

Abstract
Reaction of prostaglandin H synthase (PGHS) isoforms 1 or 2 with peroxide forms a radical at Tyr385 that is required for cyclooxygenase catalysis, and another radical at Tyr504, whose function is unknown. Both tyrosyl radicals are transient and rapidly dissipated by reductants, suggesting that cyclooxygenase catalysis might be vulnerable to suppression by intracellular antioxidants. Our initial hypothesis was that the two radicals are in equilibrium and that their proportions and stability are altered upon binding of fatty acid substrate. As a test, we examined the effects of three competitive inhibitors (nimesulide, flurbiprofen and diclofenac) on the proportions and stability of the two radicals in PGHS-2 pretreated with peroxide. Adding nimesulide after ethyl peroxide led to some narrowing of the tyrosyl radical signal detected by EPR spectroscopy, consistent with a small increase in the proportion of the Tyr504 radical. Neither flurbiprofen nor diclofenac changed the EPR linewidth when added after peroxide. In contrast, the effects of cyclooxygenase inhibitors on the stability of the preformed tyrosyl radicals were dramatic. The half-life of total tyrosyl radical was 4.1 min in the control, >10 hr with added nimesulide, 48 min with flurbiprofen, and 0.8 min with diclofenac. Stabilization of the tyrosyl radicals was evident even at substoichiometric levels of nimesulide. Thus, the inhibitors had potent, structure-dependent, effects on the stability of both tyrosyl radicals. This dramatic modulation of tyrosyl radical stability by cyclooxygenase site ligands suggests a mechanism for regulating the reactivity of PGHS tyrosyl radicals with cellular antioxidants.
Cyclooxygenase (COX)1 catalysis by PGHS-1 and -2 requires tyrosyl radical formation by oxidizing equivalents generated by reaction with peroxides (1, 2). Reaction of either PGHS isoform with peroxide generates radicals at Tyr385 and at Tyr504 (3, 4); both Tyr385 and Tyr504 are conserved in all vertebrate PGHS proteins discovered so far (5). Tyr385 is in the COX active site (6, 7) and the Tyr385 radical is well established as the immediate oxidant of fatty acid in the COX catalytic cycle for both PGHS isoforms (2). Tyr504 is far from the COX pocket where fatty acid binds (6, 7), and the Tyr504 radical is not itself capable of COX catalysis (3, 4). However, mutation of Tyr504 does decrease COX activation efficiency by ~50% in both PGHS-1 and -2 (3, 4), suggesting that the Tyr504 radical has an important regulatory role. The crystallographic data indicate that Tyr504 is buried in the protein and thus less exposed to reductants in the solvent than is Tyr385 (6, 7), and we suspect that Tyr504 serves to sequester peroxide-derived oxidant.
The peroxide-driven formation of tyrosyl radicals at both Tyr385 (giving a wide doublet, or WD, EPR signal) and at Tyr504 (giving a narrow singlet, or NS, EPR signal) (reaction 1 in Scheme 1) makes it important to understand the kinetic and mechanistic relationships between the two tyrosyl radicals. The Tyr385 radical appears more quickly than the Tyr504 radical (3, 8), but the Tyr504 radical still forms in the Y385F mutant (3, 9). Thus, oxidation of Tyr385 and Tyr504 by higher oxidation states of the heme may involve parallel reactions with different rates rather than sequential reactions with an obligatory Tyr385 radical intermediate. There is also the question of whether radical formed at Tyr385 can later be redistributed to Tyr504 or vice versa. Such redistribution, perhaps gated by fatty acid binding at the COX site, would presumably be required for Tyr504 to function as an oxidant reservoir.
Scheme 1.
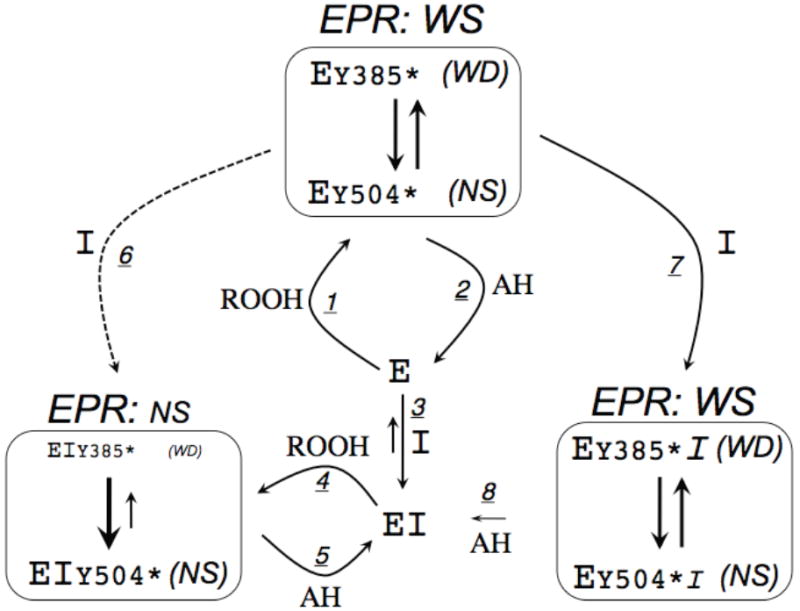
Hypothetical interconversions of PGHS-2 (E) in reactions with peroxide (ROOH), reducing compounds (AH) and COX inhibitors (I). The radical at Tyr385 (wide doublet EPR signal; WD) and Tyr504 (narrow singlet EPR signal; NS) are shown as Y385* and Y504*, respectively. The WD and NS signals combine to give a wide singlet (WS) EPR spectrum. Steps 6 and 7 represent the expected and observed outcomes, respectively, when COX inhibitor interacts with PGHS-2 containing preformed tyrosyl radicals.
Direct testing of the effects of fatty acid on the tyrosyl radicals is difficult because of the rapid oxidation of bound fatty acid to a carbon-centered radical (9). However, competitive COX inhibitors can be used as unreactive substrate analogs. Binding of some COX inhibitors, such as nimesulide, to PGHS-2 before reaction with peroxide leads to accumulation of radical at Tyr504 and not at Tyr385 (3); this is shown as steps 3 and 4 in Scheme 1. The ability of pretreatment with some COX inhibitors to change the radical distribution when added before reaction with peroxide suggested that these inhibitors might redistribute preformed radical from Tyr385 to Tyr504 (step 6 in Scheme 1) and thus demonstrate a dynamic redox connection between the two tyrosines.
Another major issue regarding the COX mechanism centers on the instability of the Tyr385 and Tyr504 radicals. The tyrosyl radicals have been found to be transient species in both PGHS isoforms, dissipating in a few minutes even under optimal conditions in reactions with stoichiometric amounts of peroxide (10-12). Moderate levels of exogenous reductants greatly deplete tyrosyl radicals in purified PGHS (10, 13) (shown as steps 2, 5 and 8 in Scheme 1), an action that is particularly paradoxical because such compounds are known to promote COX catalytic turnover (11, 13-16). The sensitivity of tyrosyl radicals in purified PGHS to quenching by exogenous reductants is also difficult to reconcile with cellular COX catalysis because PGHS-1 and -2 face the lumen of the endoplasmic reticulum (17, 18), where the redox potential is ~ -0.2 V and an efficient antioxidant system is present (19, 20).
In the present studies we tested three COX inhibitors for their actions on a preformed mixture of Tyr385 and Tyr504 radicals in hPGHS-2, using experiments in which the enzyme was reacted with peroxide before addition of inhibitor. The results indicate that inhibitor binding to PGHS-2 containing Tyr385 and Tyr504 radicals does redistribute a small amount of radical to Tyr504 (step 6 in Scheme 1). However, the major action of inhibitor binding is to generate a distinct, new complex (via step 7 in Scheme 1) whose reactivity with reductant (via step 8 in Scheme 1) is greatly decreased in the case of nimesulide and flurbiprofen, and increased in the case of diclofenac. This demonstration that COX site ligands can modulate the stability of PGHS-2 tyrosyl radicals should help to decipher how the radical-dependent COX catalysis can be sustained in a reducing cellular environment.
