

Abstract
The overall fidelity of protein synthesis has been thought to rely on the combined accuracy of two basic processes; the aminoacylation of tRNAs with their cognate amino acid by the aminoacyl-tRNA synthetases and the selection of cognate aminoacyl-tRNAs by the ribosome in cooperation with the GTPase EF-Tu. These two processes, which together ensure the specific acceptance of a correctly charged cognate tRNA into the A site, operate prior to peptide bond formation. Here we report the identification of an additional mechanism that contributes to high fidelity protein synthesis following peptidyl transfer. In this retrospective quality control step, incorporation of an amino acid from a non-cognate tRNA into the growing polypeptide chain leads to a general loss of specificity in the A site of the ribosome, and thus to a propagation of errors that results in abortive termination of protein synthesis.
The overall in vivo rate of misincorporation during protein synthesis has been estimated to be in the range of 6 × 10−4 to 5 × 10−3 per amino acid incorporated1,2. Current models for the mechanisms governing this level of accuracy focus on the accurate charging of tRNAs with their cognate amino acid by the aminoacyl-tRNA synthetases and correct tRNA selection by the ribosome facilitated by the GTPase elongation factor EF-Tu in bacteria (or eEF1A in eukaryotes). Kinetic discrimination mechanisms, driven by induced fit, have been demonstrated for the synthetases and the ribosome to facilitate accurate selection of amino acids or charged tRNAs respectively3,4. In addition, for both processes, proofreading (or editing) mechanisms have been shown to further increase the overall fidelity3,5-7. Experimental measurements of in vitro aminoacylation accuracy (~105) agree well with that observed in vivo8. In vitro protein synthesis systems (generally poly-phe synthesis on polyU) have been shown to proceed with an overall fidelity (combining the tRNA selection and proofreading steps) of as high as 10−4 3,9,10. However, fidelity measurements in our own lab conducted in the full range of published buffer systems with tRNA mixtures on heteropolymeric mRNA suggest that in vitro protein synthesis proceeds with somewhat lower fidelity (an error rate of 2 × 10−3 to 10 × 10−3, Supplementary Fig. 1), thus arguing that additional quality control mechanisms may exist.
Here we identify a previously uncharacterized ribosome-centered mechanism that contributes to translational quality control, and which may help explain discrepancies between in vitro and in vivo measured fidelity values. The surprising feature of this pathway is that it monitors the fidelity of protein synthesis after the formation of a peptide bond (retrospectively), in certain ways analogous to the exonucleolytic proofreading step in DNA replication11. We provide evidence that the ribosome recognizes errors during synthesis by evaluating the codon:anticodon helix in the P site of the small subunit of the ribosome, leading first to reduced fidelity during subsequent tRNA selection and ultimately to premature termination by release factors.
A mismatched codon:anticodon pair in the P site triggers unusual release behavior
During the course of reconstituting in vitro the translation of ribosome nascent chain complexes (RNCs), we identified an abundant miscoding event wherein Lys-tRNALys (anticodon UUU) efficiently decoded an AAU asparagine codon in a short peptide sequence, as previously documented in vivo12. In these reactions, we observed that in miscoded ribosome complexes the peptidyl-tRNA did not efficiently react to incorporate the next amino acid encoded by the mRNA, but instead appeared to be promiscuously hydrolyzed. These data suggested the existence of a quality control step that follows peptide bond formation and effectively functions to terminate translation of aberrant protein products, thus enhancing the overall fidelity of protein synthesis.
