

Abstract
Human immunodeficiency virus-1 (HIV-1) infection of the nervous system commonly results in neuroinflammation leading to overt neuronal dysfunction and subsequent cognitive and behavioral impairments. The multifaceted disease process, now referred to as HIV-1 associated neurocognitive disorders (HAND), provides a range of molecular targets for adjunctive therapies. One is CEP-1347, an inhibitor of mixed lineage kinases that elicits neuroprotective and anti-inflammatory responses in models of neurodegenerative diseases. As HAND is associated with inflammatory encephalopathy induced by virus infection and mononuclear phagocytes (perivascular macrophages and microglia) immune activation, we investigated whether CEP-1347 could ameliorate disease in laboratory models of HAND. We now demonstrate that CEP-1347 reduces the levels of secreted pro-inflammatory cytokines and chemokines in HIV-1-infected human macrophages and attenuates dose-dependent neurotoxicity in rodent cortical neurons. CEP-1347-treated mice readily achieve therapeutic drug levels in peripheral blood. HIV-1 encephalitic (HIVE) mice, where human virus-infected monocyte-derived macrophages are stereotactically injected into the basal ganglia of CB17 severe combined immunodeficient mice, received daily intraperitoneal injections of CEP-1347 to test its therapeutic benefit. Here, CEP-1347 treatment of HIVE mice showed a dose-dependent reduction in microgliosis. Dendritic integrity and neuronal loss were sustained and prevented, respectively. These results demonstrate that CEP-1347 elicits anti-inflammatory and neuroprotective responses in an HIVE model of human disease and as such warrants further study as an adjunctive therapy for human disease.
Keywords: HIV-1 associated neurocognitive disorders, monocyte-derived macrophages, CEP-1347, neuroprotection, severe combined immunodeficient mice
Human immunodeficiency virus type one (HIV-1) infection commonly leads to immune suppression and CNS disease (1–6). Commonly, cognitive, behavioral, and motor dysfunction falls into a spectrum of neurological abnormalities now termed HIV-1 associated neurocognitive disorders (HAND) (7, 8). Nonetheless and following the wide spread use of antiretroviral drugs, the severity of disease has diminished and now <10% of infected persons show signs and symptoms of substantive neurological deficits (9). Although neuropathological correlates of disease have evolved to subtle changes, in its most severe form, HIV-1 encephalitis (HIVE) dominates. This is characterized by the accumulation of virus-infected mononuclear phagocytes (MP; blood borne macrophages and microglia) in deep grey matter with myelin pallor, astrocytosis and neuronal dropout. Neuronal pathology may be characterized by dendritic pruning and vacuolation (10). Disease ensues as a result of viral infection and MP immune activation with the secretion of viral and cellular neurotoxins (11–15).
In recent years research activities focused on developing adjunctive therapies for disease (16–19). These are important for several reasons. First, the emergence of HIV-1 resistant phenotypes is a common place during chronic antiretroviral therapy (20–24). Second, toxicities of antiretroviral medicines are a frequent cause of noncompliance and treatment failures over time (20, 25). Third, HIV-1 neuropathogenesis while elicited by virus infection is fueled by paracrine immune events amplified by disordered innate immunity. This results in a metabolic encephalopathy and subsequent neuronal injury and death (26). Last, interests in alternative therapies have increased because similar immune-based pathogenic mechanisms are shared among a number of neurodegenerative disorders including amyotrophic lateral sclerosis, multiple sclerosis, Alzheimer’s and Parkinson’s diseases (27–35). Thus, discovery of adjunctive drugs that are effective in improving clinical disease outcomes will have broad applicability. Moreover, it is at least theoretically possible to interrupt disease pathogenesis by therapeutic approaches that do not directly target HIV-1 replication.
Based on these considerations, we pursued the utility of CEP-1347 as an adjunctive medicine for treatment of HAND. CEP-1347 acts as an inhibitor of multi-lineage kinases and can induce neuroprotective responses through its abilities to down-regulate p38 and Jun N-terminal kinase (JNK) phosphorylation (32–36). Macrophages are pivotal cells in an immune response through their phagocytic, antigen presentation and secretory functions. The latter occurs through macrophage secretion of immune modulatory factors including pro-inflammatory cytokines. In neurons, phosphorylation of p38 and JNK leads to apoptotic changes and apoptosis. In macrophages, phosphorylation and activation of multilineage kinases (MLK) results in transcription factor activation, leading to the production of pro-inflammatory cytokines and other immune factors that induce neurotoxic activities (37, 38). CEP-1347 modulates kinase activity following stimulation of MP and neurons by the viral proteins HIV-1 gp120 and Tat (38, 39). This led to the working hypothesis that CEP-1347 may induce neuroprotection and anti-inflammatory activities for HAND. Our results support this notion and show CEP-1347 can attenuate HIV-1ADA mediated neurotoxicity independent of anti-retroviral activities. We now human monocyte derived macrophages (MDM) and in a rodent model of viral encephalitis. The data, taken together, demonstrate that CEP-1347 treatment can lead to neuroprotective responses for HAND and as such be developed for clinical use.
Materials and Methods
Primary human monocyte isolation and HIV-1 infection
Monocytes were obtained from leukopheresis of HIV-1, 2 and hepatitis B seronegative donors and purified by countercurrent centrifugal elutriation. Cells were cultured with 10% heat-inactivated pooled human serum, 1% glutamine (Sigma Chemical Co., St. Louis, MO), 10 μg/ml ciprofloxacin (Sigma), and 1000 U/ml highly purified recombinant human MCSF (a generous gift from Genetics Institute, Inc., Cambridge, MA) in Dulbecco’s Modified Eagle’s Medium (DMEM). After 7 days, the MDM were infected with HIV-1ADA (a macrophage tropic viral strain) at a multiplicity of infection (MOI) of 0.01 (40). The ADA strain was used in these experiments as a result of prior and extensive analyses of macrophage function and neurotoxicity based on strain differences (12). We found in these systems used that the levels of viral replication not the strains per se govern the levels of neurotoxicity. Thus we used the laboratory adapted HIV-1 ADA strain as the levels of viral growth are uniform and not dependent on host cell differences. This ensured that the data acquired was reproducible from one experiment to another regardless of macrophage donor.
HIV reverse transcription assays
HIV replication was examined by measuring viral RT activity as previously described (41). In these assays, 90,000 monocytes were cultured in 96-well plates for seven days, then infected with HIV-1ADA at a MOI of 0.01 and washed 24 hr later to remove virus. Media was changed every other day. To estimate HIV-1 replication, RT activity was determined by incubating 10 mL of sample with a reaction mixture consisting of 0.05% Nonidet P-40 (Sigma) and [3H]dTTP (2 Ci/mmol; Amersham, Arlington Heights, IL) in Tris-HCl buffer (pH 7.9) for 24 hr at 37°C on days 3, 5, 7 and 10. Radiolabeled nucleotides were precipitated on paper filters in an automatic cell harvester (Skatron, Sterling, VA), and incorporated activity was measured by liquid scintillation spectroscopy.
Murine cortical neurons (MCN) cultures
Cerebral cultures, containing neurons and glia in similar proportions to that found in the brain, were derived from the cerebral hemisphere of embryonic C57Bl/6 mice (Jackson Laboratories) on day 17 of gestation and cultured as described previously (42) following dissociation in 0.027% trypsin. Neuron-enriched cells were resuspended in neurobasal medium (Invitrogen, Grand Island, NY) with heat-inactivated fetal calf serum supplemented with B-27, 500 μM glutamine, and 25 μM glutamate then seeded at a density of 2.8 × 105 cells/cm2 on poly-d-lysine-coated 96 and 24-well plates.
Cell viability assays
Ninety thousand human MDM were cultured in 96-well plates and treated with 0, 80, 160 or 220 nM CEP-1347 24 hr before, at, or 4 hr after viral infection. Media was changed every other day and appropriate concentrations of CEP-1347 added. Cultures were monitored for 10 days. Cell viability assays were performed on days 1, 3, 5, 7 and 10 by measuring mitochondrial activity by reduction of tetrazolium salt MTT as described previously (43).
MCN cytotoxicity assays
One million human MDM were cultured in 24-well plates and treated with/without CEP-1347 as outlined above, cultured for 5 days and then washed with PBS. Media was replaced with neural basal media (Invitrogen) every 24 hr. At that time, a ratio of 1:5 (supernatant:media) fluids were placed onto murine cortical neurons for 24 hrs to assess neurotoxicity by neuronal immunohistochemistry. For the neuronal antigen measurements, cells were blocked with 5% normal goat sera for 1 hr then incubated with antibodies to microtubule-associated protein 2 (MAP-2) (neural cell bodies, axons and dendrites, 1:1000) and neuron-specific nuclear protein (NeuN) (1:100) for 1 hr (Chemicon International, Inc., Temecula, CA). Cells were then washed three times with PBS and then incubated with fluorescent antibodies (1:1000 dilutions) to rabbit and mouse Alexa Fluor® 488 (green) and 594 (red) as a secondary antibody (Molecular Probes Inc., Eugene, OR) for 1 hr. Cells were washed twice with water, and water was allowed to cover the wells. Plates were read at 617 and 519 nM for MAP-2 and NeuN using a fluorescent plate reader, and the data were quantified (4 wells/group). Data were expresses as the changes in the mean of fluorescence intensity (MFI) in treated versus non-treated MCN. Lactate dehydrogenize (LDH) levels in neuronal fluids were determined by a cytotoxicity detection kit (Roche, Indianapolis, IN). In this case, MCN culture plates were read at 490 nM. Data were expressed as percent changes in MCN LDH-release amongst conditioned media treatments as compared to 1nM staurosporin (St)-treated cells (listed as 100%). For characterization of neuronal morphology double-immunofluorescence staining was carried out using Alexa Fluor® 488 (green) and 594 (red) as a secondary antibody (Molecular Probes Inc.). Confocal laser scanning imaging was used, which contains Argon, and Argon/Krypton lasers allowing for up to 4 Acousto-Optical Tunable Filter (AOTF) modulated excitation lines of 488 nm, 514 nm, 568 nm and 633 nm (Melles Griot, Prairie Technologies, Inc., Madison, WI). The lasers are connected to SweptField scanner head (Nikon Instruments, Melville, NY) attached with Cascade 512B back illuminated 12-bit CCD digital camera (Roper Scientific, Duluth, GA), which are connected to the side port of Nikon TE2000U Inverted Microscope with a high-resolution X-Y-Z-motorized stage.