EXPERIMENTAL PROCEDURES
Reagents
Heme, protease inhibitor, glutathione, imidazole and diclofenac were purchased from Sigma; nimesulide and (S)-flurbiprofen were from Cayman; arachidonic acid was from NuChek Preps; diethyldithiocarbamate and phenol were from Fisher; Tween 20 was from Anatrace; and Ni-NTA agarose was from Qiagen. The EtOOH solution was a gift from Dr. G. Barney Ellison, University of Colorado; the concentrations of working stocks were determined from the absorbance at 230 nm (43 mM-1cm-1).
Expression and purification of hPGHS-2
cDNA coding for human PGHS-2 with a hexa-histidine tag at the mature N-terminus was constructed essentially as described by Smith et al. (21) and used to generate recombinant baculovirus for expression of the protein in Sf9 cells following published procedures (22). Purification procedures were carried out at 0-4 °C. The Sf9 cell pellets were suspended in 25 mM NaPi / 20 mM imidazole / 5 mM EDTA / 5 mM diethyldithiocarbamate / 1 mM phenol, pH 7.4 containing protease inhibitor cocktail (Sigma #P2714) at 0.2 g cells / ml and homogenized by sonication. The membrane fraction was collected by centrifugation at 150,000 × g for 1 hr and resuspended in 25 mM NaPi / 100 mM NaCl / 20 mM imidazole / 0.1 mM phenol, pH 7.4 (4 ml / g cells) using a Dounce homogenizer. The hPGHS-2 was solubilized by addition of 0.2 vol of 10% Tween 20 and stirring for 90 min before residual membrane materials were removed by centrifugation at 150,000 × g for 1 hr. The detergent extract was mixed with Ni-NTA agarose (0.33 ml / g cells) and mixed by gentle inversion overnight. The mixture was poured into a chromatography column and unbound material drained out before the affinity resin was washed with six vol of 25 mM NaPi / 200 mM NaCl / 20 mM imidazole / 0.1 mM phenol / 0.1 mM glutathione / 0.1% Tween 20, pH 7.4 and then with four vol of 25 mM NaPi / 100 mM NaCl / 20 mM imidazole / 0.1 mM phenol / 0.1 mM glutathione / 0.1% Tween 20, pH 7.4. Specifically bound material was eluted with repeated, 1.0 ml aliquots of 25 mM NaPi / 100 mM NaCl / 200 mM imidazole / 0.1 mM phenol / 0.1 mM glutathione / 0.1% Tween 20, pH 7.4. The active fractions were pooled and concentrated to 2.0 ml by ultrafiltrattion on a PM-30 membrane (Millipore) before gel filtration on a 10DG column (BioRad) equilibrated and eluted with 50 mM KPi, pH 7.2, containing 100 mM NaCl, 0.1% Tween 20 and 0.01% NaN3. The active fractions were pooled, mixed with 0.25 vol of glycerol, and stored at -70 °C.
Enzyme assay
Cyclooxygenase activity was assayed with a Clark-type polarographic oxygen electrode at 30 °C in 3.0 ml of 100 mM KPi, pH 7.2, containing 1.0 mM phenol, 100 μM arachidonate, 1.0 μM heme and 0.1% Tween-20; one unit of activity gives a velocity of 1 nmol O2 / min (22). The cyclooxygenase specific activity of purified hPGHS-2 used in these studies was 29 kunits/mg.
Tyrosyl radical kinetics
The preformed mixture of Tyr385 and Tyr504 radicals was generated by adding 5 μl of 1.14 mM EtOOH solution to 250 μl of 11 μM PGHS-2 holoenzyme in a quartz EPR tube at 0 °C, and mixing for 6-8 s with a prechilled nichrome wire loop. Where desired, cyclooxygenase inhibitor was added next, in a few microliters of ethanol, and mixing was continued for a further 6-8 s, still at 0 °C. Each initial sample produced in this fashion was then quickly frozen in a dry ice/ethanol bath before storage in liquid nitrogen until EPR analysis. Reaction time courses were generated by putting the initial samples through repeated cycles of warming and refreezing. For each cycle, the sample was transferred from liquid nitrogen to the dry ice/ethanol bath for 1-2 min, then moved to the ice bath for the desired length of time, then refrozen in the dry ice/ethanol bath, and finally chilled in liquid nitrogen for EPR analysis.
EPR spectrometry
EPR spectra were recorded with a Bruker EMX spectrometer at 9.2-9.3 GHz with a modulation amplitude of 2.0 G, a modulation frequency of 100 kHz, a time constant of 328 ms, a microwave power of 1.0 mW, and a temperature of 118-125 K. Radical concentrations were determined by double integration of the EPR signals with reference to a 1 mM copper sulfate standard (23).
RESULTS
Tyrosyl radicals in PGHS-2 pretreated with cyclooxygenase inhibitors
As noted above, pretreatment of PGHS-2 with nimesulide before reaction with peroxide is known to favor accumulation of radical at Tyr504 relative to Tyr385 (3, 8). Based on prior experience with PGHS-1 (24), it was anticipated that other COX inhibitors might not affect the distribution of radical, and would thus be useful as negative controls for the planned experiments where inhibitor is added to PGHS-2 with preformed tyrosyl radicals. To identify appropriate compounds, several COX inhibitors were tested for their effects on the tyrosyl radical EPR when added to PGHS-2 before reaction with a large excess of peroxide (Fig. 1). A wide singlet EPR signal (WS), 29 G peak-trough, was observed in control hPGHS-2. As expected (8), pretreatment of PGHS-2 with nimesulide resulted in a much narrower peroxide-induced singlet, 25 G peak to trough, which originates from enrichment in the Tyr504 radical (3). The results for the other inhibitors tested fell into two groups (Fig. 1). Pretreatment with diclofenac, flufenamate, meclofenamate, and niflumate resulted in singlet EPR signals that were narrower than the WS in the control. On the other hand, pretreatment with indomethacin or flurbiprofen did not noticeably narrow the peroxide-induced radical signal. Thus, preincubation with diclofenac, flufenamate, meclofenamate, and niflumate increased the proportion of Tyr504 radical relative to Tyr385 radical upon subsequent reaction with peroxide, whereas preincubation with indomethacin or flurbiprofen did not alter the proportions of the two tyrosyl radicals.
Figure 1.
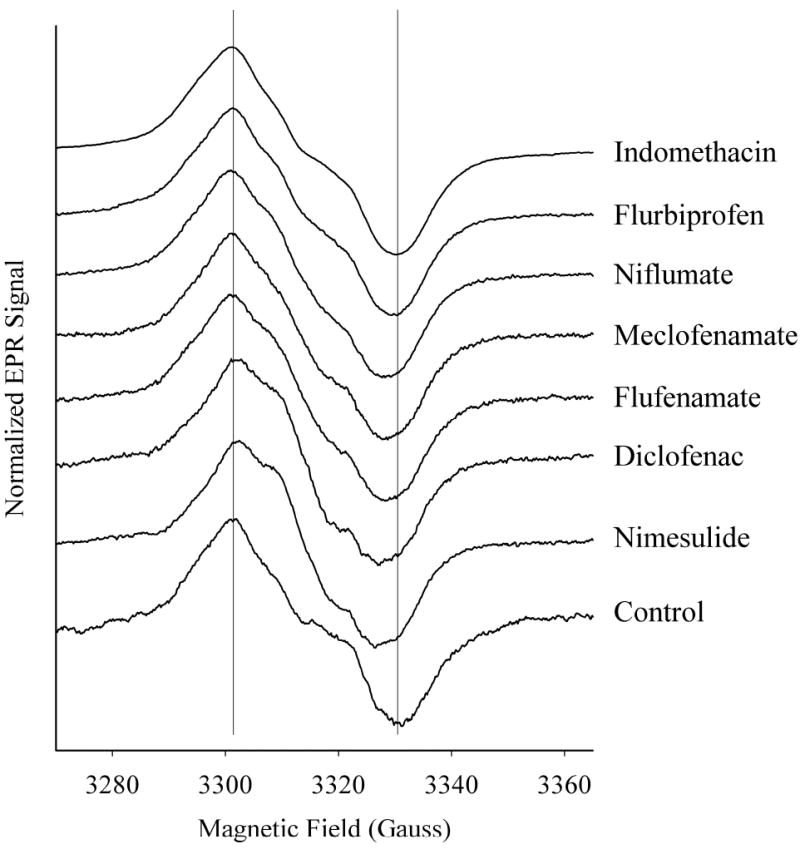
Effects of cyclooxygenase inhibitors on tyrosyl radical EPR spectrum when added to PGHS-2 before reaction with peroxide. Aliquots of hPGHS-2 holoenzyme (11 μM in 0.1 M KPi, pH 7.2 containing 11 μM phenol, 5% glycerol and 0.1% Tween 20) were incubated with 1.5 eq of the indicated inhibitor on ice for at least 30 min before reaction with 10 eq of EtOOH for 6-8 s. The mixtures were frozen in a dry ice/ethanol bath and their EPR spectra were acquired at 120 K. The EPR signals were normalized to give the same amplitude for all samples. The peak-trough widths indicated in parentheses were determined from the zero-crossings of the first derivative of the EPR signals.