To characterize this unusual observation, we produced RNCs carrying a dipeptidyl-tRNA in the P site with either a matched or mismatched codon:anticodon helix and a variety of different codons in the A site. We started with a pair of RNCs containing a stop codon in the A site (mRNAs encoding MKX (AUG-AAA-UGA) or MNX (AUGAAU-UGA)) to check anticipated RF2 properties on these complexes. Complexes were assembled in a simplified reaction mixture containing initiation factors (IFs 1-3) and fMet-tRNAfMet, and were reacted with ternary complex (Lys-tRNALys:EF-Tu:GTP) in the presence of EF-G to yield ribosome complexes with fMet-Lys-tRNALys in the P site (Fig. 1b and Supplementary Fig. 2), followed by purification over a sucrose cushion. As anticipated, RF2 reacted efficiently with both complexes (MKX and MNX), releasing the dipeptide with a rate constant (kcat) close to those previously reported in buffer A (~0.05 s−1, Fig. 1c)13,14. Interestingly, titration experiments indicated that less RF2 was required to promote the maximal rate of catalysis on the mismatched P site complex than on the matched one (K1/2 values of ~75 nM and 800 nM respectively, Fig. 1d). These data suggested that RF2 interacts differently with these two complexes that differ by a single mismatch in the P site. We note that the maximal rate of release on other matched stop codon-programmed complexes was, as expected, dependent on the buffer used and on the source of RF2 (over- or chromosomally-expressed) reaching a maximum of 10 s−1, close to numbers reported previously15 (Supplementary Fig. 3). Though maximal rates of release are achieved in buffer D, we chose to complete the study in buffer A because background release rates were minimal under these conditions.
Figure 1. Unusual behavior by release factor 2 (RF2) following a miscoding event.

a, Schematic of core steps of elongation and termination on the ribosome. During the elongation cycle, a ternary complex comprised of aminoacylated tRNA, EF-Tu and GTP enters the A site, and reacts with the peptidyl-tRNA elongating the nascent peptide by one amino acid. When a stop codon enters the A site, it is recognized by a class I RF resulting in the hydrolysis of peptidyl-tRNA. b, Schematic representation of the matched MKX and mismatched MNX (mismatched base-pair shown in red) dipeptidyl-tRNA ribosome complexes (dipeptidyl-RNCs), which have a stop codon in the A site. c, Rate constants for release (khyd) measured on the P-site-matched (MKX) and mismatched (MNX) complexes with saturating RF2. d, K1/2 values for the same complexes. e, An example of matched (MKI) and mismatched (MNI) dipeptidyl-RNCs with a sense codon (I) in the A site. f, Rate constants for release (khyd) measured on several matched (MKI, MKF, MFK) or mismatched (MNI, MNF, MLK) RNCs with saturating RF2. Rate constants for MKI and MKF complexes are immeasurably low. Error bars indicate the standard error obtained from the non-linear regression fit of the data.
We next prepared a number of dipeptidyl-tRNA ribosomal complexes that instead carried sense codons in the A site and assessed their behavior in pre-steady state release assays. In the first set, complexes MKI and MNI carrying fMet-Lys-tRNALys in the P site and the Ile codon AUC in the A site were employed (Fig. 1e). RF2-catalyzed release on the sense codon was immeasurably slow for the matched (MKI) complex (kcat <0.0002 s−1), as expected16, but was markedly faster (kcat ~0.002 s−1) for the P-site-mismatched complex (Fig. 1f). We next prepared similar matched and mismatched ribosome complexes, MKF and MNF, carrying a different A-site codon (Phe, UUU). As above, the kcat of the mismatched P site complex (MNF) was markedly faster than that of the matched complex (MKF) (0.0025 s−1 vs. <0.0001 s−1 respectively, Fig. 1f). We note further that the second order rate difference (kcat/Km) was considerably greater for the mismatched complex MNF relative to MKF (>300 fold, Supplementary Fig. 4).
To determine whether the increased reactivity of the P-site-mismatched complex depended on particular decoding mistakes or if the phenomenon was more general, we forced a different near-cognate pairing interaction to prepare matched and mismatched dipeptidyl tRNA complexes (MFK and MLK). These complexes contained multiple differences compared to the previous set (P-site tRNA, P-site mismatch and A-site codon). Again, RF2-catalyzed peptide release was considerably faster for the P-site-mismatched complex than for the matched version (0.0048 s−1 vs. 0.0004 s−1 respectively, Fig. 1f). The related release factor, RF1, exhibited similar promiscuous release activity (data not shown), but will not be further characterized in this study.