Antibody array and flow cytometric analyseis of cytokines and chemokines
One million human MDM were cultured in 24-well plates and infected with HIV-1ADA at an MOI of 0.01 for 1 day, then washed with PBS. Infection was allowed to grow for 5 days. On day 5, cells were treated with 220 nM CEP-1347 for 45 min, washed with PBS, and phenol-red free and serum free media were added. Supernatants were harvested and assayed 24 hr later by Panomic antibody array 3.0 (Panomics, Inc., Fremont, CA) for human cytokines. Panomic antibody array 3.0 measured human cytokines: ApoI/Fas, Leptin, RANTES, ICAM-1, IL-2, IL-7, CTLA, MIP1α, TGFβ, VCAM-1, IL-3, IL-8, Eotaxin, MIP1β, IFNγ, VEGF, IL-4, IL-10, GM-CSF, MIP4, TNF-α, IL-1α, IL-5, IL-12(p40), EGF, MIP-5, TNFRI, IL-1β, IL-6, IL-15, IP-10, MMP3, TNFRII, IL-IRα, IL-6R, and IL-17. For the Cytometric Bead Array (CBA) flow cytometric of secreted chemokine analysis, one million human MDM were cultured in 24-well plates and infected with HIV-1ADA at an MOI of 0.01 for 1 day, then washed with PBS. At time of infection, cells were treated with 80, 160 or 220 nM CEP-1347. Half media exchanges were carried out on day 2 and 4 with respective drug concentrations. On days 3 and 5, 50 μL of supernatant was assayed for each group by CBA Human Inflammation Kit and Chemokine Kit (BD Biosciences, San Diego, CA). Human Inflammation and Chemokine Kit measures IL-8, IL-1β, IL-6, IL-10, TNF-α, IL-12p70 and IL-8, RANTES, MIG, MCP-1, and IP-10, respectively.
Western blot analysis
MDM were infected and cultured with CEP-1347 for 5 days then lysed using RIPA lysis buffer (Fischer Scientific, Pittsburgh, PA) containing additional protease and phosphatase inhibitors (Calbiochem, San Diego, CA). Protein concentrations of whole cell lysates were estimated using BSA assay and stored at −80°C. Cell lysates were allowed to thaw on ice then processed for Western by boiling with SDS laemmli buffer (Bio-Rad Laboratories, Hercules, CA). Whole cell lysate (40 μg protein) was loaded into each well of the Bio-Rad SDS gradient PAGE ranging from 4–15 % and run at 100 V for 1 hour 30 minutes. Protein was transferred to polyvinylidene fluoride (PVDF) membrane (Millipore Billerica, MA) using Bio-Rad semidry transfer apparatus at 25 V for 1 h. The membrane was then blocked with 5% BSA in TBS. Primary antibodies used were anti-phosphorylated-MLK3 (1:1000, Cell Signaling Technology, Inc., Danvers, MA), anti-MLK3 (1:1000, Cell Signaling Technology, Inc.), anti-JNK (1:1000, Cell Signaling), anti-phosphorylated-JNK (1:2000, Cell Signaling Technology, Inc.), anti-p38 (1:1000, Cell Signaling), anti-phosphorylated-p38 (1:1000, Cell Signaling Technology, Inc.), anti-phosphorylated- nuclear factor-kappa beta (NF-κB) p65 (Ser 468) (1:1000, Cell Signaling) anti-NF-κB p65 (1:1000, Cell Signaling), anti-phosphorylated-NF-κB p105 (Ser933) (1:1000, Cell Signaling), anti- NF-κB p105/50 (1:1000, Cell Signaling) and GAPDH (1:1000, Cell Signaling Technology, Inc.). Primary antibodies were detected with HRP-linked secondary antibodies, anti-mouse or anti-rabbit as per antibody used (1:20,000, Chemicon International) followed by detection by ECL femto-detection reagent (Pierce Biotechnology, Rockford, IL) and subsequent exposure to X-ray films for 5–30 min (for phosphoproteins up to 6h). Blots were quantified by inverting scanned images of the blots using the program ImageJ (Naitonal Institutes of Health, NIH) and measuring intensity. Readings were normalized to respective GAPDH expression levels and compared to uninfected, untreated MDM.
Pharmacokinetic study
Five-week old male CB17/scid mice and C57Bl/6 mice were purchased from Charles River Laboratory, Wilmington, WA. Animals were administered i.p. injections either 1.5 or 15.0 mg/kg CEP-1347. The C57B1/6 mice received both drug dosages while the CB17/scid received only the lower 1.5 mg/kg dosage. Blood was extracted by cheek puncture at 0, 0.5, 1, 2, 4, 6 or 8 hr. Blood samples were centrifuged at 5000 rpm for 10 min at 4°C. Plasma was collected and stored at −20°C pending analysis. The plasma samples were prepared for bioanalysis by adding ten volumes of acetonitrile containing an internal standard (alprenolol). After the samples were vortexed and centrifuged, the supernatant was transferred to a 96-well plate for analysis by liquid chromatography/mass spectrometry. The amount of CEP-1347 in the sample was quantified using a plasma standard curve made via serial dilution in a concentration range from 10 to 5000 ng/mL.
SCID mouse model of HIVE
Four-week old male C.B-17/IcrCrl-SCIDbr (CB17/scid) mice were purchased from Charles River Laboratory. Animals were maintained in sterile microisolator cages under pathogen-free conditions in the Laboratory of Animal Medicine at the University of Nebraska Medical Center in accordance with ethical guidelines for care of laboratory animals set forth by the National Institutes of Health. HIV-1ADA-infected MDM (1.5 × 105 cells infected at an MOI of 0.1 in 5 μl) were stereotactically injected intracranially (i.c.) after 1 day of viral infection and referred to as HIVE mice (44). The higher multiplicity used in the animal studies as compared to the in vitro experiments reflected the need to infect a larger proportion of cells prior to cell injections. CEP-1347 was then administered i.p. daily for 7 days at doses 0.5, 1.0, 1.5, 5.0 and 15.0 mg/kg/d (n = 4 mice/treatment group). Vehicle only was the control. CB17/scid mice received i.c. injections of media (sham-operated) and served as additional controls. Seventeen animals were included in each group. Animals were treated with vehicle or CEP-1347 starting 1day post-i.c. injection and for 7 days following MDM injections and CEP-1347 treatments.
Histopathology and image analysis
Brain tissue was collected at necropsy, fixed in 4% phosphate-buffered paraformaldehyde and embedded in paraffin. Paraffin blocks were cut until the injection site of the human MDM was identified. HIV-1 p24 antigen (clone Kal-1; Dako, Carpinteria, CA) was used to test for virus infected human MDM. For each mouse, 30–100 serial (5-μm-thick) sections were cut from the injection site and 3–7 sections (10 sections apart) analyzed. Antibodies to vimentin intermediate filaments (clone VIM 3B4; Boehringer Mannheim, Indianapolis, IN) were used for detection of human cells in mouse brains. Mouse microglia were detected by antibodies to Iba-1 (WAKO, Osaka, Japan), and astrocytes were detected by antibodies for glial fibrillary acidic protein (GFAP; Dako). NeuN, MAP-2 (both from Chemicon International Inc.) and heavy chain (200kDa) neurofilaments (Dako) were used for detection of neurons. Appropriate secondary antibodies and the Vectastain Elite ABC kit (Vector Laboratories, Berlingame, CA) were used to complete the immunohistochemical tests. All sections were counterstained with Mayer’s hematoxylin. The numbers of human MDM and HIV-1 p24 antigen-positive cells were counted with a Nikon Microphot-FXA microscope. All obtained images were imported into Image-Pro Plus, v. 4.0 (Media Cybernetics, Silver Spring, MD) for quantifying area (%) of GFAP, Iba-1, MAP-2 and NeuN positive staining.
Real time polymerase chain reaction (PCR)
The levels of HIV RNA in the injected hemisphere were determined by real time PCR using ABI 7000 prism (Perkin-Elmer, Applied Biosystems, Foster City, CA). Briefly, total RNA was extracted from the brain tissues. The HIV-1gag RNA specific primers and probe were used as previously described (45) and are: forward, 5′-ACA TCA AGC CAT GCA AAT -3′; reverse, 5′-ATC TGG CCT GGT GCA ATA GG -3′; and probe, 5′-CAT CAA TGA GGA AGC TGC AGA ATG GGA TAG A -3′. The reverse primer was used to make cDNA from RNA, which was further amplified using primers and probe at 50°C for 2 min., 95°C for 10 min., and 40 cycles at 95°C for 15 sec. and 60°C for 1 min. Separate GAPDH amplifications were used as an endogenous control to ensure that equal amounts of RNA were used. For GAPDH, Mac-1, TNF, GFAP and IL-10 Taqman gene expression assays were used (Applied Biosystems). Results were expressed as mean copy number ± SEM.
Statistical analysis
Data was analyzed using Excel (Macintosh, 1994) with Student t-test for comparisons. P<0.05 was designated statistically significant.