COX activity in hPGHS-2 treated with inhibitors
The compounds tested in the experiments in Fig. 1 were all expected to be time dependent inhibitors, with rapid initial binding (E + I <=> EI) and subsequent slower conversion to a tighter complex (EI <=> EI’) (25, 26). One possible explanation for the observed lack of effect of indomethacin or flurbiprofen on the tyrosyl radical EPR after subsequent peroxide addition (Fig. 1) is that these compounds bind poorly to PGHS-2 or undergo the EI => EI’ transition slowly under the low-temperature incubation conditions. To examine this possibility, the cyclooxygenase assay was used to screen for incomplete binding of inhibitors under the conditions used for the experiments in Fig. 1. Small aliquots of hPGHS-2 were incubated on ice with the inhibitors and two aliquots were taken for COX assay, one relatively early (16-59 min) in the incubation and one later (159-192 min) (Table 1). The control COX activity was not significantly affected by extended incubation on ice. Nimesulide, diclofenac, flufenamate, niflumate, and flurbiprofen showed similar degrees of COX inhibition at the early and later time points, indicating that these compounds bound to PGHS-2 and established an equilibrium between the EI and EI’ complexes by the time of the initial assay. The residual COX activity observed with these compounds ranged from 0-62%, indicating that there were large differences between the compounds in the position of the EI <=> EI’ equilibrium. Two compounds, meclofenamate and indomethacin, showed an increase in inhibition between the early and later time points, indicating that EI and EI’ were not in equilibrium by the time of the first assay.
Table 1.
Effects of inhibitors on PGHS-2 cyclooxygenase activitya.
| Inhibitor | 1st time point | 2nd time point | COX site Occupancyb | ||
|---|---|---|---|---|---|
| (min) | COX activity (units/μg) | (min) | COX activity (units/μg) | (EI’/Et) | |
| None | 16 | 26.0 | 159 | 28.1 | 0 |
| Nimesulide | 23 | 4.9 | 192 | 5.0 | 0.81 |
| Diclofenac | 30 | 0.1 | 186 | 0.0 | 1 |
| Flufenamate | 59 | 16.7 | 182 | 17.0 | 0.38 |
| Meclofenamate | 48 | 3.4 | 177 | 0.1 | 1 |
| Niflumate | 44 | 9.4 | 172 | 9.5 | 0.65 |
| Flurbiprofen | 39 | 1.8 | 168 | 1.8 | 0.93 |
| Indomethacin | 23 | 25.0 | 163 | 20.1 | ~0.3 |
The holoenzyme (11 μM in 0.1 M KPi, pH 7.2, containing 0.1% Tween 20) was incubated on ice with 1.5 eq of inhibitor and aliquots were withdrawn at the two time points indicated for assay of cyclooxygenase activity. The concentration of inhibitor in the assay was 35 nM.
Calculated as (1 – f), where f is the fraction of control activity remaining at the second time point; assumes that I dissociates from EI during assay.
Effects of inhibitors on EPR linewidth of preformed tyrosyl radicals in PGHS-2
Nimesulide, diclofenac, and flurbiprofen exhibited stable levels of COX inhibition (Table 1), confirming that each bound to the protein and established stable proportions of EI and EI’ reasonably quickly. Nimesulide and diclofenac pretreatment led to altered radical distribution in the screening above (indicated by narrowing of the EPR signal in Fig. 1), whereas pretreatment with flurbiprofen did not, thus providing positive and negative effects for comparison. Accordingly, these three compounds were selected to probe the effects of COX site ligands on preformed tyrosyl radicals in PGHS-2.
The design of these experiments was to first react PGHS-2 with 2 eq of EtOOH for 6-8 s on ice to generate the WS EPR signal that reflects a mixture of Tyr385 and Tyr504 radicals, add 20 eq of one of the inhibitors, and then monitor the EPR spectrum over time for changes that might reflect alteration in the relative proportions or stability of the Tyr385 and Tyr504 radicals. The level of peroxide used was equimolar with the sum of the concentrations of hPGHS-2 heme and phenol in the reaction, minimizing multiple turnovers while generating a usable level of tyrosyl radicals. The 20-fold molar excess of inhibitor was chosen to favor rapid binding and equilibration.
In control PGHS-2, reaction with peroxide produced a WS that was 29 G peak-to-trough, with a small central inflection (Fig. 2A), essentially the same as the signal observed with higher levels of peroxide (Fig. 1). The signal intensity declined with further incubation on ice, but the overall line width did not change significantly, averaging 28.6 G (Table 2). Addition of 20 eq of nimesulide to hPGHS-2 after reaction with 2 eq of peroxide produced a slightly narrower singlet with a less pronounced central feature (Fig. 2B). Further incubation on ice did not change the peak-to-trough width, which averaged 27.3 G and was significantly less than the control (Table 2); the radical intensity was remarkably stable. Addition of flurbiprofen after the peroxide resulted in a signal that slowly decreased in intensity with time but maintained a peak-to-trough width indistinguishable from the control value (Fig. 2C and Table 2). Addition of diclofenac after reaction with peroxide produced an EPR signal whose intensity decayed but whose overall line width remained constant, averaging 28.3 G, which is not significantly different from the control value (Fig. 2D and Table 2). Regarding linewidth changes, the most striking observation is that only nimesulide produced some narrowing of the tyrosyl radical EPR signal when added after radical formation by peroxide, and the narrowing was distinctly less than that seen when the ligands were added before the peroxide (Table 2). This result indicates that nimesulide, but not diclofenac or flurbiprofen, partially redistributed preformed radical from Tyr385 to Tyr504. The binding of nimesulide, flurbiprofen or diclofenac to PGHS-2 before reaction with 2 eq of EtOOH decreased the accumulation of tyrosyl radical to just 12-29% of the control value (Table 2).
Figure 2.
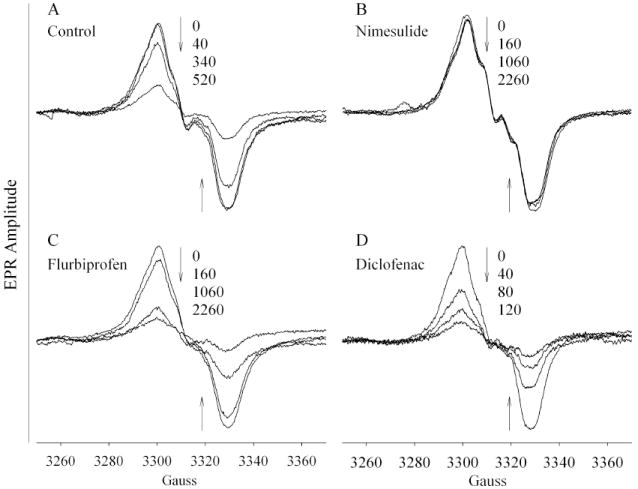
Effects of COX inhibitors on EPR spectra of preformed tyrosyl radicals in PGHS-2. The holoenzyme (11 μM) was reacted at 0 °C with 2 eq of EtOOH for 6-8 s before addition of 20 eq of inhibitor and 6-7 s of further reaction before freezing and recording the initial EPR spectrum (0s). The control sample was frozen after reaction with peroxide, without addition of inhibitor. The subsequent spectra were recorded after warming the samples in an ice bath for the indicated cumulative lengths of time and refreezing. The arrows indicate the direction of amplitude change with time.