Given the apparent generality of the observed RF-promiscuity on sense codons, we performed a number of experiments to determine whether this phenomenon is an authentic ribosome-based event. One concern was that the mismatch in the P site might destabilize the complex and permit a frame-shifting or hopping event on the mRNA17 that might re-position an authentic stop codon in the A site. While the mRNAs were designed to avoid this potential complication, we nevertheless examined the positioning of the ribosome on the mRNA in the matched and mismatched ribosome complexes using a primer-extension based toe-printing assay18. The primer extension reactions on MKI and MNI exhibit indistinguishable toe-printing patterns, consistent with a 3 nucleotide shift of the complex following a round of elongation (Supplementary Fig. 5). Further controls established that hydrolysis cannot be attributed to P-site tRNA drop-off followed by hydrolysis by potential contaminating peptidyl hydrolase (Supplementary Fig. 6). Additionally, the promiscuous release activity, like authentic release, was inhibited by paromomycin14,19 (Supplementary Fig. 7). These experiments together argue that the observed activity on the P-site-mismatched complexes reports on an authentic RF-mediated ribosomal event.
Effects of RF3 on promiscuous RF2 activity support in vivo relevance
Though the observed stimulation of premature peptide release on the sense codons of P-site-mismatched complexes was substantial (>20-fold), the resulting rate constant for the reaction (from 0.002 to 0.005 s−1) still lagged behind that of authentic peptide release (~0.05 s−1) and the competing tRNA selection/peptidyl transfer processes (~2 s−1)20,21. Class II release factor RF3 is a GTPase integrally involved in the removal of the class I RF following peptide release but has no effect on the rate constants for peptide release on authentic termination complexes (Fig. 2a and ref 22). Interestingly, when RF2 and RF3 were added together to a variety of P-site-mismatched complexes (with 1st, 2nd, 3rd position mismatches in the P site), release activity was substantially accelerated (~20-to 50-fold, Fig. 2a). We note that the resulting rate constants for the release reaction can be remarkably fast for some complexes (~0.1 s−1), in a range where this promiscuity could influence the fidelity of protein synthesis in vivo. Interestingly, Ehrenberg and co-workers found that RF3 could stimulate release on certain ribosome complexes containing a near-cognate stop codon in the A site16.
Figure 2. Abortive termination reaction is stimulated by the class II RF3 and is general for all P-site mismatches.
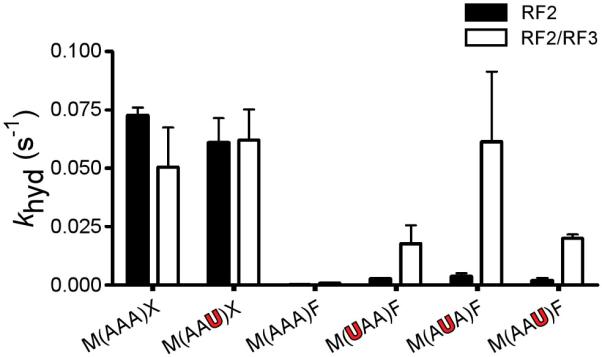
Rate constants for release (khyd) for the indicated P-site-matched (black) and mismatched (red) dipeptidyl-RNC complexes with RF2 only or with RF2 and RF3, all at saturating concentrations. f-Met-Lys-tRNALys occupies the P site in each case, either on cognate (AAA) or on 1st (UAA), 2nd (AUA) or 3rd (AAU) position mismatches. Error bars indicate the standard error obtained from the non-linear regression fit of the data.
A related question is whether the mechanism that monitors miscoding is sensitive to the identity of the mismatch found at each of the three positions of the codon. The U:U mismatch and U:G wobble resulted in similar release activity when located at the 1st or 2nd codon positions; in contrast, at the 3rd codon position, while the U:U mismatch strongly stimulated peptide release, the U:G wobble had little consequence (Supplementary Fig. 8). These data are fully consistent with expectations based on the permissivity of the genetic code at the 3rd codon position and argue that the system for monitoring fidelity in the P site can identify a wide range of errors during translation.
Effects on the fidelity of tRNA selection specified by a mismatched codon:anticodon pair in the P site
Class I release factors must naturally compete during translation with the cognate and near-cognate tRNA species that sample the same ribosomal A site. Given the apparent substantial effects of the P-site mismatch on RF activity, we next looked at the peptidyl transfer (PT) activity of the P-site-matched and mismatched complexes. In two different examples (MKI vs. MNI and MKF vs. MNF), we observed that the rate constant for PT (for cognate Ile-tRNAIle and Phe-tRNAPhe, respectively) was unaffected by the mismatch in the P site (Fig. 3a). We did observe however that catalysis of peptidyl transfer is diminished by 2- to 6-fold for the P-site-mismatched complexes relative to the matched complexes in the presence of a full complement of competitor tRNA (Fig. 3a). We suggest that this reduced overall PT rate results from the increased dwell time of near-cognate tRNAs on the P-site-mismatched complexes with inefficient progression to the irreversible peptidyl transfer reaction.