Results
CEP-1347 Modulates Cytokine and Chemokine Release by MDM
To explore the effects of CEP-1347 on human HIV-1-infected MDM, we performed in vitro studies and evaluated effects of the drug on MDM viability (MTT), viral replication (RT activity), and secretory profile (cytokines). Human MDM were treated from 0 to 220 nM CEP-1347 either 24 hr before, at, or 4 hr post HIV-1 infection for up to 10 days. Cell viability was not affected and no morphological changes were seen (data not shown). The levels of viral replication were measured and also showed no significant change (Figure 1B); only results derived from simultaneous treatment of the cells with CEP-1347 for 24 hr are shown, but similar results were obtained in the cells treated with CEP-1347 before or after infection). To explore the immediate effect of CEP-1347 on cytokine production by human MDM, cells were infected with HIV-1ADA for 24 hr and then cultured for 5 days. Cells were treated with 220 nM CEP-1347 for 45 min, washed and incubated for 24 hr without CEP-1347, at which time supernatants were removed and analyzed using a commercially available human cytokine/chemokine antibody array. Treatment with CEP-1347 reduced the release of macrophage inflammatory proteins [MIP-1β (CCL4), MIP-4 (CCL18), IL-12 p40, and Stromelysin-1 (MMP-3)] by HIV-1-infected macrophages (Figure 1A). Up-regulation of proteins with anti-inflammatory properties, such as IL-4 and TGF-®, TNFR1, and IL-1R antagonist, was also detected (Figure 1A). Three experiments using three donors were used, but results were not pooled. Results are from one experiment but are representative of all three experiments. Each antibody on the array was comprised of two dots; statistics were not used. Although there may be differences between groups, it cannot be stated if these differences are significant. The quantitative cytometric bead array performed on supernatants collected during the course of 5 day viral infection with CEP-1347 treatment (Figure 1C) further revealed that CEP-1347 caused a down regulation of CXCL10/(IP-10) and IL-8 secretion by infected MDM. These data demonstrate that CEP-1347 treatment of HIV-1-infected MDM markedly down-regulates their pro-inflammatory phenotype. Three separate experiments using three donors were carried out. Data presented is from one experiment but is representative of all three experiments
Figure 1.
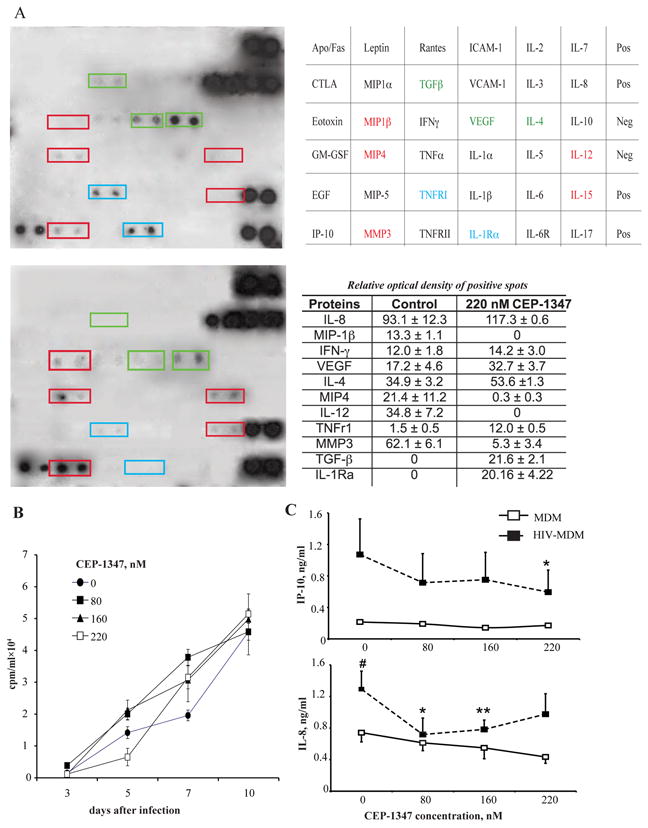
CEP-1347 and MDM secretory and viral replication activities. Effect of CEP-1347 on HIV-1 replication, growth factor, chemokine, pro-inflammatory cytokine and enzyme secretion by human monocyte derived macrophages (MDM). A, TransSignal human cytokine antibody array 3.0 by Panomics and results table. At 5-day post-HIV-1ADA infection, human MDM were treated with 220 nM CEP-1347 (upper blot) or vehicle (lower blot) for 45 min. Cells were washed to remove CEP-1347, and supernatants were harvested 24 hr later. Each cytokine on array is composed of 2 horizontal dots. Data is representative of 3 independent experiments (mean ± SEM). B, CEP-1347 was administered to cultures at a concentration of 0 – 220 nM 24 hrs before, at the time of, and 4 hrs after HIV-1ADA infection. Monocytes were cultivated for seven days, then infected at a MOI of 0.01. Supernatant fluids were collected and assayed for RT activities from zero to 10 days after viral infection. Values represent counts per minute (mean ± SEM) of samples from six independent cultures and are representative of three independent separate experiments when CEP-1347 was added simultaneously with infection. The same results were obtained with drug administration before and after infection. C, In addition, Cytometric Bead Array showed down-regulation of IL-8 and CXCL10/IP-10. Values represent concentrations of chemokines in supernatants collected from three independent experiments, when CEP-1347 was added simultaneously with infection (mean ± SEM). # - P < 0.05 difference between uninfected and HIV-1-infected MDM. *P < 0.05, differences between untreated and treated infected cells. **P < 0.01, differences between untreated and treated infected cells.
CEP-1347 Neuroprotective Activities in HIV-1ADA -infected Human MDM
The data presented above suggested that CEP-1347 treatment reduces the neurotoxicity mediated by HIV-1-infected MDM in vitro. To analyze this possibility, we used primary MCN cultures. Supernatants were collected from human MDM that were treated 24 hr before, at, or 4 hr post HIV-1ADA infection with CEP-1347 concentrations of 0, 80, and 160 nM and cultured for 5 days in the presence of CEP-1347. On day 4, cells were washed, and media replaced with neural basal media (Invitrogen) for 24 hr without CEP-1347. On day 5, supernatants were harvested. These conditioned supernatants were placed on 10-day-old cultures of primary MCN for 24 hr, and neurotoxicity was assessed by measuring MAP-2 and NeuN staining. The results were then verified by conducting LDH release assays. The morphological changes for MCN are shown in Figures 2 and 3 include quantitative MFI measurements for MFI for dendrites and neuronal nuclei (MAP-2 at 617 nm and NeuN at 519 nm wavelength, respectively). Conditioned media-treated MCN was subtracted in all evaluations. In untreated MCN cultures, neurons were evenly distributed and connected with each other with a high density of dendritic branchpoints and long neuritic processes; these cells also contained prominent cell bodies (Figure 2A). This morphology was unaltered by treatment with CEP-1347 (data not shown). After exposure to HIV-1 conditioned media, neurons (Figure 2B) displayed a low density of dendritic nodes, shorter neurites, and a loss of connected processes. In contrast, conditioned media from CEP-1347-treated, HIV-1 infected MDM did not elicit these neurotoxic effects (Figure 2C and D, Figure 3A). In this latter case, neurites were retained and dendritic nodes showed long processes in high density, at similar levels to controls (untreated and uninfected cells); prominent cell bodies were also seen. The greatest increase in MAP-2 immunostaining was seen in cultures that were exposed to supernatant fluids from HIV-1 infected MDM treated with 160 nM CEP-1347 (P < 0.0001, compared to cultures exposed to conditioned media from untreated HIV-1 infected MDM). MCN that were exposed to conditioned supernatants from HIV-1 infected MDM treated with 80 nM CEP-1347 also showed increased levels of MAP-2, but this result did not achieve statistical significance (P < 0.087). NeuN immunostaining was also significantly increased in MCN cultures that were exposed to conditioned supernatants from HIV-1 infected MDM that had been treated with 160 nM of CEP-1347, versus untreated HIV-1 infected MDM (P < 0.0005). MCN exposed to conditioned media from HIV-1 infected MDM treated with 80 nM CEP-1347 also showed elevated numbers of NeuN reactive cells, compared to cultures exposed to supernatants from untreated HIV-1 infected MDM, although this result did not achieve statistical significance. Neuronal protection was also assessed by measuring LDH levels in the extracellular milieu. LDH release from MCN subjected to different MDM conditioned medias were compared to 1nM St-mediated neuronal destruction (100%, Figure 3B). LDH release from neurons treated with conditioned media collected from non-treated MDM was subtracted in these evaluations. Conditioned media collected from HIV-1 infected MDM elicited high levels of LDH release in MCN cultures. Levels of LDH release were lower in cultures exposed to conditioned media from HIV-1 infected MDM cultures that were treated with both 80 and 160 nM CEP-1347 (P = 0.05 and P = 0.013, respectively, Figure 3B).
Figure 2.
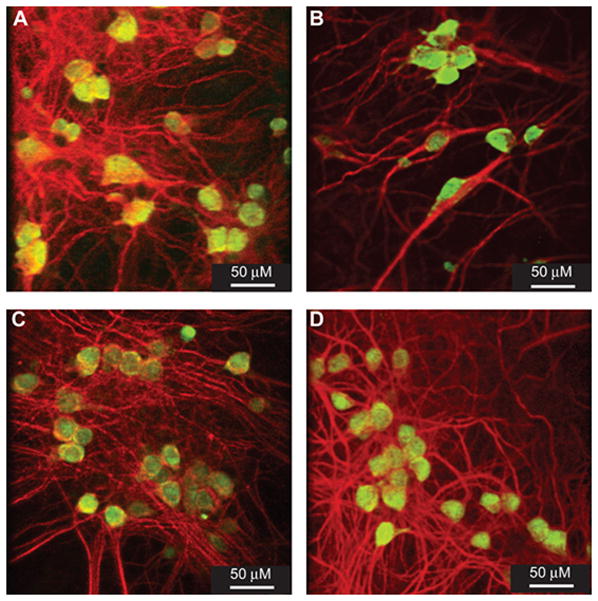
CEP-1347 neuroprotective activities. Human MDMs were infected with HIV-1ADA (MOI of 0.01) and treated simultaneously with CEP-1347 (80 or 160 nM). Conditioned media was collected at day 5 after infection. Twenty four hrs before conditioned media harvest MDM were washed to remove CEP-1347 and neurobasal media was added to the cultures. Conditioned media thus collected were added to primary MCN to measure neurotoxicity levels. Neurons were stained immunocytochemically for MAP-2 (red) and NeuN (green) to assess neuronal integrity. A, Primary MCN culture treated with conditioned media from control MDM. B, Primary MCN cultures cultivated with HIV-1 infected MDM conditioned media. These neuronal cultures demonstrate both decreased dendritic density (MAP-2 immunostaining) and reduced neural density (number of NeuN positive cells). C, HIV-1 infected MDM conditioned media that received 80 nM CEP-1347 show partial protection of dendritic and neural density. D, Conditioned media from HIV-1 infected MDM treated with 160 nM CEP-1347 show protection of dendritic and neural densities. Original magnification x400.
Figure 3.