Table 2.
Inhibitor effects on tyrosyl radical EPR characteristics.
| Inhibitor | Linewidth (G) | Linewidth (G) | Relative Half-lifec | Relative Initial Yieldd | Relative Initial Yieldd |
|---|---|---|---|---|---|
| I firsta | ROOH firstb | ROOH firstb | I firste | ROOH firstb | |
| None | - | 28.6 ± 0.4 (n=8) | 1.0 | 1.0 | 1.0 |
| Nimesulide | 25.2 ± 0.5 (n=4) | 27.3 ± 0.4 (n=11) | 170 | 0.12 | 1.4 |
| Flurbiprofen | 28.6 ± 0.2 (n=3) | 29.0 ± 0.3 (n=9) | 12 | 0.29 | 1.1 |
| Diclofenac | 25.0 ± 0.5 (n=4) | 28.3 ± 0.7 (n=5) | 0.20 | 0.17 | 0.8 |
Preincubated with 2 eq of inhibitor before reaction with 10 eq of EtOOH for 6-8 s.
Reacted with 2 eq EtOOH for 6-8 s before addition of 20 eq of inhibitor (if any).
Normalized to the tyrosyl radical half-life in the control reacted with only EtOOH. Values are the averages of duplicate determinations.
Normalized to the initial tyrosyl radical intensity in the control reacted with only EtOOH. Values are the averages of duplicate determinations.
Preincubated with 2 eq of inhibitor before reaction with 2 eq of EtOOH for 6-8 s.
Effects of inhibitors on stability of preformed tyrosyl radicals in PGHS-2
The decay kinetics of the mixtures of Tyr385 and Tyr504 radicals in the four samples in Fig. 2, along with a duplicate set of samples and some additional controls, are presented in more detail in Fig. 3. The tyrosyl radicals’ intensity in the control samples declined with first order kinetics, with a t1/2 value of 4.1 min. Addition of nimesulide after reaction with peroxide slowed the radicals’ decay rate by a factor of 170, increasing the t1/2 value to some 11 hr. Addition of flurbiprofen after peroxide slowed the decay by a factor of 12, giving a t1/2 of 48 min. In contrast, addition of diclofenac after peroxide increased the decay rate 5-fold, giving a t1/2 value of just 48 s. Thus, all three cyclooxygenase inhibitors altered the lifetime of preformed tyrosyl radicals in PGHS-2, with two of them strongly stabilizing and one destabilizing. It is important to note that the overall linewidths of the EPR signals did not change over time after addition of the inhibitors, indicating that the stabilizing/destabilizing effects of the inhibitors did not discriminate between the Tyr385 and Tyr504 radicals or that oxidizing equivalent rapidly equilibrates between the two tyrosines.
Figure 3.
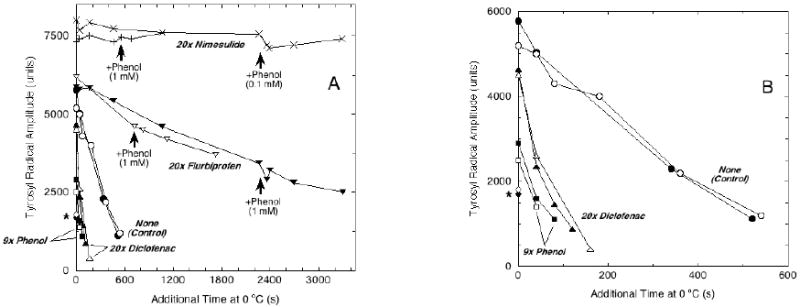
Effects of COX inhibitors and reducing cosubstrate on PGHS-2 tyrosyl radical decay kinetics. Panel A: Peak-to-trough amplitudes of the g = 2 EPR signals from samples from the experiment in Fig. 2 are plotted as a function of the cumulative length of incubation at 0 °C after acquisition of the initial spectrum. Quantitation of radical intensity by double integration of the initial control samples indicated that an amplitude of 7500 arbitrary units corresponded to ~ 0.15 spins/heme. Two samples were run for each inhibitor. Extra phenol was added to the nimesulide and flurbiprofen samples at the times indicated by arrows. The time scale is expanded in panel B to show details from early time points. The asterisk indicates the data points from the two PGHS-2 samples reacted with nimesulide before EtOOH.
The PGHS-2 stocks contained equimolar levels of phenol, added to prevent inactivation during reconstitution of the holoenzyme. The susceptibility of the PGHS-2 tyrosyl radicals to higher levels of reducing cosubstrate was tested by addition of more phenol. In the control hPGHS-2 samples, addition of 100 μM (9 eq) phenol a few seconds after hPGHS-2 was reacted with peroxide in the absence of inhibitor accelerated the decay of the radical by some 5-fold (t1/2 = 52 s) (Fig. 3). In contrast, addition of 100 μM phenol to one nimesulide-treated sample after 38 min of incubation on ice did not change the radical decay rate (Fig. 3). Addition of a much higher level of phenol (1.0 mM, 90 eq) to the other nimesulide-treated sample after 7.7 min of incubation on ice also had essentially no effect on the radical decay rate (Fig. 3). This higher concentration of phenol is 4.5-fold that of the nimesulide, which rules out a direct chemical action of nimesulide on phenol. Comparison of the radical decay rates in the presence of 100 μM phenol for control (~10-2 s-1) and nimesulide-treated (~10-5 s-1) PGHS-2 indicates that the binding of nimesulide to PGHS-2 after reaction with peroxide stabilizes the tyrosyl radicals by three orders of magnitude when a modest level of cosubstrate is present. This COX site ligand thus renders the both PGHS-2 tyrosyl radicals essentially unreactive with phenol.
Two samples of PGHS-2 were reacted with 20 eq of nimesulide for 7 s before reaction with 2 eq of EtOOH for another 7 s. The singlet radical signal formed in both cases was of very low intensity (indicated by an asterisk in Fig. 3B) and had a peak-to-trough width of 26 G (not shown). The low initial radical intensity precluded analysis of the decay kinetics.
Inhibitor stoichiometry and stabilization of preformed tyrosyl radical
The stability of the EtOOH-induced tyrosyl radical EPR signal was also examined at lower levels of nimesulide. This experiment was performed under the same conditions used for the experiments in Fig. 3, except that the concentration of nimesulide was varied to give inhibitor/PGHS-2 molar ratios ranging from 0.5 to 10. The results are shown in Fig. 4. When 0.5 eq of nimesulide was added after reaction with peroxide, the decay of the radical followed that of the control for the first 240 s, but then slowed to reach a stable plateau level. Addition of 1.0 eq of inhibitor after the peroxide decreased the amplitude of the decay phase, roughly doubling the plateau level of stable radical. With 2.0 eq of nimesulide, the decay amplitude was further decreased and the plateau level increased. There was little or no decay phase observed in the samples with 5-10 eq of inhibitor, and progressively higher plateau levels of stable radical were seen. Thus, large increments in the level of stabilized radical are evident for the samples with nimesulide/PGHS ratios of 0.5, 1.0 or 2.0; smaller increments are seen for the higher inhibitor stoichiometries. Each PGHS-2 monomer has one high-affinity binding site for inhibitors such as nimesulide, located in the COX channel (7). The observation of tyrosyl radical stabilization even at a substoichiometric level of nimesulide links the radical stabilizing action of the inhibitor to its binding in the COX channel rather than elsewhere on the protein. The radical stabilization appeared to be complete during the mixing and initial reaction at higher nimesulide ratios but required further incubation on ice for the lower nimesulide ratios. This is consistent with stabilization occurring upon nimesulide binding, with the rate of binding increasing with inhibitor level.
Figure 4.