Figure 3. A single miscoding event promotes iterated errors in tRNA selection.

a, Rate constants for peptidyl transfer (kPT) for the indicated dipeptidyl-RNCs with either the cognate aminoacyl-tRNA (for MKI and MNI, total tRNA mix aminoacylated only with isoleucine, and for MKF and MNF, purified Phe-tRNAPhe) or with bulk aminoacyltRNA. Error bars indicate the standard error obtained from the non-linear regression fit of the data. b, Two-dimensional TLCs resolve the peptidyl transfer reaction products resulting from reaction of the MKI (left panel) or MNI (right panel) dipeptidyl-RNC with bulk aminoacylated tRNA; the reaction was incubated for 0.5 s, short of its endpoint.
A more complete understanding of the observed competition from total tRNA mix comes from analysis of the actual peptide products. We compared product purity of two different sets of P-site-matched and mismatched complexes, MKI vs. MNI and MKF vs. MNF, using a two-dimensional TLC format. As anticipated based on the high fidelity of protein synthesis, the P-site-matched complexes (MKI and MKF) yielded a predominant tripeptide product corresponding to the encoded sequence (Fig. 3b, and Supplementary Fig. 9). However, for both mismatched complexes (MNI and MNF) we observed striking losses of fidelity such that a wide range of miscoded products is observed following the initial forced miscoding event. An estimate of partitioning to correct product (relative to partitioning to incorrect products) in the P-site-matched complex is higher than 96%, whereas for the mismatched complex that same value falls in a range between 10 and 30%. Indeed, we can show that specifically chosen near-cognate tRNAs react more readily with the mismatched complex than with the matched one (Supplementary Fig. 10). The observed losses in fidelity for tRNA selection coupled with the promiscuity of release factors documented above, both following a simple miscoding event, suggest that the A site itself is generally perturbed.
Amplification of errors during tRNA selection leads ultimately to fast termination
The previous sections have described a series of experiments suggesting that a single misincorporation during translation leads to marked changes in A-site behavior including elevated miscoding and accelerated release of polypeptides on sense codons. Elevating the rate of miscoding is not terminal, but it does serve at a minimum to extend the window of opportunity for release factors to abort translation. We tested whether a second sequential miscoding event might generate a complex with even more unusual properties because it carries a mismatch in both the E- and P-site regions of the small ribosomal subunit. We explored the consequences of the iterated miscoding by programming tripeptidyl-tRNA ribosome complexes with either a single mismatch in the P site (MKNF, MEDP, or MFLK), a single mismatch in the E site (MNKF, MDEP, or MLFK), or a combination of both mismatches (MNNF, MDDP, or MLLK) as might result from repeated miscoding events (the MKKF series is depicted in Fig. 4a). As we saw previously, the P-site-mismatched complexes are robust substrates for RF2/RF3, yielding rate constants for peptide release in the range of 0.005 to 0.01 s−1. For the singly mismatched E-site complexes, there was no stimulation of release activity in two of the complexes (MDEP and MLFK) while the results were buffer dependent for the MNKF complex (Fig. 4b and Supplementary Fig. 11). These differences will be discussed below. Most striking however was the increased rate of premature peptide release on each of the doubly mismatched complexes with rate constants for release ranging from 0.07 to 2 s−1 (Fig. 4b-d). These values are in a range comparable to tRNA selection and thus should compete in the ongoing process of protein synthesis. The robust nature of these findings was confirmed by observing very similar relative rates of release on the MKKF RNC series in a different buffer (polymix, D) and with chromosomally-expressed (fully methylated) RF2 (Supplementary Fig. 11). Control experiments were also performed to ensure that RNCs carrying a miscoding event that has fully exited the E site are no longer identified by RF2 as aberrant (Supplementary Fig. 12).
Figure 4. Iterated miscoding results in doubly mismatched complexes, where release catalysis is dramatically promoted.