Quantitative analyses of CEP-1347 induced neuroprotective activities. Quantitative analyses of MAP-2 and NeuN staining in primary MCN culture treated with conditioned media from CEP-1347 treated, HIV-1-infected MDM. A, Analysis of dendritic density, as assessed by MAP-2 staining and neural density, measured by NeuN staining. Data demonstrate MFI at 617 nm (MAP-2) and 519 nm (NeuN) for culture wells containing treated MCN. Values are mean ± SEM for quadruplicate cultures. Data are representative of three independent experiments using three different donors. B, LDH concentration from replicate primary cortical neural cultures with CM from CEP-1347 treated, HIV-1-infected MDM; values are mean ± SEM of quadruplicate cultures. Data expressed are percentage changes in LDH release compared to MCN treated with 1nM St, 100%. Data are representative of three independent experiments using three different donor conditioned medias.
CEP-1347 Pharmacokinetic Analyses
In order to gain insight into the metabolism of CEP-1347 in our SCID mouse model of HIVE, we measured plasma concentrations of CEP-1347 following i.p. administration at dosages of 1.5 mg/kg and 15.0 mg/kg in CB17/scid mice (n = 4 mice/treatment/time point) and collected blood at varying time points as depicted in Figure 4.
Figure 4.
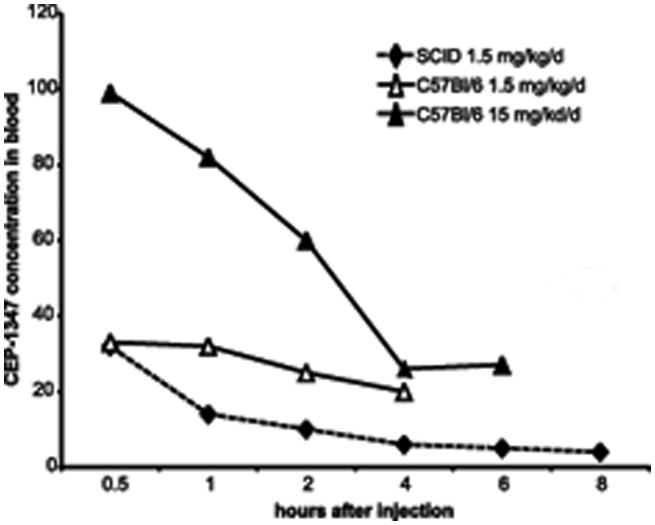
Pharmacokinetic analyses of CEP-1347 levels in plasma of experimentally treated mice. The plasma concentration of CEP-1347 was measured following i.p. dosing of mice with 1.5 or 15.0 mg/kg of CEP-1347. For this experiment, both C57Bl/6 and CB17 SCID mice were treated. However, the CB17 SCID mice were treated only with the lower 1.5 mg/kg dose of CEP-1347. Blood samples were collected at selected time points (0.5, 1, 2, 4, 6 and 8 h), and plasma was separated for analyses. Samples were analyzed by liquid chromatography/mass spectrometry. Values are mean ± SEM from 4 animals. Data are represented as a line graph (n = 4 mice/treatment).
CEP-1347 affects Neuroinflammatory Responses in HIVE Mice
Human HIV-1ADA infected MDM were stereotactically injected into the basal ganglia of CB17/scid mice. Histopathological changes observed in murine brain tissue paralleled those seen for human HIVE. This included HIV-1 infection in perivascular and parenchymal human MDM, the formation of multinucleated giant cells, astrocytosis, and neuronal dropout. Therapeutic efficacy of different dosages of CEP-1347 was evaluated using immunohistochemistry in HIVE scid mice after administration. Human HIV-1ADA-infected MDM were stereotactically injected in the basal ganglia of CB17/scid mice (n = 4 mice/treatment group). CEP-1347 was then administered i.p. daily for 7 days at doses of 0 mg/kg/d (vehicle only), 0.5 mg/kg/d, 1.0 mg/kg/d, 1.5 mg/kg/d, 5.0 mg/kg/d and 15.0 mg/kg/d. Morphological changes in astrocytes surrounding the lesion site were evaluated using GFAP immunostaining. Quantitation of GFAP expression used three sections from each animal: immediately before, at, and immediately after the lesion site. GFAP expression was quantified by determining GFAP positive area as a percentage of total image area per microscopy field. Astrogliosis was not unaltered in CEP-1347 treated HIVE mice, when compared to vehicle-only treated HIVE mice (Figure 5E to H). Serial brain sections were stained with Iba-1 to determine activation of microglia. Microglia stained area was calculated as a percentage of the area of the entire microscopy field. Microglial activation was significantly decreased in all CEP-1347 treated groups when compared to vehicle-only treated HIVE mice (2.71 ± 0.61). Results of microglial activation for 0.5 mg/kg/d, 1.0 mg/kg/d, 1.5 mg/kg/d, 5.0 mg/kg/d and 15.0 mg/kg/d were 0.91 ± 0.16 (P < 0 .0001 compared to HIVE mice treated with vehicle alone), 1.13 ± 0.26 (P < 0 .01), 0.87 ± 0.40 (P < 0 .001), 0.75 ± 0.17 (P < 0 .0001), and 0.41 ± 0.06 (P < 0 .00001), respectively (Figure 5A to C). Microglial activation decreased with increasing concentrations of CEP-1347 (Figure 5D).
Figure 5.

CEP-1347 suppresses neuroinflammatory responses in murine HIVE. SCID mice were injected with HIV-1ADA infected MDM with/out CEP-1347 treatment. MDM were propagated in suspension cultures and infected with HIV-1ADA at an MOI of 0.1 for 5 days prior to brain injections. All animals were sacrificed seven days after i.c. cell injection. Immunohistological images of the basal ganglia are shown. Serial 5-μM sections containing the injection area were stained for Iba-1 (A, B, C) and GFAP (E, F, G). A, Immunohistological stain for Iba-1 in sham-operated murine brain. B, Replicate image to (A) of area adjacent to injection site in HIVE murine brain stained for Iba-1; note the increased in Iba-1 reactive microglia. C, Immunohistological stain for Iba-1 in tissue adjacent to the injection site from HIVE murine brain treated with 15 mg/kg/d CEP-1347; note the reduction in Iba-1 staining compared to B. D, Quantitation of Iba-1 staining. Values are mean ± SEM obtained from analyzing four brain tissue sections for each experimental animal (4 mice/group). E, Immunohistological image of uninfected murine brain stained for GFAP. F, Untreated HIVE brain and G, HIVE brain treated with 15 mg/kg/d CEP-1347. H, Quantification of GFAP staining. Values are mean ± SEM obtained from analyzing three brain tissue sections in each experimental animal (4 mice/group).
CEP-1347 Elicits Neuroprotective Responses in HIVE mice
To determine if CEP-1347 was neuroprotective in HIVE mice, brain tissue from treated animals was subjected to immunostaining with anti-NeuN and anti-MAP-2 antibodies. The area analyzed corresponded to the same area used to assess astrocytosis and microglial activation. Significant neuronal loss was present beyond the lesion site in vehicle-only treated HIVE mice; however, CEP-1347 was neuroprotective in a dose-dependent manner (Figure 6). Neurons stained with anti-NeuN were counted using several brain sections from each mouse surrounding the lesion area. HIVE mice receiving CEP-1347 had significantly increased numbers of neurons surrounding the lesion when compared to vehicle-only treated HIVE animals (13.9 ± 3.3). HIVE mice receiving 0.5, 1.0, 1.5, 5.0 and 15.0 mg/kg/d of CEP1347 had neuron counts of 29.3 ± 3.6 (P < 0.05 compared to vehicle-treated HIVE mice), 37.0 ± 4.0 (P < 0.001), 52.7 ± 3.7 (P < 0.00001), 51.8 ± 4.6 (P < 0.0001), and 51.1 ± 5.2 (P < 0.0002), respectively. Dendritic processes, stained with anti-MAP-2 from the same field as neurons were counted, and were quantified as a percentage of the area of the entire microscopy field. Several brain sections were used to determine dendritic process loss surrounding the lesion area. Dendritic loss was significantly decreased in HIVE mice receiving CEP-1347 compared to HIVE mice receiving vehicle-only treatment (19.5 ± 1.5). Mice receiving 0.5, 1.0, 1.5, 5.0, and 15.0 mg/kg/d had 28.6 ± 1.4 (P < 0.0001 compared to vehicle-treated HIVE mice), 29.3 ± 1.5 (P < 0.0001), 43.2 ± 1.1 (P < 0.00001), 45.5 ± 1.1 (P < 0.00001), and 47.0 ± 1.2 (P < 0.00001), respectively. CEP-1347 treated HIVE mice showed decreased neuronal loss and dendritic processes when compared to vehicle-only treated HIVE mice. A dose-dependent protective effect was observed at CEP-1347 levels up to 1.5 mg/kg/d, but no increased therapeutic advantage was observed for doses above 1.5 mg/kg/d (Figure 6).
Figure 6.

CEP-1347 elicits a neuroprotective effect in HIVE mice. Tissue sections from HIVE mice were stained for MAP-2 (pink) and NeuN (brown) (A, B, C). Neuronal integrity was assessed in areas of focal encephalitis in CEP-1347 treated mice. A, Immunohistochemical stain for MAP-2 and NeuN in control sham-operated mice. B, Immunohistological image of HIV-1-infected murine brain stained for MAP-2 and NeuN, 7 days post injection. C, Immunohistological image of the brain from an HIV-1-infected mouse treated with 15 mg/kg/d of CEP-1347, stained for MAP-2 and NeuN, 7 days post injection. Original magnification 200×. D, Quantitation of MAP-2 immunoreactivity with increasing doses of CEP-1347. Values are mean ± SEM from multiple images obtained at original magnification 200× per mouse (4 mice/group). E, Quantitation of NeuN immunoreactivity with increasing doses of CEP-1347. Values are mean ± SEM from multiple images per mouse (4 mice/group).
CEP-1347 was further assessed in the same HIVE model for tests that included both histology and quantitative real time RT-PCR. In this study 1.5 mg/kg/d was used for treatments (n = 17 mice/treatment group). SCID mice were stereotactically injected in the basal ganglia with human HIV-1ADA-infected MDM and administered CEP-1347 i.p. daily for 7 days. Animals were then sacrificed, and brain tissue removed for immunohistology or RT-PCR. Histopathological changes observed included formation of multi-nucleated giant cells, astrocytosis and neuronal dropout (Table 1).