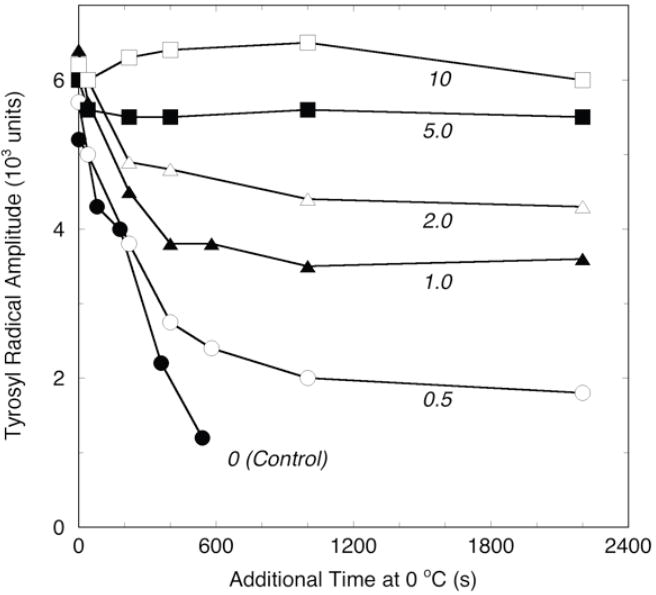
Effect of nimesulide stoichiometry on stabilization of preformed PGHS-2 tyrosyl radical. PGHS-2 was reacted with 2 eq of EtOOH for 6-8 s at 0 °C before reaction with nimesulide at the indicated inhibitor/PGHS-2 molar ratios for a further 6-8 s before freezing for the initial EPR analysis. Additional spectra were recorded after the indicated lengths of extra incubation at 0 °C and refreezing. EPR signal amplitudes were measured as described in the legend to Fig. 3.
Heme redox state in PGHS-2 incubated with inhibitor after reaction with peroxide
Optical spectroscopy was used to look for changes in heme redox state over the first 10 min of tyrosyl radical stabilization by flurbiprofen. Flurbiprofen was used instead of nimesulide to avoid complications from the visible absorbance of the latter. PGHS-2 (11 μM in 0.1 M KPi, pH 7.2, containing 5% glycerol and 0.1% Tween 20) was equilibrated at 2-3 °C in a cuvette positioned in the diode array spectrophotometer. Absorbance spectra (250-700 nm) were acquired every 1 s before and after addition of 2 eq of EtOOH followed 6 s later by 20 eq of flurbiprofen. The manual mixing process was complete in a few seconds in both of the duplicate experiments. No obvious spectral shifts were observed during 600 s of either reaction (not shown). Detailed examination of the kinetic traces for A410 (absorbance peak for ferric PGHS-2 heme) did not show any changes once mixing was complete; the A424 kinetic traces similarly showed no evidence for formation or decay of ferryl PGHS-2 heme content. These results indicate that the peroxide-driven redox changes in the PGHS-2 heme were rapid and completed in the dead time of these manually mixed reactions, as expected from earlier results (27). Taken with the results in Fig. 3, it is evident that the flurbiprofen-treated PGHS-2 with stabilized tyrosyl radical has heme in the ferric state, and that the slow decay of the tyrosyl radical is not accompanied by significant accumulation of heme in other oxidation states. In other words, redox changes at the heme appear to be decoupled from redox changes at Tyr385 and Tyr504 after formation of the radicals.
DISCUSSION
Effects of NSAID binding on subsequent tyrosyl radical formation
A major goal of the present study was to characterize the effects of COX inhibitors on the distribution and stability of preformed tyrosyl radicals in PGHS-2. Early studies with PGHS-1 and -2 found that binding of some inhibitors narrowed the tyrosyl radical EPR signals produced upon subsequent reaction with peroxide, and decreased the intensity of the signals (12, 24). Mutagenic experiments later revealed that the peroxide-induced EPR signals are mixtures of a WD from a Tyr385 radical and a NS from a Tyr504 radical, and that the narrowing of the overall EPR signal reflects decreased accumulation of Tyr385 radical relative to Tyr504 radical (3, 4). Many COX inhibitors are substrate analogs whose binding site is adjacent to Tyr385 and some distance from Tyr504 (28), so the altered distribution of radical between the two tyrosines is likely due to slower formation or faster dissipation of the Tyr385 radical. The absence of a transient WD signal in rapid quench EPR experiments with PGHS-1 preincubated with indomethacin or PGHS-2 preincubated with nimesulide (8) suggests that the inhibitors block formation of the Tyr385 radical, though a very rapid dissipative action on the radical cannot be excluded.
COX inhibitors vary considerably in their ability to affect the distribution of radical in PGHS-1 and -2 when bound before reaction with peroxide. For example, preincubation with flurbiprofen or aspirin led to altered radical distribution in PGHS-1, whereas meclofenamate and flufenamate had no effect on radical distribution (24); this pattern is reversed for PGHS-2, where both fenamates altered radical distribution but flurbiprofen and aspirin did not (Fig. 1) (12). This variation presumably reflects the ability of particular combinations of inhibitor and PGHS isoform to perturb the local structure near Tyr385.
Most previous studies of tyrosyl radical in PGHS-2 pretreated with COX inhibitor have used large excesses of peroxide (10-fold or more), conditions that sustain multiple turnovers and make it difficult to accurately assess radical dissipation kinetics. However, in one study using 5-fold peroxide (12), control and nimesulide-pretreated PGHS-2 had half-lives of ~50 and ~30, respectively. This indicates that pretreatment with nimesulide destabilized the subsequent PGHS-2 tyrosyl radical, consistent with the very low radical intensity at the initial time point in the present experiments using 2-fold peroxide (Table 2), and emphasizing the very different actions of nimesulide when added before and after formation of radical.
Effects of tyrosyl radical formation on PGHS-2 interaction with COX inhibitors
The observation that the effects of competitive inhibitor on the PGHS-2 tyrosyl radicals are very different when the inhibitor binds after the radicals are formed is one of the major surprises in the present study. These differences were manifested both in the distribution of radical between Tyr385 and Tyr504 and the stability of the radicals, and one or both aspects were evident with each of the three inhibitors examined in detail (Figs. 2-5 and Table 2). The clear implication is that the interaction between PGHS-2 and COX inhibitor is fundamentally different when tyrosyl radical is already formed. As the inhibitor structure is unchanged, the presence of tyrosyl radical must alter the structure of PGHS-2 residues that interact, directly or indirectly, with the inhibitor. This alteration of COX channel structure upon radical formation at Tyr385 is depicted as a doming of the COX pocket in Scheme 2. The molecular nature of this structural change remains to be established, but it could plausibly involve modification of the hydrogen bonding between Tyr385 and Tyr348 that is implied by crystallographic and mutagenic results (7, 29, 30).
Figure 5.
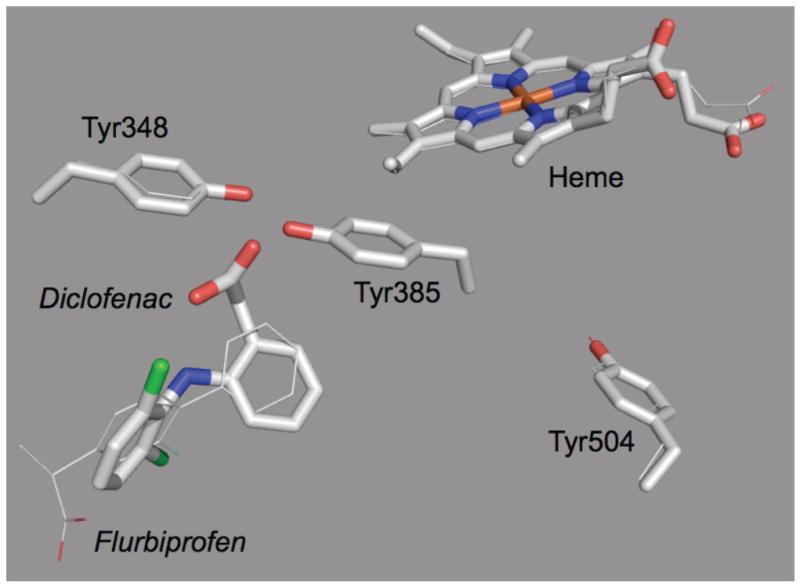
Comparison of the orientations of diclofenac and flurbiprofen in crystallographic data for complexes of the inhibitors with murine PGHS-2 (PDB codes 1PXX and 3PGH, respectively) (7, 47).
Scheme 2.