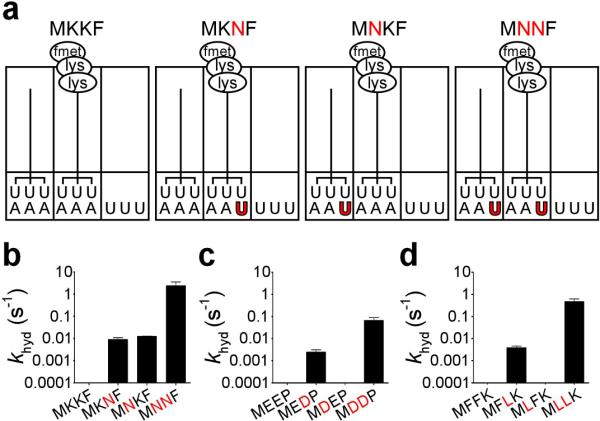
a, Schematic representation of one of the tripeptidyl-RNC series used to address E-site effects on release; shown is the MKKF series. The MKKF complex carries no mismatches, MKNF contains a P-site mismatch, MNKF has an E-site mismatch, and MNNF bears both E- and P-site mismatches. Rate constants for release (khyd) on a log scale with saturating RF2 and RF3 for the b, MKKF series, c, MEEP series, and d, MFFK series. Error bars indicate the standard error obtained from the non-linear regression fit of the data.
To further explore the molecular mechanism responsible for the disparate E-site effects, we performed a toe-printing experiment to determine the positioning of the tripeptidyl-tRNA RNCs on several mRNA species. For the MEEP series, the tripeptidyl-tRNA complexes appear rather uniform and fully extended with a toeprint positioned primarily at +6 relative to their starting position (Supplementary Fig. 13a). Strikingly, for the MKKF series, while the P-site-mismatched complex (MKNF) was positioned as anticipated on the mRNA, for both complexes carrying an E-site mismatch (MNKF and MNNF), a substantial fraction of the complex exhibits a heterogeneous banding pattern (Supplementary Fig. 13b). These latter data are consistent with proposals suggesting that codon:anticodon interactions in the E site can be critical for reading frame maintenance during translation23. We can not explain in molecular terms why the E-site mismatch alone typically has no effect on release activity (MDEP, MLFK, and MNKF in buffer D), but strongly stimulates release in all cases when combined with a P-site mismatch, though the synthetic E-site effects are reminiscent of E-site allostery models proposed by Nierhaus and colleagues24. We suggest that the effects of E-site mismatches on the A site are in great part manifested through structural perturbations of the proximal P-site decoding helix, an idea nicely supported by studies in yeast demonstrating that P-site mismatches can affect frame maintenance25.
In vivo relevance of A-site promiscuity following miscoding during translation
To evaluate the potential contribution of retrospective quality control to the fidelity of translation in the cell, we estimated the partitioning between premature release, inaccurate and accurate PT following a first miscoding event (Fig. 5a) based on rough cellular estimates of tRNA and release factor concentrations (50-200 μM and 6-25 μM, respectively, ref 26) and the measured kcat/Km values of PT, incorrect PT and release following a single miscoding event (Figs 3, 4, and Supplementary Fig. 3). We predict from these calculations (see Supplementary Information) that the net effect of the iterated partitioning steps (detailed in Fig. 5a) is that a single initial miscoding event results in a dramatic increase in premature chain termination (highlighted by green arrows).
Figure 5. An initial miscoding event results in an overall drop in yield of full-length peptides.

a, Proposed model for the events following a miscoding event with the steps contributing to the quality control described here highlighted with green arrows. b, Mock in vivo experiment recapitulates predictions of model. The indicated series of mRNAs (MKX through MKIFHKX for the matched series and MNX through MNIFHKX for the mismatched series) were used in complete translation reactions to observe the consequences of competition between tRNAs and RFs for peptide synthesis. Peptides initiated with the cognate dipeptide MK from the matched mRNA series are assigned the color black (MK-matched), peptides initiated with the cognate MN from the mismatched mRNA series are assigned the color blue (MN-matched), while peptides resulting from an incorrect decoding by Lys-tRNALys on the Asn (N) codon are assigned the color red (MN-mismatched). c, Yield was quantified as the fractional radioactivity in each product band relative to the whole lane. The plot represents the average of three independent experiments, with error bars representing the standard deviation from the mean.