Table 1.
CEP-1347 affects on neuroinflammation and neuroprotection in HIVE mice*
| Treatment groups | ||
|---|---|---|
| Parameters | Control | CEP-1347a |
| Astrogliosis (GFAP immunostaining) | 21.4 ± 1.16 | 27.1 ± 0.66 |
| Microgliosis (Iba-1 immunostaining) | 8.56 ± 0.24 | 3.61 ± 0.17b |
| Dendritic density (MAP-2 immunostaining) | 21.1 ± 3.0 | 29.0 ± 2.3c |
| Neuron density (NeuN immunostaining) | 32.6 ± 2.2 | 35.0 ± 2.13 |
| Degenerating neurons number (Neurofilament immunostaining) | 6.5 ± 0.81 | 2.1 ± 0.65d |
Data was assessed by immunohistochemical assays and shown as percent stained brain subregions for GFAP, Iba, MAP-2, NeuN (ImagePro program) and for numbers of neuronal bodies as stained by neurofilament antibodies.
CEP-1347 was administered by the i.p. route at a dose of 1.5 mg/kg/d for 7 days following stereotactic intracranial injection of human HIV-1ADA-infected MDM into the basal ganglia CB17/scid mice (n = 17 mice/treatment group).
P <0.00001;
P < 0.05;
P <0.01.
Human MDM were identified by immunostaining with vimentin and were present in the area adjacent to the stereotactic injection site. In HIVE mice treated with 1.5 mg/kg/d CEP-1347 the mean number of MDM was 271.3 ± 121.0 compared with 275.2 ± 123.0 MDMs for HIVE vehicle-only treated animals. HIV-1 infected MDM in brain were immunostained with anti-p24 antigen and quantified. CEP-1347 treated mice had 35.4 ± 19.0 HIV-1 infected MDM; whereas, HIVE vehicle-only treated mice had 61.8 ± 28.4 HIV-1 infected MDM (the difference was not statistically significant). Antiretroviral activity was assessed as a percentage of MDM infected with HIV-1 and compared between CEP-1347 treated HIVE mice (13.0% ± 15.7%) and vehicle-only treated HIVE mice (22.5% ± 23.1%); once again, the difference was not statistically significant.
Using quantitative RT-PCR analysis revealed no difference in HIV-1 RNA expression between groups (data not shown). GFAP expression was also unaltered, as determined by both histologic and RT-PCR analyses (data not shown). In contrast, microglial activation, as assessed by Iba-1 immunostaining, was significantly decreased in CEP-1347 treated HIVE mice as compared to vehicle-only treated HIVE mice (3.61 ± 0.17, 8.56 ± 0.24, respectively; P < 0.00001). This result was confirmed by analysis of RNA transcript levels for Mac-1, another marker for activated microglia. HIVE mice treated with CEP-1347 have reduced levels of Mac-1 mRNA, when compared to vehicle-only treated animals (0.09 ± 0.01 and 0.20 ± 0.03, respectively; P < 0.05). We also measured the mRNA expression levels of TNF-α, a pro-inflammatory cytokine that plays a major role in inflammation, and IL-10, an anti-inflammatory cytokine, in the brains by RT-PCR. TNF-α is up-regulated in many neurodegenerative disorders including HIV and is elevated in HIVE mice (46). Neither TNF-α, nor IL-10 mRNA expression levels were significantly different in CEP-1347 treated HIVE mice compared to vehicle-only treated HIVE mice.
CEP-1347 and MDM Signaling by Western Blot Assays
We next assessed the relationships between HIV-1-infected MDM secreted proteins and cell signaling pathways that are known to be modified by inhibition of MLK activity (47, 48). These experiments were done based on the fact that perivascular brain macrophages are the principal target cell in disease and responsible for much of the neuronal impairments observed (1). Mechanisms for CEP-1347 actions were sought. To this end, we performed Western blot assays for the kinases ERK1/2, p38 and JNK. These are involved in apoptotic pathways and previously shown to have lower phosphorylation levels in HIV-1gp120 or Tat-stimulated MP following CEP-1347 treatment (38, 39). Levels of phosphorylated to total protein were used to analyze differences between infected and uninfected groups. CEP-1347 (220 nM) treatment groups all showed reduced levels of phosphorylated ERK and p38. However, such reductions were limited. Phosphorylated pJNK (pJNK) was increased in both HIV and further increased in the HIV/LPS treated cells. CEP reduced the level of pJNK in both conditions but modestly so. The ratio of phosphorylated to total ERK was unchanged in uninfected groups treated with CEP-1347 (Figure 7). While HIV infection leads to increased pMLK3, the effect of CEP 1347 is less clear.
Figure 7.
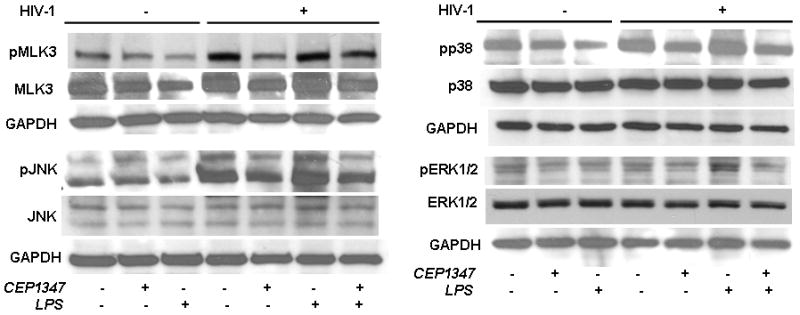
MAPK kinase pathways in HIV-1ADA infected human MDM treated with CEP-1347. Human monocytes collected after centrifugal elutriation were cultured for seven days in 1,000 U/ml MCSF then infected with HIV-1ADA viral stock at an MOI of 0.01 for 6 h with/out 220 nM CEP-1347. CEP-1347 was continued through the experiment. Uninfected MDM with or without CEP-1347 was used as controls. Viral replication continued for 5 days. Viral infection at 5 days showed RT activity 10-fold above background levels. Replicate control and HIV-1 infected MDM were treated with LPS at 100 ng/ml for 24 h when all cells were harvested for Western blot assays. Cell lysates were analyzed using Abs specific MLK3, phospho-MLK3, ERK1/2, phospho-ERK1/2, p38, phospho-p38, JNK and phospho-JNK. The data presented here is a representative of four independent experiments.
In parallel with MLK3 pathway, we also pursued analysis for the NF-κB pathway to determine the potential for the robust anti-inflammatory responses seen by CEP1347. In this context we analyzed both the cytoplamic and nuclear fractions of the HIV-1 infected macrophages as well as controls with/without LPS treatment. We observed (1) - a modest increase in the p65 and phospho-p105 level in the nuclear fractions with HIV infection as compared to uninfected control, and (2) - a significant increase in case of LPS treated HIV infected sample (Figure 8). We also observed a limited reduction in the level of p65 in CEP1347 treated HIV infected/LPS stimulated cells (Figure 8). However, we did not see significant differences in the phospho-p65 and p65 levels in the cytoplasmic fractions. CEP1347 failed to elicit specific response for NF-κB signaling pathway.
Figure 8.
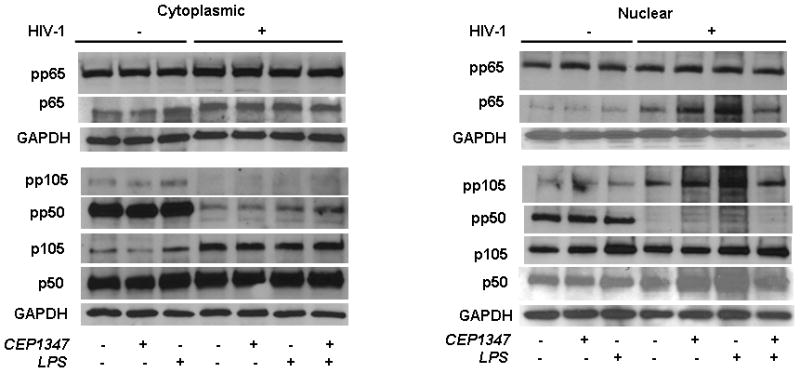
CEP-1347 does not affect NF-κB pathways in HIV-1 infected human MDM. Human monocytes collected after centrifugal elutriation were cultured for seven days in 1,000 U/ml MCSF then infected with HIV-1ADA viral stock at an MOI of 0.01 for 6 h with or without CEP-g1347 at 220 nM. CEP-1347 was continued through the experiment. Uninfected MDM with or without CEP-1347 was used as controls. Viral infection at 5 days showed RT activity 10-fold above background levels. Replicate control and HIV-1 infected MDM were treated with LPS at 100 ng/ml for 24 h when all cells were harvested for Western blot assays. Cells were fractionated into cytosolic and nuclear fractions using Nuclear/Cytosol fractionation Kit (BioVision, Cat. No- K266-25). The fractionation was performed according to the manufacturers instructions. Both cytosolic and nuclear fractions were then analyzed by using specific Ab to p65, p105/p50 and its phophorylated derivatives (Cell Signaling Technology). For all analyses GAPDH was used as loading control. The data presented here is a representative of three independent experiments.
Discussion
Progressive HIV-1 infection commonly elicits neurological impairments despite aggressive anti-retroviral regimens, and significant improvements in the quality and duration of life may be balanced with demonstrable cognitive decline (49–54). Such decline is linked, in part, to variable penetration of anti-retroviral drugs across the blood-brain barrier (BBB), difficulties with drug toxicity and compliance, and viral mutation (50–57).
