Ringe D, Petsko GA. Malonate: a versatile cryoprotectant and stabilizing solution for salt-grown macromolecular crystals. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 12):2356–2358. doi: 10.1107/s0907444903021784. [DOI] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40(4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu X, Janson CA. Structure of apo acyl carrier protein and a proposal to engineer protein crystallization through metal ions. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 9):1545–1554. doi: 10.1107/S0907444904015422. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26(2):283–291. [Google Scholar]
- 29.Petrek M, Otyepka M, Banas P, Kosinova P, Koca J, Damborsky J. CAVER: a new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinformatics. 2006;7:316. doi: 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38(3):305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 31.Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR, Jr, Fidock DA. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods. 2006;3(8):615–621. doi: 10.1038/nmeth904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trager W, Jensen JB. Continuous culture of Plasmodium falciparum: its impact on malaria research. Int J Parasitol. 1997;27(9):989–1006. doi: 10.1016/s0020-7519(97)00080-5. [DOI] [PubMed] [Google Scholar]
- 33.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma AK, Sharma SK, Surolia A, Surolia N, Sarma SP. Solution structures of conformationally equilibrium forms of holo-acyl carrier protein (PfACP) from Plasmodium falciparum provides insight into the mechanism of activation of ACPs. Biochemistry. 2006;45(22):6904–6916. doi: 10.1021/bi060368u. [DOI] [PubMed] [Google Scholar]
- 35.Roujeinikova A, Simon WJ, Gilroy J, Rice DW, Rafferty JB, Slabas AR. Structural studies of fatty acyl-(acyl carrier protein) thioesters reveal a hydrophobic binding cavity that can expand to fit longer substrates. J Mol Biol. 2007;365(1):135–145. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 36.Cryle MJ, Schlichting I. Structural insights from a P450 Carrier Protein complex reveal how specificity is achieved in the P450(BioI) ACP complex. Proc Natl Acad Sci U S A. 2008;105(41):15696–15701. doi: 10.1073/pnas.0805983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu BN, Zhang YM, Rock CO, Zheng JJ. Structural modification of acyl carrier protein by butyryl group. Protein Sci. 2009;18(1):240–246. doi: 10.1002/pro.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roujeinikova A, Baldock C, Simon WJ, Gilroy J, Baker PJ, Stuitje AR, Rice DW, Slabas AR, Rafferty JB. X-ray crystallographic studies on butyryl-ACP reveal flexibility of the structure around a putative acyl chain binding site. Structure. 2002;10(6):825–835. doi: 10.1016/s0969-2126(02)00775-x. [DOI] [PubMed] [Google Scholar]
- 39.Parris KD, Lin L, Tam A, Mathew R, Hixon J, Stahl M, Fritz CC, Seehra J, Somers WS. Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Structure. 2000;8(8):883–895. doi: 10.1016/s0969-2126(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 40.Kosower NS, Kosower EM. Diamide: an oxidant probe for thiols. Methods Enzymol. 1995;251:123–133. doi: 10.1016/0076-6879(95)51116-4. [DOI] [PubMed] [Google Scholar]
- 41.Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc Natl Acad Sci U S A. 2009;106(2):422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han JC, Han GY. A procedure for quantitative determination of tris(2-carboxyethyl)phosphine, an odorless reducing agent more stable and effective than dithiothreitol. Anal Biochem. 1994;220(1):5–10. doi: 10.1006/abio.1994.1290. [DOI] [PubMed] [Google Scholar]
- 43.Houk J, Singh R, Whitesides GM. Measurement of thiol-disulfide interchange reactions and thiol pKa values. Methods Enzymol. 1987;143:129–140. doi: 10.1016/0076-6879(87)43023-1. [DOI] [PubMed] [Google Scholar]
- 44.Milani M, Balconi E, Aliverti A, Mastrangelo E, Seeber F, Bolognesi M, Zanetti G. Ferredoxin-NADP+ reductase from Plasmodium falciparum undergoes NADP+-dependent dimerization and inactivation: functional and crystallographic analysis. J Mol Biol. 2007;367(2):501–513. doi: 10.1016/j.jmb.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Kimata-Ariga Y, Kurisu G, Kusunoki M, Aoki S, Sato D, Kobayashi T, Kita K, Horii T, Hase T. Cloning and characterization of ferredoxin and ferredoxin-NADP+ reductase from human malaria parasite. J Biochem. 2007;141(3):421–428. doi: 10.1093/jb/mvm046. [DOI] [PubMed] [Google Scholar]
- 46.Nickel C, Rahlfs S, Deponte M, Koncarevic S, Becker K. Thioredoxin networks in the malarial parasite Plasmodium falciparum. Antioxid Redox Signal. 2006;8(7–8):1227–1239. doi: 10.1089/ars.2006.8.1227. [DOI] [PubMed] [Google Scholar]
- 47.Toler S. The plasmodial apicoplast was retained under evolutionary selective pressure to assuage blood stage oxidative stress. Med Hypotheses. 2005;65(4):683–690. doi: 10.1016/j.mehy.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 48.Jocelyn PC. The standard redox potential of cysteine-cystine from the thiol-disulphide exchange reaction with glutathione and lipoic acid. Eur J Biochem. 1967;2(3):327–331. doi: 10.1111/j.1432-1033.1967.tb00142.x. [DOI] [PubMed] [Google Scholar]
- 49.Balconi E, Pennati A, Crobu D, Pandini V, Cerutti R, Zanetti G, Aliverti A. The ferredoxin-NADP reductase/ferredoxin electron transfer system of Plasmodium falciparum. Febs J. 2009 doi: 10.1111/j.1742-4658.2009.07100.x. [DOI] [PubMed] [Google Scholar]
- 50.Modak R, Sinha S, Surolia N. Isothermal unfolding studies on the apo and holo forms of Plasmodium falciparum acyl carrier protein. Role of the 4′-phosphopantetheine group in the stability of the holo form of Plasmodium falciparum acyl carrier protein. Febs J. 2007;274(13):3313–3326. doi: 10.1111/j.1742-4658.2007.05856.x. [DOI] [PubMed] [Google Scholar]
- 51.Cronan JE., Jr Molecular properties of short chain acyl thioesters of acyl carrier protein. J Biol Chem. 1982;257(9):5013–5017. [PubMed] [Google Scholar]
- 52.Gong H, Murphy A, McMaster CR, Byers DM. Neutralization of acidic residues in helix II stabilizes the folded conformation of acyl carrier protein and variably alters its function with different enzymes. J Biol Chem. 2007;282(7):4494–4503. doi: 10.1074/jbc.M608234200. [DOI] [PubMed] [Google Scholar]
- 53.Sharma S, Sharma SK, Modak R, Karmodiya K, Surolia N, Surolia A. Mass spectrometry-based systems approach for identification of inhibitors of Plasmodium falciparum fatty acid synthase. Antimicrob Agents Chemother. 2007;51(7):2552–2558. doi: 10.1128/AAC.00124-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chan DI, Stockner T, Tieleman DP, Vogel HJ. Molecular dynamics simulations of the Apo-, Holo-, and acyl-forms of Escherichia coli acyl carrier protein. J Biol Chem. 2008;283(48):33620–33629. doi: 10.1074/jbc.M805323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.