There are two predictions of the proposed model: 1) that the yield of full-length product will diminish following a miscoding event and 2) that there will be evidence of prematurely truncated, multiply miscoded peptides. To test these predictions under competitive conditions, we evaluated the translation of a heteropolymeric mRNA sequence containing an AAU codon at position two for targeted decoding and miscoding by Asn-tRNAAsn and Lys-tRNALys, respectively, followed by sequence coding for abundant aminoacyl-tRNAs in the mixture. Translation of this mRNA in an S100 extract under limiting concentrations of Asn-tRNAAsn allowed us to follow the consequences of cognate and near-cognate decoding during translation in an in vivo like setting. In this competition experiment (Fig. 5b/c), we observe that the yield of full-length product following an initial miscoding event was decreased nearly four-fold (relative to two different controls). When the experiment was repeated in buffer D, the observed reduction in product following a miscoding event was even more impressive (~ 10-fold, Supplementary Fig. 15). This overall drop in yield is markedly consistent with the partitioning that we predict in Fig. 5a. While we see no strong signature of prematurely-released miscoded polypeptides, we expect that such a diffuse set of products would be very difficult to detect in our TLC system. We also have looked directly at partitioning for a defined (biochemically isolated) tripeptidyl tRNA RNC (MNNF) in the presence of S100 and aminoacyl-tRNA mix, and observe an extent of release consistent with estimates from our purified system (Supplementary Fig. 14).
With some hindsight, early in vivo studies of translation processivity likely provided evidence for the post-peptidyl transfer editing process described here, though it was not possible at the time to identify the root cause of the premature termination events27. Later studies by Kurland and colleagues characterized connections between ribosome accuracy and processivity28, but did not predict that there might be a direct consequence of miscoding on termination. In these studies, we note that low fidelity ribosome (ram) strains exhibited poor processivity, as would be predicted by our model.
Another intriguing in vivo correlation for our studies comes from the known auto-regulation of RF2. Translation of RF2 in the cell depends on a programmed frame-shifting event that occurs when a UGA codon encountered in the A site results in a stalled complex due to the low abundance of RF229. As with many frame-shifting events, the decoding helix found in the P site following the programmed frame-shifting event is perturbed (with RF2, for example, a G:U mismatch in the 1st position of the codon) and thus should normally be a prime candidate for recognition by the surveillance system described here. Paradoxically, in this case, RF2 concentration is low and thus is not likely to contribute to substantial premature chain termination until overall RF2 levels in the cell are increased. We suggest that this feedback loop has evolved to effectively evade the quality control mechanism described here.
Conclusions
Quality control mechanisms are key throughout the cell in ensuring the faithful replication of the genome and its expression into functional components. Like DNA replication, the quality control system that we describe here for protein synthesis depends on recognition of error following chemical incorporation of the building block into the growing polymer. However, unlike DNA replication, where extension of the growing strand is completed after hydrolytic action of the polymerase removes the misincorporated nucleotides, the quality control described here results in termination of protein synthesis.
In light of the competition experiments described earlier, we argue that the post-peptidyl transfer process described here might contribute close to an order of magnitude to fidelity in vivo under standard conditions. Moreover, under conditions of starvation, where amino acids become limiting in the cell and miscoding events are increasingly likely, we suggest that this surveillance system could play an even more substantial role in specifying the fidelity of translation. We note that the experiments here were conducted on very early stage RNCs (almost initiation complexes) and that premature release might play a distinct role during the elongation phase of translation. Nevertheless, the effects we have measured in this study are striking. To give some perspective, while false termination at a sense codon normally occurs with a frequency of less than once per 100,000 codons (measured here and in ref 30) in fully matched complexes, in one of the doubly mismatched complexes, false termination occurs half the time. These dramatic changes in the biochemical activity of the ribosome are triggered by single mismatches positioned in the P and E sites of the small ribosomal subunit, highlighting the existence of another intricate molecular system within the ribosome that precisely dictates perfection in the transmission of the genetic code.
METHODS SUMMARY
RNCs were essentially prepared as described in ref 13, and subsequent release or peptidyl transfer assays are described in the online methods.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Material
Acknowledgements
We thank Brendan Cormack, Ali Nahvi, Allen Buskirk and Randy Reed for helpful comments on the manuscript and members of the laboratory for useful discussions. This work was supported by the NIH with salary support from HHMI.