The complex, multifactorial pathogenesis of HAND presents therapeutic opportunities with respect to the development of novel adjunctive therapies. For example, viral infection and immune activation of MP play an essential role in mediated neuronal damage in disease and can be exploited for therapeutic gain (58–60). Medicines that interfere with neuroinflammation or that protect neurons from damage can be expected to have a positive effect on the pathogenesis of HAND. CEP-1347 is a semi-synthetic indolocarbazole that inhibits MLK by acting as a competitive adenosine triphosphate (ATP) site inhibitor (61). MLK regulate neuronal-programmed cell death through the MAPK cascade by phosphorylation and activation of the transcription factor c-jun (32, 62). CEP-1347 is also neuroprotective by down-regulating p38 and JNK activation in neurons (63) and can prevent the activation of human monocytes following exposure to HIV-1 gp120 and Tat (39). Prevention of phosphorylation of p38 and JNK may lead to decreased secretion of cytokines and inflammatory factors from MP as well as the overall activation of MP in response to HIV. Thus, we hypothesized CEP-1347 could attenuate HIV-1 associated neuroinflammation and in so doing protect against neuronal damage and apoptosis seen as a consequence of MP infection and immune activation.
In this report, we demonstrate that CEP-1347 is a potent inhibitor of the neurotoxic secretome for HIV-1 infected macrophages. First, we found that the drug did not alter the levels of viral replication. Second, we showed that secretion of macrophage inflammatory proteins CCL4/MIP-1β, CCL18/MIP-4, IL-12, and Stromelysin-1 (MMP-3) was reduced by CEP-1347 after 45 min. This may reduce neurotoxicity both by preventing the release of pro-inflammatory cytokines and chemokines within the brain and by preventing the entry of monocytes into the CNS (64–66). Up-regulation of anti-inflammatory cytokines was also observed following exposure of MDM to CEP-1347. Levels of IL-4, TGF-β, IL-1R antagonist, and soluble TNFR1 secretion were all increased in CEP-1347 treated HIV-1-infected MDM, as compared to vehicle-treated HIV-1 infected MDM. These anti-inflammatory molecules may possess neuroprotective activity (67–73). CEP-1347’s upregulation of vascular endothelial growth factor (VEGF) production may also have neuroprotective significance (74).
Productive HIV-1 infection in human macrophages results in increased secretion of pro-inflammatory cytokines linked to secondary disease processes (11, 12) and suggesting the engagement of multiple kinases (75–77). In prior studies, CEP-1347 was shown to affect the balance between phosphorylation/dephosphorylation of downstream inflammatory eventsincluding ERK1/2, p38, and JNK (32, 61, 77–79). Thus, we performed cell based phosphorylation assays for these three kinases. Activation of ERK is known to be associated with cell growth and differentiation (80); whereas, activation of JNK and p38 MAPK are associated with growth arrest, apoptosis and oncogenic transformation (81). HIV-1-infection was also shown to increase JNK phosphorylation, a pro-apoptotic pathway (82). In the current report, CEP-1347 showed some effect on JNK activation, consistent with CEP-1347’s activity as a potent inhibitor of JNK activation. HIV-1 infection also increased the phosphorylation (activation) of p38 MAPK, and CEP-1347 reduced this level of phosphorylation although the effects were quite modest. We also examined the effect of HIV-1ADA infection on activation of MLK3. Unexpectedly, CEP-1347 treatment resulted in a decline in the total level of MLK3 within the cells. This suggests the possibility that MLK3 may positively autoregulate its own expression; studies will be required to further examine this hypothesis.
The modulation of anti-inflammatory responses by CEP-1347 also suggested that engagement of MLK was linked to reductions in neurotoxicity mediated by HIV-1-infected MDM. This hypothesis was tested by adding conditioned supernatants from HIV-1 infected MDM that were treated with CEP-1347 or vehicle, to 10 day old cultures of primary MCN as a commonly used target for HIV-1-mediated toxicity (42, 83). These experiments showed that culture media from CEP-1347 treated HIV-1 infected MDM elicited lower levels of neuronal death and dendritic damage than conditioned supernatants from untreated HIV-1 infected MDM, as assessed by MAP-2 and NeuN immunostaining. Thus, CEP-1347 proved to elicit neuroprotective responses in HIVE mice model as previously was shown for antiretrovirals and anti-inflammatory drugs (84, 85).
We examined the morphology, activation status and survival of key CNS cell populations in this model, either in the presence or absence of CEP-1347, including astrocytes, microglia and neurons. HIVE mice treated with CEP-1347 were found to have lower levels of microglial activation and diminished neuronal loss when compared to untreated HIVE mice in a dose-dependent fashion. The in vivo studies with CEP-1347 involved an initial analysis of the bio-distribution and pharmacokinetics of the drug in CB17/scid mice. This showed relatively low steady-state plasma levels of drug in mice given 1.5 mg/kg/d [a dose similar to that used in human subjects enrolled in the CEP-1347 PRECEPT trial (86)]. The measured plasma levels of 4–32 ng/mL of CEP-1347 in mice over 8 hours suggest that the biodistribution and/or pharmacokinetics of CEP-1347 is markedly different in animals treated i.p., as compared to human subjects receiving an oral dose of 10 to 50 mg CEP-1347, which resulted in plasma levels between 20 to 200 ng/mL. As a result, we performed experiments at a second dose of CEP-1347 (1.5 mg/kg/d), since this was expected to elicit plasma levels of drug equivalent to those in human subjects treated with the CEP-1347. Our experiments also revealed significant differences in the pharmacokinetics of CEP-1347 in C57BL/6 versus CB17/scid mice. The basis for this is unclear at present.
In light of our analysis of the plasma levels of CEP-1347, we were somewhat surprised to observe that even a low in vivo dose of CEP-1347, capable of eliciting relatively modest steady-state plasma levels of drug (< 20 nM), had a striking and statistically significant ability to reduce microglial activation and increase neuronal protection in vivo. This unexpected finding suggests that CEP-1347’s direct neuroprotective activity, may work together with its ability to induce anti-inflammatory responses thereby eliciting a very robust in vivo neuroprotective effect. This in vivo synergy may depend on CEP-1347’s ability to interfere with deleterious feedback interactions between inflammatory MP and vulnerable neurons. Interestingly, however, such affects had little to do with the NF-κB pathways.
Our in vitro analyses examined the effects of CEP-1347 only on purified cultures of MDM. This may explain why the dose levels of CEP-1347 necessary to achieve similar biological effects were higher in our in vitro model systems (80 nM or greater), than in vivo. MP infected with and activated by HIV also may require higher concentrations of CEP-1347 in vitro to effect any change in regards to more robust alterations in cytokine and inflammatory factors. Regardless of these in vitro considerations, the major take-home message of this study is that CEP-1347 elicits significant neuroprotective effects in vivo, providing support for the use of mixed lineage kinase inhibitors in HAND treatment. In conclusion, we demonstrate that the mixed lineage kinase inhibitor, CEP-1347, elicits an anti-inflammatory phenotype in HIV-1 infected human MDM and that this results in a reduction in neurotoxic activities. Our data also show that, in a murine model for HIVE, CEP-1347 reduces microglial activation and dendritic damage and speeds neuronal survival. Collectively, these findings support the idea that CEP-1347 can be of therapeutic potential for HAND.
Acknowledgments
We thank Ms. Robin Taylor for outstanding administrative and computer support. We thank Drs. Deborah Galinas and Lisa Aimone and Ms. Rebecca Morey for plasma sample and pharmacokinetic analyses and Dr. Donna Bozyczko-Coyne for thoughtful discussions and research design. We thank Ms. Nan Gong for cortical neuron culture and Mr. Michael T. Jacobson for confocal microscopy.
Footnotes
Supported by P01 NS43985, P20RR15635, R37 NS36126, PO1 NS31492, R01NS034239 (H.E.G.) and P01MH64570 and R01MH56838 (H.A.G.).