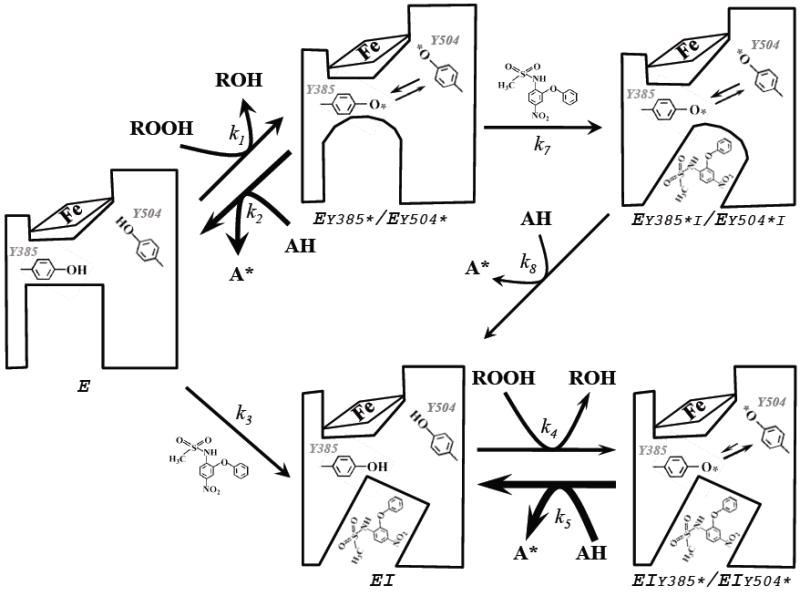
Structural interpretation of interconversions of PGHS-2 subunit (E) reacted with peroxide (ROOH) before nimesulide (k1 and k7 steps at top), or with nimesulide before peroxide (k3 and k4 steps at bottom). The POX site is indicated by the heme icon and Tyr385 is adjacent to the COX site. Formation of the Tyr385 radical is depicted as altering the nearby structure in the COX site. The time-dependent protein conformational change proposed to follow nimesulide binding (26) is depicted as a tilting of the COX site. The labeling of enzyme intermediates and reaction steps follows Scheme 1.
The dichotomy between the effects of nimesulide when added before and after tyrosyl radical formation may have implications for interactions of PGHS-1 and -2 with fatty acid substrates. In anaerobic experiments, adding arachidonate to PGHS-1 before reaction with peroxide produced a very different EPR spectrum from that seen when the enzyme was reacted with a mixture of peroxide and arachidonate (Tsai, unpublished; note that this experiment focused on EPR lineshape rather than radical stability). Reaction of PGHS-1 with peroxide is very rapid (31, 32), so adding a mixture of arachidonate and peroxide approximates a reaction with peroxide before addition of fatty acid. The result thus suggests that arachidonate may bind differently to PGHS that has tyrosyl radical present.
Effects of COX inhibitor binding on stability of preformed tyrosyl radicals
The most striking finding of the present studies is that COX inhibitor binding can markedly change the stability of preformed tyrosyl radicals. Previous in vitro studies generally have found the PGHS tyrosyl radicals to be quite transient (10, 12, 13, 23, 33), in contrast with the much more stable tyrosyl radicals observed in ribonucleotide reductase (34). The decay of the PGHS tyrosyl radicals is due to direct or indirect redox reactions of the radicals with reductants, as accumulation of substrate-induced tyrosyl radical in microsomal and purified PGHS-1 and -2 has been found to be attenuated or dissipated by relatively low levels of exogenous reductant (11, 13, 35, 36). Some small molecules known to act as PGHS peroxidase reductants, such as ascorbate, lipoate, urate, NADH and NADPH, are ubiquitous cellular components and represent potential quenchers of PGHS tyrosyl radicals in vivo (37-39).
The marked sensitivity of PGHS tyrosyl radicals to quenching by small molecule reductants in vitro has raised the question of how tyrosyl radical-based COX catalysis can be sustained in cellular environments where such reductants are abundant. An additional complication is that low levels of many small molecule reductants that quench the PGHS tyrosyl radicals actually increase COX catalytic turnover of the purified enzymes (14, 15, 40, 41). This paradoxical contrast between the effects of reductants on PGHS tyrosyl radicals and on COX catalysis contributed to some early skepticism regarding the validity of the tyrosyl radical mechanism (13). A satisfactory resolution to the paradox has not been forthcoming even though the preponderance of accumulated evidence now strongly supports the tyrosyl radical mechanism (2). The present finding that some COX site ligands increase the tyrosyl radical lifetime by two orders of magnitude (Table 2) demonstrates that tyrosyl radicals actually can be remarkably stable when particular ligands bind in the COX channel, even in the presence of small molecule reductants. This stabilization is depicted in Scheme 2 by having the k8 step much slower than the k2 and k5 steps. We suspect that binding of fatty acid in the COX site stabilizes the tyrosyl radicals towards small molecule reductants in the same fashion observed with nimesulide and flurbiprofen. It is worth noting that differences in fatty acid structure may impact the degree of tyrosyl radical stabilization, just as observed for differences in inhibitor structure (Fig. 3, Table 2). Differences in tyrosyl radical stabilization might help explain the wide variation in COX activation efficiency among substrate fatty acids (42).
COX inhibitors and tyrosyl radical dynamics
A working structural interpretation of PGHS-2 tyrosyl radical dynamics is depicted in Scheme 2, using nimesulide as the example. Reaction of peroxide with resting enzyme (E) in the k1 step produces radical either at Tyr385 (EY385*; WD) or Tyr504 (EY504*; NS). The Tyr385 radical accumulates faster than the Tyr504 radical, resulting in a transition in the EPR spectrum over time from a signal dominated by the WD component to a WS signal with comparable contributions from WD and NS (8). Subsequent reaction with endogenous or exogenous reductant (AH in the k2 step) dissipates tyrosyl radical and returns the enzyme to resting state. The WS lineshape does not change appreciably as the EPR signal intensity declines (9, 12), indicating that the proportions of the two radicals hold steady during the dissipation process. This behavior suggests that the equilibration of radical between Tyr385 and Tyr504 (depicted in Scheme 2) is faster than dissipation of radical, presumably by reaction of reductant with Tyr385 radical. Equilibration between Tyr385 and Tyr504 radicals is consistent with earlier electron transfer pathway calculations (3), which predicted that the coupling of Tyr504 with Tyr385 is five-fold greater than coupling of Tyr504 with the heme. It’s important to note that such interconversion of the two radicals would be required for the Tyr504 radical to function as a radical reservoir, furnishing oxidizing equivalents for formation of the catalytic Tyr385 radical.
Mechanisms for alteration of tyrosyl radical stability
The basic elements of tyrosyl radical formation in PGHS-1 and -2 are well studied (1, 2), but the process of dissipation of the Tyr385 and Tyr504 radicals has not been characterized in detail for either PGHS isoform. The ability of nimesulide and flurbiprofen to interfere with radical dissipation makes them useful tools for elucidating the process. The degree of radical stabilization is very large, especially for nimesulide, where the half-life increases by 170-fold, and addition of even millimolar phenol after nimesulide does not appreciably accelerate radical decay (Fig. 3 and Table 2). Radical stabilization can be seen even at low inhibitor stoichiometries (Fig. 4), consistent with specific binding at the COX site. Taken together, these results indicate that nimesulide or flurbiprofen binding at the COX site effectively blocks all routes for electron transfer from small molecule reductants in the medium to either Tyr385 or Tyr504.
Time dependent COX inhibitors are thought to trigger a non-covalent protein conformational change (26, 43). Scheme 2 depicts this conformational change as an overall tilting limited to the COX channel, although structural effects of inhibitor binding far from the COX pocket have been demonstrated in PGHS-1 (43). Nimesulide, flurbiprofen and diclofenac are all time-dependent inhibitors of COX-2 (44-46) and thus all might be expected to trigger a generally similar conformational shift. However, diclofenac’s effect on tyrosyl radical stability is opposite to that of the other two inhibitors (Fig. 3 and Table 2). This argues against attributing the modulation of tyrosyl radical stability to a general inhibitor-triggered protein conformational change, pointing instead to more localized differences in the interactions between individual inhibitors and key COX channel residues.