METHODS
Materials
Buffers used were as follows; buffer A [50 mM Tris-HCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2, 1 mM DTT]31, buffer B (HiFi) [50 mM Tris-HCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 3.5 mM MgCl2, 0.5 mM spermidine, 8 mM putrescine, and 2 mM DTT]3, buffer C (Polyamine) [20 mM HEPES-KOH pH 7.6, 150 mM NH4Cl, 4.5 mM MgCl2, 2 mM spermidine, 0.05 mM spermine, 4 mM BME]32, buffer D (Polymix) [95 mM KCl, 5 mM NH4Cl, 5 mM Mg(OAc)2, 0.5 mM CaCl2, 8 mM putrescine, 1 mM spermidine, 5 mM potassium phosphate pH 7.5, 1 mM DTT]10.
E. coli MRE600 (ATCC29417) tight couple 70S ribosomes were prepared as described previously33. Over-expressed native IF1 and IF3 and His-tagged IF2 were purified as described34. N-terminally His-tagged RF1 and the 20 aminoacyl-tRNA synthetases were expressed and purified as previously described35. His6-tagged EF-Tu and EF-G were purified over Ni-NTA resin, the His tag was later removed by tobacco etch virus (TEV) protease, which was followed by a second passage over Ni-NTA column36. Over-expressed His-tagged RF2 and RF3 were purified as described15. Chromosomally-expressed RF2 was purified using a procedure similar to the one described by Dincbas-Renqvist et al.15 except for the following modifications. After the ammonium sulfate precipitation following the first gel-filtration step, fractions containing RF2 were resuspended in 25 mM sodium phosphate buffer pH 6.8 and dialyzed against the same buffer over night. The protein was then applied to a hydroxyapatite column (0.7 cm × 5.2 cm), and eluted with a 50 mL linear phosphate gradient (25 mM-500 mM). The purified protein was finally dialyzed in a buffer comprised of 40 mM Tris-HCl pH 7.5, 10 mM MgCl2, 100 mM KCl, 1 mM DTT, 50 % glycerol.
tRNALys, tRNAPhe, tRNAfMet (all from E. coli) and rabbit muscle pyruvate kinase were purchased from Sigma-Aldrich. Total E. coli tRNA was purchased from Roche. mRNA templates were prepared from double-stranded DNA templates using run-off transcription by T7 RNA polymerase37, and purified by PAGE. mRNAs used for dipeptidyl complexes had the following sequence – GGGUGUCUUGCGAGGAUAAGUGCAUU AUG (X) (Y) UGA UUUGCCCUUCUGUAGCCA - the initiator Met codon is in bold, while X and Y denote codons occupying the P and A site, respectively. The tripeptidyl RNCs were programmed with similar mRNAs that had an additional codon, Z, after the Y codon. The mRNA coding for fMet-Phe (AUG UUC) used for fidelity measurements (Supplementary Fig. 1) was chemically synthesized (Dharmacon).
tRNA charging
Aminoacylation and formylation of the initiator tRNAfMet with radio-labeled [35S]-methionine using an S100 extract was performed as described previously38. Pure tRNAs were charged by incubating the tRNA at 10 μM with the corresponding aminoacyl-tRNA synthetase (~1 μM) in the presence of the appropriate amino acid and ATP (100 μM and 2 mM, respectively) in the following buffer; 20 mM Tris-HCl pH 7.5, 20 mM MgCl2, 1 mM DTT. After incubation at 37°C for 30 minutes, the aminoacylated tRNA was purified by phenol and chloroform extraction followed by ethanol precipitation and resuspended in 20 mM potassium acetate pH 5.1 buffer with 1 mM DTT. Total tRNA was charged using a similar procedure except for the following; the tRNA concentration was increased to 100 μM and all 20 aminoacyl-tRNA synthetases were added (1 μM each) and all 20 amino acids were added (100 μM each). In cases where a single tRNA in the complete tRNA mixture was aminoacylated, the same basic reaction was set up, but only the desirable synthetase and amino acid were supplied (e.g. for the Ile-tRNAIle in Fig. 3a).