References
- 1.Lipton SA, Gendelman HE. Dementia associated with the acquired immunodeficiency syndrome. New Engl J Med. 1995;16:934–940. doi: 10.1056/NEJM199504063321407. [DOI] [PubMed] [Google Scholar]
- 2.Michaels J, Sharer LR, Epstein LG. Human immunodeficiency virus type 1 (HIV-1) infection of the nervous system: a review. Immunodefic Rev. 1988;1:71–104. [PubMed] [Google Scholar]
- 3.Everall IP, Luthert PJ, Lantos PL. Neuronal loss in the frontal cortex in HIV infection [see comments] Lancet. 1991;337:1119–1121. doi: 10.1016/0140-6736(91)92786-2. [DOI] [PubMed] [Google Scholar]
- 4.Swindells S, Zheng J, Gendelman HE. HIV-associated dementia: new insights into disease pathogenesis and therapeutic interventions. Aids Patient Care STDS. 1999;13:153–163. doi: 10.1089/apc.1999.13.153. [DOI] [PubMed] [Google Scholar]
- 5.Kure K, Llena JF, Lyman WD, Soeiro R, Weidenheim KM, Hirano A, Dickson DW. Human immunodeficiency virus-1 infection of the nervous system: an autopsy study of 268 adult, pediatric, and fetal brains. Hum Pathol. 1991;22:700–710. doi: 10.1016/0046-8177(91)90293-x. [DOI] [PubMed] [Google Scholar]
- 6.An SF, Scaravilli F. Early HIV-1 infection of the central nervous system. Arch Anat Cytol Pathol. 1997;45:94–105. [PubMed] [Google Scholar]
- 7.Villa GSA, Moro E, Tavolozza M, Antinori A, De Luca A, Murri R, Tamburrini E. Cognitive impairment in asymptomatic stages of HIV infection. A longitudinal study. Eur Neurol. 1996;36:125–133. doi: 10.1159/000117228. [DOI] [PubMed] [Google Scholar]
- 8.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Letendre S, Ances B, Gibson S, Ellis RJ. Neurologic complications of HIV disease and their treatment. Top HIV Med. 2007;15:32–39. [PubMed] [Google Scholar]
- 10.Masliah E, Ge N, Achim CL, DeTeresa R, Wiley CA. Patterns of neurodegeneration in HIV encephalitis. NeuroAIDS. 1996;1:161–173. doi: 10.1300/j128v01n01_08. [DOI] [PubMed] [Google Scholar]
- 11.Anderson E, Zink W, Xiong H, Gendelman HE. HIV-1-associated dementia: a metabolic encephalopathy perpetrated by virus-infected and immune-competent mononuclear phagocytes. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S43–54. doi: 10.1097/00126334-200210012-00004. [DOI] [PubMed] [Google Scholar]
- 12.Nukuna A, Gendelman HE, Limoges J, Rasmussen J, Poluektova L, Ghorpade A, Persidsky Y. Levels of human immunodeficiency virus type 1 (HIV-1) replication in macrophages determines the severity of murine HIV-1 encephalitis. J Neurovirol. 2004;10(Suppl 1):82–90. doi: 10.1080/753312757. [DOI] [PubMed] [Google Scholar]
- 13.Nottet H, Gendelman H. Unraveling the neuroimmune mechanisms for the HIV-1-associated cognitive/motor complex. Immunology Today. 1995;16:441–448. doi: 10.1016/0167-5699(95)80022-0. [DOI] [PubMed] [Google Scholar]
- 14.Nath A, Geiger J. Neurobiological aspects of human immunodeficiency virus infection: neurotoxic mechanisms. Prog Neurobiol. 1998;54:19–33. doi: 10.1016/s0301-0082(97)00053-1. [DOI] [PubMed] [Google Scholar]
- 15.Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J. 1996;320:595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry SW, Norman JP, Gelbard HA. Adjunctive therapies for HIV-1 associated neurologic disease. Neurotox Res. 2005;8:161–166. doi: 10.1007/BF03033827. [DOI] [PubMed] [Google Scholar]
- 17.Schifitto G, Peterson DR, Zhong J, Ni H, Cruttenden K, Gaugh M, Gendelman HE, Boska M, Gelbard H. Valproic acid adjunctive therapy for HIV-associated cognitive impairment: a first report. Neurology. 2006;66:919–921. doi: 10.1212/01.wnl.0000204294.28189.03. [DOI] [PubMed] [Google Scholar]
- 18.Schifitto G, Yiannoutsos CT, Simpson DM, Marra CM, Singer EJ, Kolson DL, Nath A, Berger JR, Navia B. A placebo-controlled study of memantine for the treatment of human immunodeficiency virus-associated sensory neuropathy. J Neurovirol. 2006;12:328–331. doi: 10.1080/13550280600873835. [DOI] [PubMed] [Google Scholar]
- 19.Schifitto G, Sacktor N, Marder K, McDermott MP, McArthur JC, Kieburtz K, Small S, Epstein LG. Randomized trial of the platelet-activating factor antagonist lexipafant in HIV-associated cognitive impairment. Neurological AIDS Research Consortium. Neurology. 1999;53:391–396. doi: 10.1212/wnl.53.2.391. [DOI] [PubMed] [Google Scholar]
- 20.Tozser J. HIV inhibitors: problems and reality. Ann N Y Acad Sci. 2001;946:145–159. doi: 10.1111/j.1749-6632.2001.tb03909.x. [DOI] [PubMed] [Google Scholar]
- 21.Deeks SG, Hoh R, Neilands TB, Liegler T, Aweeka F, Petropoulos CJ, Grant RM, Martin JN. Interruption of treatment with individual therapeutic drug classes in adults with multidrug-resistant HIV-1 infection. J Infect Dis. 2005;192:1537–1544. doi: 10.1086/496892. [DOI] [PubMed] [Google Scholar]
- 22.Cunningham PH, Smith DG, Satchell C, Cooper DA, Brew B. Evidence for independent development of resistance to HIV-1 reverse transcriptase inhibitors in the cerebrospinal fluid. Aids. 2000;14:1949–1954. doi: 10.1097/00002030-200009080-00010. [DOI] [PubMed] [Google Scholar]
- 23.Sacktor N, Lyles RH, Skolasky R, Kleeberger C, Selnes OA, Miller EN, Becker JT, Cohen B, McArthur JC. HIV-associated neurologic disease incidence changes:: Multicenter AIDS Cohort Study, 1990–1998. Neurology. 2001;56:257–260. doi: 10.1212/wnl.56.2.257. [DOI] [PubMed] [Google Scholar]
- 24.Dore GJ, Correll PK, Li Y, Kaldor JM, Cooper DA, Brew BJ. Changes to AIDS dementia complex in the era of highly active antiretroviral therapy. Aids. 1999;13:1249–1253. doi: 10.1097/00002030-199907090-00015. [DOI] [PubMed] [Google Scholar]
- 25.Gebo KA. HIV and aging: implications for patient management. Drugs Aging. 2006;23:897–913. doi: 10.2165/00002512-200623110-00005. [DOI] [PubMed] [Google Scholar]
- 26.Gendelman HE, Diesing Scott, Gelbard Harris, Swindells Susan. The Neuropathogenesis of HIV-1 Infection. Elsevier; London: 2004. [Google Scholar]
- 27.Bellizzi MJ, Lu SM, Gelbard HA. Protecting the synapse: evidence for a rational strategy to treat HIV-1 associated neurologic disease. J Neuroimmune Pharmacol. 2006;1:20–31. doi: 10.1007/s11481-005-9006-y. [DOI] [PubMed] [Google Scholar]
- 28.Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA. Glycogen synthase kinase 3 beta (GSK-3 beta) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007;2:93–96. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- 29.Gorantla S, Liu J, Wang T, Holguin A, Sneller HM, Dou H, Kipnis J, Poluektova L, Gendelman HE. Modulation of innate immunity by copolymer-1 leads to neuroprotection in murine HIV-1 encephalitis. Glia. 2008;56:223–232. doi: 10.1002/glia.20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, Takikawa O, Munn DH, Gendelman HE, Persidsky Y. Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood. 2005;106:2382–2390. doi: 10.1182/blood-2005-04-1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang LH, Paden AJ, Johnson EM., Jr Mixed-lineage kinase inhibitors require the activation of Trk receptors to maintain long-term neuronal trophism and survival. J Pharmacol Exp Ther. 2005;312:1007–1019. doi: 10.1124/jpet.104.077800. [DOI] [PubMed] [Google Scholar]
- 33.Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK. Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochim Biophys Acta. 2004;1697:89–101. doi: 10.1016/j.bbapap.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 34.Bozyczko-Coyne D, O’Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL, Scott RW. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J Neurochem. 2001;77:849–863. doi: 10.1046/j.1471-4159.2001.00294.x. [DOI] [PubMed] [Google Scholar]
- 35.Namgung U, Xia Z. Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N-terminal protein kinase 3 and p38 mitogen-activated protein kinase. J Neurosci. 2000;20:6442–6451. doi: 10.1523/JNEUROSCI.20-17-06442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, Goetz M, Lucius R, Herdegen T, Hanisch UK. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia. 2005;50:235–246. doi: 10.1002/glia.20173. [DOI] [PubMed] [Google Scholar]
- 37.Lund S, Porzgen P, Mortensen AL, Hasseldam H, Bozyczko-Coyne D, Morath S, Hartung T, Bianchi M, Ghezzi P, Bsibsi M, Dijkstra S, Leist M. Inhibition of microglial inflammation by the MLK inhibitor CEP-1347. J Neurochem. 2005;92:1439–1451. doi: 10.1111/j.1471-4159.2005.03014.x. [DOI] [PubMed] [Google Scholar]
- 38.Bodner A, Maroney AC, Finn JP, Ghadge G, Roos R, Miller RJ. Mixed lineage kinase 3 mediates gp120IIIB-induced neurotoxicity. J Neurochem. 2002;82:1424–1434. doi: 10.1046/j.1471-4159.2002.01088.x. [DOI] [PubMed] [Google Scholar]
- 39.Sui Z, Fan S, Sniderhan L, Reisinger E, Litzburg A, Schifitto G, Gelbard HA, Dewhurst S, Maggirwar SB. Inhibition of mixed lineage kinase 3 prevents HIV-1 Tat-mediated neurotoxicity and monocyte activation. J Immunol. 2006;177:702–711. doi: 10.4049/jimmunol.177.1.702. [DOI] [PubMed] [Google Scholar]
- 40.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kalter DC, Nakamura JA, Turpin JA, Baca LM, Hoover DL, Dieffenbach C, Ralph P, Gendelman HE, Meltzer MS. Enhanced HIV replication in macrophage colony-stimulating factor- treated monocytes. J Immunol. 1991;146:298–306. [PubMed] [Google Scholar]
- 42.Zheng J, Ghorpade A, Niemann D, Cotter RL, Thylin MR, Epstein L, Swartz JM, Shepard RB, Liu X, Nukuna A, Gendelman HE. Lymphotropic virions affect chemokine receptor-mediated neural signaling and apoptosis: implications for human immunodeficiency virus type 1-associated dementia. J Virol. 1999;73:8256–8267. doi: 10.1128/jvi.73.10.8256-8267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan DM. Tetrazolium (MTT) assay for cellular viability and activity. Methods Mol Biol. 1998;79:179–183. doi: 10.1385/0-89603-448-8:179. [DOI] [PubMed] [Google Scholar]