COX inhibitors are known to bind near the top of the COX channel (28), providing plausible opportunities for altering interactions with the nearby Tyr385. It is noteworthy that crystallographic models of PGHS-2 show that the binding orientation of diclofenac is inverted compared to that of flurbiprofen (7, 47), as illustrated in Fig. 5. The carboxyl group of diclofenac is within three angstroms of the Tyr385 hydroxyl. Such an arrangement places a strong charge next to a Tyr385 radical and could explain the decreased radical lifetime observed after diclofenac addition (Fig. 3). On the other hand, the carboxyl of flurbiprofen is positioned near Arg120 in the crystal structure, with the hydrophobic phenyl ring of the inhibitor near the Tyr385 hydroxyl group. This arrangement could explain the radical stabilization observed upon flurbiprofen addition (Fig. 3). Unfortunately, crystallographic data on the binding orientation of nimesulide or a close structural analog are not available. Nimesulide does have a phenyl group (Scheme 2) that could plausibly stabilize a Tyr385 radical if positioned similarly to the phenyl group of flurbiprofen in Fig. 5. It remains to be seen if the difference in binding orientation in ground state inhibitor complexes of diclofenac and flurbiprofen is relevant to the differences in tyrosyl radical stability when these compounds bind to PGHS-2 with preformed radical.
In addition to the mechanistic implications discussed above, the results of the present study may have some bearing on COX pharmacology. The wide variation between COX inhibitors in tyrosyl radical stabilization may be a factor in the potency of these agents as COX inhibitors. Specifically, inhibitors that destabilize the tyrosyl radicals, thus necessitating reactivation by peroxide, would have an additional mode of COX inhibition. Screening for effects on tyrosyl radical stability in PGHS-1 and -2 may be useful in identifying improved COX inhibitors.
Conclusions
The present finding that the tyrosyl radicals in PGHS-2 can be dramatically stabilized is a major conceptual break from the previous picture of the PGHS radicals as transient species, vulnerable to interception by small molecule reductants. The ability of some competitive COX inhibitors to render the tyrosyl radicals unreactive towards reductants provides a novel mechanism for regulation of PGHS activity by physiological COX site ligands in cellular environments where reductants are abundant. The modulation of tyrosyl radical dynamics by COX inhibitors may also have practical applications for functional screening of improved inhibitors and for spectroscopic characterization of PGHS-1 and -2 tyrosyl radicals.
Acknowledgments
We thank Dr. Wen Liu for construction of the baculovirus for expression of his-tagged hPGHS-2.
Footnotes
This work was supported by NIH grants GM52170 (RJK) and GM44911 (ALT).
The abbreviations used are: COX, cyclooxygenase; PGHS, prostaglandin H synthase; hPGHS-2, human PGHS-2; EtOOH, ethyl hydrogen peroxide; WD, wide doublet tyrosyl radical; WS, wide singlet tyrosyl radical; NS, narrow singlet tyrosyl radical; and EPR, electron paramagnetic resonance spectroscopy.
Contributor Information
Gang Wu, Email: Gang.Wu@uth.tmc.edu.
Ah-Lim Tsai, Email: Ah-Lim.Tsai@uth.tmc.edu.
Richard J. Kulmacz, Email: Richard.J.Kulmacz@uth.tmc.edu.
References
- 1.Tsai A, Kulmacz RJ. Tyrosyl radicals in prostaglandin H synthase-1 and -2. Prostaglandins Other Lipid Mediat. 2000;62:231–254. doi: 10.1016/s0090-6980(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 2.Rouzer CA, Marnett LJ. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem Rev. 2003;103:2239–2304. doi: 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- 3.Rogge CE, Liu W, Wu G, Wang LH, Kulmacz RJ, Tsai AL. Identification of Tyr504 as an alternative tyrosyl radical site in human prostaglandin H synthase-2. Biochemistry. 2004;43:1560–1568. doi: 10.1021/bi035717o. [DOI] [PubMed] [Google Scholar]
- 4.Rogge CE, Liu W, Kulmacz RJ, Tsai AL. Peroxide-induced radical formation at TYR385 and TYR504 in human PGHS-1. J Inorg Biochem. 2009;103:912–922. doi: 10.1016/j.jinorgbio.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Havird JC, Miyamoto MM, Choe KP, Evans DH. Gene duplications and losses within the cyclooxygenase family of teleosts and other chordates. Mol Biol Evol. 2008;25:2349–2359. doi: 10.1093/molbev/msn183. [DOI] [PubMed] [Google Scholar]
- 6.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 7.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 8.Tsai A, Wu G, Palmer G, Bambai B, Koehn JA, Marshall PJ, Kulmacz RJ. Rapid kinetics of tyrosyl radical formation and heme redox state changes in prostaglandin H synthase-1 and -2. J Biol Chem. 1999;274:21695–21700. doi: 10.1074/jbc.274.31.21695. [DOI] [PubMed] [Google Scholar]
- 9.Tsai A, Palmer G, Xiao G, Swinney DC, Kulmacz RJ. Structural characterization of arachidonyl radicals formed by prostaglandin H synthase-2 and prostaglandin H synthase-1 reconstituted with mangano protoporphyrin IX. J Biol Chem. 1998;273:3888–3894. doi: 10.1074/jbc.273.7.3888. [DOI] [PubMed] [Google Scholar]
- 10.Karthein R, Dietz R, Nastainczyk W, Ruf HH. Higher oxidation states of prostaglandin H synthase. EPR study of a transient tyrosyl radical in the enzyme during the peroxidase reaction. Eur J Biochem. 1988;171:313–320. doi: 10.1111/j.1432-1033.1988.tb13792.x. [DOI] [PubMed] [Google Scholar]
- 11.Tsai AL, Palmer G, Kulmacz RJ. Prostaglandin H synthase. Kinetics of tyrosyl radical formation and of cyclooxygenase catalysis. J Biol Chem. 1992;267:17753–17759. [PubMed] [Google Scholar]
- 12.Xiao G, Tsai AL, Palmer G, Boyar WC, Marshall PJ, Kulmacz RJ. Analysis of hydroperoxide-induced tyrosyl radicals and lipoxygenase activity in aspirin-treated human prostaglandin H synthase-2. Biochemistry. 1997;36:1836–1845. doi: 10.1021/bi962476u. [DOI] [PubMed] [Google Scholar]
- 13.Lassmann G, Odenwaller R, Curtis JF, DeGray JA, Mason RP, Marnett LJ, Eling TE. Electron spin resonance investigation of tyrosyl radicals of prostaglandin H synthase. Relation to enzyme catalysis. J Biol Chem. 1991;266:20045–20055. [PubMed] [Google Scholar]
- 14.Miyamoto T, Ogino N, Yamamoto S, Hayaishi O. Purification of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. J Biol Chem. 1976;251:2629–2636. [PubMed] [Google Scholar]
- 15.Hemler M, Lands WE. Purification of the cyclooxygenase that forms prostaglandins. Demonstration of two forms of iron in the holoenzyme. J Biol Chem. 1976;251:5575–5579. [PubMed] [Google Scholar]
- 16.Van der Ouderaa FJ, Buytenhek M, Nugteren DH, Van Dorp DA. Purification and characterisation of prostaglandin endoperoxide synthetase from sheep vesicular glands. Biochim Biophys Acta. 1977;487:315–331. doi: 10.1016/0005-2760(77)90008-x. [DOI] [PubMed] [Google Scholar]
- 17.Otto JC, Smith WL. The orientation of prostaglandin endoperoxide synthases-1 and -2 in the endoplasmic reticulum. J Biol Chem. 1994;269:19868–19875. [PubMed] [Google Scholar]
- 18.Ren Y, Walker C, Loose-Mitchell DS, Deng J, Ruan KH, Kulmacz RJ. Topology of prostaglandin H synthase-1 in the endoplasmic reticulum membrane. Arch Biochem Biophys. 1995;323:205–214. doi: 10.1006/abbi.1995.0027. [DOI] [PubMed] [Google Scholar]
- 19.Banhegyi G, Benedetti A, Csala M, Mandl J. Stress on redox. FEBS Lett. 2007;581:3634–3640. doi: 10.1016/j.febslet.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 20.Dixon BM, Heath SH, Kim R, Suh JH, Hagen TM. Assessment of endoplasmic reticulum glutathione redox status is confounded by extensive ex vivo oxidation. Antioxid Redox Signal. 2008;10:963–972. doi: 10.1089/ars.2007.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith T, Leipprandt J, DeWitt D. Purification and characterization of the human recombinant histidine-tagged prostaglandin endoperoxide H synthases-1 and -2. Arch Biochem Biophys. 2000;375:195–200. doi: 10.1006/abbi.1999.1659. [DOI] [PubMed] [Google Scholar]
- 22.Bambai B, Rogge CE, Stec B, Kulmacz RJ. Role of Asn-382 and Thr-383 in activation and inactivation of human prostaglandin H synthase cyclooxygenase catalysis. J Biol Chem. 2004;279:4084–4092. doi: 10.1074/jbc.M304762200. [DOI] [PubMed] [Google Scholar]
- 23.Kulmacz RJ, Tsai AL, Palmer G. Heme spin states and peroxide-induced radical species in prostaglandin H synthase. J Biol Chem. 1987;262:10524–10531. [PubMed] [Google Scholar]
- 24.Kulmacz RJ, Palmer G, Tsai AL. Prostaglandin H synthase: perturbation of the tyrosyl radical as a probe of anticyclooxygenase agents. Mol Pharmacol. 1991;40:833–837. [PubMed] [Google Scholar]
- 25.Rome LH, Lands WE. Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc Natl Acad Sci U S A. 1975;72:4863–4865. doi: 10.1073/pnas.72.12.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Copeland RA, Williams JM, Giannaras J, Nurnberg S, Covington M, Pinto D, Pick S, Trzaskos JM. Mechanism of selective inhibition of the inducible isoform of prostaglandin G/H synthase. Proc Natl Acad Sci U S A. 1994;91:11202–11206. doi: 10.1073/pnas.91.23.11202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu G, Tsai AL, Van Wart HE, Kulmacz RJ. Comparison of the peroxidase reaction kinetics of prostaglandin H synthase-1 and -2. J Biol Chem. 1999;274:16162–16167. doi: 10.1074/jbc.274.23.16162. [DOI] [PubMed] [Google Scholar]
- 28.Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu Rev Biophys Biomol Struct. 2003;32:183–206. doi: 10.1146/annurev.biophys.32.110601.141906. [DOI] [PubMed] [Google Scholar]
- 29.Rogge CE, Ho B, Liu W, Kulmacz RJ, Tsai AL. Role of Tyr348 in Tyr385 radical dynamics and cyclooxygenase inhibitor interactions in prostaglandin H synthase-2. Biochemistry. 2006;45:523–532. doi: 10.1021/bi051235w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hochgesang G, Jr, Rowlinson S, Marnett L. Tyrosine-385 is critical for acetylation of cyclooxygenase-2 by aspirin. J Am Chem Soc. 2000;122:6514–6515. [Google Scholar]
- 31.Lambeir AM, Markey CM, Dunford HB, Marnett LJ. Spectral properties of the higher oxidation states of prostaglandin H synthase. J Biol Chem. 1985;260:14894–14896. [PubMed] [Google Scholar]
- 32.Kulmacz RJ. Prostaglandin H synthase and hydroperoxides: peroxidase reaction and inactivation kinetics. Arch Biochem Biophys. 1986;249:273–285. doi: 10.1016/0003-9861(86)90003-2. [DOI] [PubMed] [Google Scholar]
- 33.Gunther MR, Hsi LC, Curtis JF, Gierse JK, Marnett LJ, Eling TE, Mason RP. Nitric oxide trapping of the tyrosyl radical of prostaglandin H synthase-2 leads to tyrosine iminoxyl radical and nitrotyrosine formation. J Biol Chem. 1997;272:17086–17090. doi: 10.1074/jbc.272.27.17086. [DOI] [PubMed] [Google Scholar]
- 34.Pesavento RP, van der Donk WA. Tyrosyl radical cofactors. Adv Protein Chem. 2001;58:317–385. doi: 10.1016/s0065-3233(01)58008-0. [DOI] [PubMed] [Google Scholar]
- 35.Egan RW, Paxton J, Kuehl FA., Jr Mechanism for irreversible self-deactivation of prostaglandin synthetase. J Biol Chem. 1976;251:7329–7335. [PubMed] [Google Scholar]
- 36.Lassmann G, Curtis J, Liermann B, Mason RP, Eling TE. ESR studies on reactivity of protein-derived tyrosyl radicals formed by prostaglandin H synthase and ribonucleotide reductase. Arch Biochem Biophys. 1993;300:132–136. doi: 10.1006/abbi.1993.1018. [DOI] [PubMed] [Google Scholar]
- 37.Ogino N, Yamamoto S, Hayaishi O, Tokuyama T. Isolation of an activator for prostaglandin hydroperoxidase from bovine vesicular gland cytosol and its identification as uric acid. Biochem Biophys Res Commun. 1979;87:184–191. doi: 10.1016/0006-291x(79)91664-4. [DOI] [PubMed] [Google Scholar]
- 38.Markey CM, Alward A, Weller PE, Marnett LJ. Quantitative studies of hydroperoxide reduction by prostaglandin H synthase. Reducing substrate specificity and the relationship of peroxidase to cyclooxygenase activities. J Biol Chem. 1987;262:6266–6279. [PubMed] [Google Scholar]
- 39.Strittmatter P, Machuga ET, Roth GJ. Reduced pyridine nucleotides and cytochrome b5 as electron donors for prostaglandin synthetase reconstituted in dimyristyl phosphatidylcholine vesicles. J Biol Chem. 1982;257:11883–11886. [PubMed] [Google Scholar]
- 40.Hsuanyu Y, Dunford HB. Prostaglandin H synthase kinetics. The effect of substituted phenols on cyclooxygenase activity and the substituent effect on phenolic peroxidatic activity. J Biol Chem. 1992;267:17649–17657. [PubMed] [Google Scholar]
- 41.Percival MD, Ouellet M, Vincent CJ, Yergey JA, Kennedy BP, O’Neill GP. Purification and characterization of recombinant human cyclooxygenase-2. Arch Biochem Biophys. 1994;315:111–118. doi: 10.1006/abbi.1994.1478. [DOI] [PubMed] [Google Scholar]
- 42.Liu W, Cao D, Oh SF, Serhan CN, Kulmacz RJ. Divergent cyclooxygenase responses to fatty acid structure and peroxide level in fish and mammalian prostaglandin H synthases. Faseb J. 2006;20:1097–1108. doi: 10.1096/fj.05-5273com. [DOI] [PubMed] [Google Scholar]
- 43.Kulmacz RJ. Topography of prostaglandin H synthase. Antiinflammatory agents and the protease-sensitive arginine 253 region. J Biol Chem. 1989;264:14136–14144. [PubMed] [Google Scholar]
- 44.Barnett J, Chow J, Ives D, Chiou M, Mackenzie R, Osen E, Nguyen B, Tsing S, Bach C, Freire J, et al. Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system. Biochim Biophys Acta. 1994;1209:130–139. doi: 10.1016/0167-4838(94)90148-1. [DOI] [PubMed] [Google Scholar]
- 45.Guo Q, Wang LH, Ruan KH, Kulmacz RJ. Role of Val509 in time-dependent inhibition of human prostaglandin H synthase-2 cyclooxygenase activity by isoform-selective agents. J Biol Chem. 1996;271:19134–19139. doi: 10.1074/jbc.271.32.19134. [DOI] [PubMed] [Google Scholar]
- 46.Houtzager V, Ouellet M, Falgueyret JP, Passmore LA, Bayly C, Percival MD. Inhibitor-induced changes in the intrinsic fluorescence of human cyclooxygenase-2. Biochemistry. 1996;35:10974–10984. doi: 10.1021/bi960053m. [DOI] [PubMed] [Google Scholar]
- 47.Rowlinson SW, Kiefer JR, Prusakiewicz JJ, Pawlitz JL, Kozak KR, Kalgutkar AS, Stallings WC, Kurumbail RG, Marnett LJ. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem. 2003;278:45763–45769. doi: 10.1074/jbc.M305481200. [DOI] [PubMed] [Google Scholar]