- 44.Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, Patil A, Nottet HS, Epstein L, Gelbard H, Flanagan E, Reinhard J, Pirruccello SJ, Gendelman HE. Human immunodeficiency virus encephalitis in SCID mice. Am J Pathol. 1996;149:1027–1053. [PMC free article] [PubMed] [Google Scholar]
- 45.Poluektova L, Gorantla S, Faraci J, Birusingh K, Dou H, Gendelman HE. Neuroregulatory Events Follow Adaptive Immune-Mediated Elimination of HIV-1-Infected Macrophages: Studies in a Murine Model of Viral Encephalitis. J Immunol. 2004;172:7610–7617. doi: 10.4049/jimmunol.172.12.7610. [DOI] [PubMed] [Google Scholar]
- 46.Persidsky Y, Buttini M, Limoges J, Bock P, Gendelman HE. An analysis of HIV-1-associated inflammatory products in brain tissue of humans and SCID mice with HIV-1 encephalitis. J Neurovirol. 1997;3:401–416. doi: 10.3109/13550289709031186. [DOI] [PubMed] [Google Scholar]
- 47.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 48.Roux PP, Dorval G, Boudreau M, Angers-Loustau A, Morris SJ, Makkerh J, Barker PA. K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem. 2002;277:49473–49480. doi: 10.1074/jbc.M203428200. [DOI] [PubMed] [Google Scholar]
- 49.Dore GJ, Hoy JF, Mallal SA, Li Y, Mijch AM, French MA, Cooper DA, Kaldor JM. Trends in incidence of AIDS illnesses in Australia from 1983 to 1994: the Australian AIDS cohort. J Acquir Immune Defic Syndr Hum Retrovirol. 1997;16:39–43. doi: 10.1097/00042560-199709010-00006. [DOI] [PubMed] [Google Scholar]
- 50.Enting RH, Hoetelmans RM, Lange JM, Burger DM, Beijnen JH, Portegies P. Antiretroviral drugs and the central nervous system. Aids. 1998;12:1941–1955. doi: 10.1097/00002030-199815000-00005. [DOI] [PubMed] [Google Scholar]
- 51.Clifford DB. Human immunodeficiency virus-associated dementia. Arch Neurol. 2000;57:321–324. doi: 10.1001/archneur.57.3.321. [DOI] [PubMed] [Google Scholar]
- 52.Lipton SA. Treating AIDS dementia. Science. 1997;276:1629–1630. doi: 10.1126/science.276.5319.1629b. [DOI] [PubMed] [Google Scholar]
- 53.Major EO, Rausch D, Marra C, Clifford D. HIV-associated dementia. Science. 2000;288:440–442. doi: 10.1126/science.288.5465.439d. [DOI] [PubMed] [Google Scholar]
- 54.Ferrando S, van Gorp W, McElhiney M, Goggin K, Sewell M, Rabkin J. Highly active antiretroviral treatment in HIV infection: benefits for neuropsychological function. Aids. 1998;12:F65–70. doi: 10.1097/00002030-199808000-00002. [DOI] [PubMed] [Google Scholar]
- 55.Lipton SA, Gendelman HE. Seminars in medicine of the Beth Israel Hospital, Boston. Dementia associated with the acquired immunodeficiency syndrome. N Engl J Med. 1995;332:934–940. doi: 10.1056/NEJM199504063321407. [DOI] [PubMed] [Google Scholar]
- 56.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 57.Gartner S. HIV infection and dementia. Science. 2000;287:602–604. doi: 10.1126/science.287.5453.602. [DOI] [PubMed] [Google Scholar]
- 58.Lipton S, Kieburtz K. Prospects for neuropharmacological intervention in AIDS dementia. In: Gendelman H, Lipton S, Epstein L, Swindells S, editors. The Neurology of AIDS. Chapman-Hall; N Y: 1998. pp. 377–381. [Google Scholar]
- 59.Lipton SA. Neuronal injury associated with HIV-1: approaches to treatment. Annu Rev Pharmacol Toxicol. 1998;38:159–177. doi: 10.1146/annurev.pharmtox.38.1.159. [DOI] [PubMed] [Google Scholar]
- 60.Navia BA, Dafni U, Simpson D, Tucker T, Singer E, McArthur JC, Yiannoutsos C, Zaborski L, Lipton SA. A phase I/II trial of nimodipine for HIV-related neurologic complications. Neurology. 1998;51:221–228. doi: 10.1212/wnl.51.1.221. [DOI] [PubMed] [Google Scholar]
- 61.Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem. 2001;276:25302–25308. doi: 10.1074/jbc.M011601200. [DOI] [PubMed] [Google Scholar]
- 62.Silva RM, Kuan CY, Rakic P, Burke RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson’s disease. Mov Disord. 2005;20:653–664. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- 63.Lannuzel A, Barnier JV, Hery C, Tan HV, Guibert B, Gray F, Vincent JD, Tardieu M. Human immunodeficiency virus type 1 and its coat protein gp120 induce apoptosis and activate JNK and ERK mitogen-activated protein kinases in human neurons. Ann Neurol. 1997;42:847–856. doi: 10.1002/ana.410420605. [DOI] [PubMed] [Google Scholar]
- 64.Xiong H, Boyle J, Winkelbauer M, Zeng YC, Zheng J, Gendelman HE. IL-8 Inhibits Neuronal Long-term Potentiation: A Potential Mechanism for Cognitive Decline in HIV-1-Associated Dementia. 7th Conference on Retroviruses and Opportunistic Infections; San Francisco: Foundation for Retrovirology and Human Health; 2000. [Google Scholar]
- 65.Xiong H, Zeng YC, Lewis T, Zheng J, Persidsky Y, Gendelman HE. HIV-1 infected mononuclear phagocyte secretory products affect neuronal physiology leading to cellular demise: relevance for HIV-1-associated dementia. J Neurovirol. 2000;6(Suppl 1):S14–23. [PubMed] [Google Scholar]
- 66.Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, Fiala M, Way D, Kim KS, Witte MH, Weinand M, Carhart L, Gendelman HE. Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155:1599–1611. doi: 10.1016/S0002-9440(10)65476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park KW, Lee DY, Joe EH, Kim SU, Jin BK. Neuroprotective role of microglia expressing interleukin-4. J Neurosci Res. 2005;81:397–402. doi: 10.1002/jnr.20483. [DOI] [PubMed] [Google Scholar]
- 68.Clarke RM, Lyons A, O’Connell F, Deighan BF, Barry CE, Anyakoha NG, Nicolaou A, Lynch MA. A pivotal role for interleukin-4 in atorvastatin-associated neuroprotection in rat brain. J Biol Chem. 2008;283:1808–1817. doi: 10.1074/jbc.M707442200. [DOI] [PubMed] [Google Scholar]
- 69.Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- 70.Krieglstein K, Strelau J, Schober A, Sullivan A, Unsicker K. TGF-beta and the regulation of neuron survival and death. J Physiol Paris. 2002;96:25–30. doi: 10.1016/s0928-4257(01)00077-8. [DOI] [PubMed] [Google Scholar]
- 71.Terrado J, Monnier D, Perrelet D, Vesin D, Jemelin S, Buurman WA, Mattenberger L, King B, Kato AC, Garcia I. Soluble TNF receptors partially protect injured motoneurons in the postnatal CNS. Eur J Neurosci. 2000;12:3443–3447. doi: 10.1046/j.1460-9568.2000.00240.x. [DOI] [PubMed] [Google Scholar]
- 72.Bartfai T, Sanchez-Alavez M, Andell-Jonsson S, Schultzberg M, Vezzani A, Danielsson E, Conti B. Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Ann N Y Acad Sci. 2007;1113:173–177. doi: 10.1196/annals.1391.022. [DOI] [PubMed] [Google Scholar]
- 73.Laske C, Oschmann P, Tofighi J, Kuehne SB, Diehl H, Bregenzer T, Kraus J, Bauer R, Chatzimanolis N, Kern A, Traupe H, Kaps M. Induction of sTNF-R1 and sTNF-R2 by interferon beta-1b in correlation with clinical and MRI activity. Acta Neurol Scand. 2001;103:105–113. doi: 10.1034/j.1600-0404.2001.103002105.x. [DOI] [PubMed] [Google Scholar]
- 74.Gora-Kupilas K, Josko J. The neuroprotective function of vascular endothelial growth factor (VEGF) Folia Neuropathol. 2005;43:31–39. [PubMed] [Google Scholar]
- 75.Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164:355–362. doi: 10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nguyen DG, Yin H, Zhou Y, Wolff KC, Kuhen KL, Caldwell JS. Identification of novel therapeutic targets for HIV infection through functional genomic cDNA screening. Virology. 2007;362:16–25. doi: 10.1016/j.virol.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 77.Falsig J, Porzgen P, Lotharius J, Leist M. Specific modulation of astrocyte inflammation by inhibition of mixed lineage kinases with CEP-1347. J Immunol. 2004;173:2762–2770. doi: 10.4049/jimmunol.173.4.2762. [DOI] [PubMed] [Google Scholar]
- 78.Muller GJ, Geist MA, Veng LM, Willesen MG, Johansen FF, Leist M, Vaudano E. A role for mixed lineage kinases in granule cell apoptosis induced by cytoskeletal disruption. J Neurochem. 2006;96:1242–1252. doi: 10.1111/j.1471-4159.2005.03590.x. [DOI] [PubMed] [Google Scholar]
- 79.Harris C, Maroney AC, Johnson EM., Jr Identification of JNK-dependent and -independent components of cerebellar granule neuron apoptosis. J Neurochem. 2002;83:992–1001. doi: 10.1046/j.1471-4159.2002.01219.x. [DOI] [PubMed] [Google Scholar]
- 80.Otsuka M, Goto K, Tsuchiya S, Aramaki Y. Phosphatidylserine-specific receptor contributes to TGF-beta production in macrophages through a MAP kinase, ERK. Biol Pharm Bull. 2005;28:1707–1710. doi: 10.1248/bpb.28.1707. [DOI] [PubMed] [Google Scholar]
- 81.Harper SJ, LoGrasso P. Signalling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cell Signal. 2001;13:299–310. doi: 10.1016/s0898-6568(01)00148-6. [DOI] [PubMed] [Google Scholar]
- 82.Mishra S, Mishra JP, Kumar A. Activation of JNK-dependent pathway is required for HIV viral protein R-induced apoptosis in human monocytic cells: involvement of antiapoptotic BCL2 and c-IAP1 genes. J Biol Chem. 2007;282:4288–4300. doi: 10.1074/jbc.M608307200. [DOI] [PubMed] [Google Scholar]
- 83.Dou H, Birusingh K, Faraci J, Gorantla S, Poluektova LY, Maggirwar SB, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J Neurosci. 2003;23:9162–9170. doi: 10.1523/JNEUROSCI.23-27-09162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Limoges J, Persidsky Y, Poluektova L, Rasmussen J, Ratanasuwan W, Zelivyanskaia M, McClernon D, Lanier E, Gendelman H. Evaluation of antiretroviral drug efficacy for HIV-1 encephalitis in SCID mice. Neurol. 2000;54:379–389. doi: 10.1212/wnl.54.2.379. [DOI] [PubMed] [Google Scholar]
- 85.Persidsky Y, Limoges J, Rasmussen J, Zheng J, Gearing A, Gendelman HE. Reduction in glial immunity and neuropathology by a PAF antagonist and an MMP and TNFalpha inhibitor in SCID mice with HIV-1 encephalitis. J Neuroimmunol. 2001;114:57–68. doi: 10.1016/s0165-5728(00)00454-9. [DOI] [PubMed] [Google Scholar]
- 86.Wang LH, Johnson EM, Jr, Shoulson I, Lang AE, Bozyczko-Coyne D P. I. On behalf of the Parkinson Study Group. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2008;71:462–463. doi: 10.1212/01.wnl.0000324506.93877.5e. [DOI] [PubMed] [Google Scholar]