RNC ribosome complex formation
Initiation complexes (ICs) were first prepared by incubating 70S ribosomes (2 μM) with IF1, IF2, IF3, f-[35S]-Met-tRNAfMet (3 μM each), and mRNA (6 μM) in buffer C (or buffer D for experiments in Supplementary Figs 3, 11 and 15) in the presence of GTP at 2 mM at 37°C for 45 minutes. RNCs were then obtained by adding equivalent volumes of ICs and a pre-incubated elongation mixture containing EF-Tu (15 μM), charged tRNA (6 μM for dipeptidyl and 10 μM for tripeptidyl complexes, respectively), EF-G (6 μM), and GTP (2 mM) in buffer C (or buffer D, as above) and incubating at 37°C for 10 minutes. Buffer C was utilized to form RNC complexes that were ultimately assayed in buffer A because of its permissivity in allowing near cognate tRNAs to react. Buffer A was considerably less promiscuous for certain near cognate pairings, thus making RNC complex formation difficult. To purify RNCs away from unincorporated tRNAs and factors, the reaction mixture was layered over a 1300 μL sucrose cushion (1.1 M sucrose, 20 mM Tris-HCl pH 7.5, 500 mM NH4Cl, 10 mM MgCl2, 0.5 mM EDTA) and spun at 69,000 rpm in a TLA100.3 rotor for 2 hours. The resulting pellet was resuspended in buffer A (or buffer D, as above), aliquoted and stored at −80°C. Electrophoretic TLC analysis of the complexes (see below) was used to determine the efficiency of dipeptide or tripeptide formation on the matched and mismatched mRNA templates. The typical yield for RNCs was as follows; dipeptide and tripeptide matched complexes > 80% of fMet was converted to the appropriate peptide, mismatched complexes involving Lys-tRNALys or Glu-tRNAGlu, yield was > 60% for dipeptides and > 40% for tripeptides, while mismatched complexes involving Phe-tRNAPhe had dipeptide and tripeptide yields of 40% and 10% respectively. In addition, the amount of f-[35S]-Met that pellets provides additional information about the stability of the RNC complexes. We note that mismatched templates typically yield less radioactivity in pelleting, likely because of the somewhat increased off-rates of the peptidyl-tRNA (Supplementary Fig. 4). We can easily follow the particular RNC of interest, despite the range in yield that we observe, because the relevant peptide product is well resolved in our electrophoretic TLC system (e.g. Fig. 5b)
Release assays
Peptidyl RNCs (both di- and tripeptidyl) at 25 to 150 nM were incubated with RF2 at 30 μM (determined to be saturating for mismatched complexes, see Supplemental Fig. 3) in buffer A at 37°C. Where indicated, RF3 was added to a final concentration of 30 μM with 2 mM GTP. Time-points were obtained by taking aliquots at different time intervals and stopping the reaction with 1/4th the volume of 25% formic acid. Released peptides of various lengths and identity were separated from unreacted peptidyl-tRNA using cellulose TLC plates that were electrophoresed in pyridine- acetate at pH 2.821. Reactions with relatively fast rate constants (e.g. > 0.05 s−1), such as the doubly mismatched RNCs in Fig. 4, were performed on a quench-flow instrument (RQF-3 quench-flow, KinTek Corporation). Fraction of peptide released at each time-point was quantified using ImageQuant v5.2 (Molecular Dynamics) and plotted against time. The data were fit to the first-order rate equation; F = Fmax (1-e−kt), where F is the fraction hydrolyzed, to obtain the rate constant (k) and the fraction of the population (Fmax) that could react. In most cases, Fmax was found to be between 60% and 90%. To determine K1/2 values, release time-courses were conducted with RNCs (25 nM) at varying concentrations of RF2 (0.025 μM to 30 μM). The kobs values were obtained from individual fits at a given concentration of RF2 and the K1/2 was derived from the hyperbolic fit of the kobs vs. [RF2] curve (Michaelis-Menten). The background rate - in the absence of RF2 - was determined for all complexes and subtracted from the rate observed in the presence of RF2. For certain P-site-matched complexes, the rate of peptide release was immeasurably low, and so in these cases an upper limit for the rate constant is provided (e.g. MKI and MKF complexes in Fig 1f).
Peptidyl transferase (PT) assays
EF-Tu at 100 μM was first incubated with 2 mM GTP in buffer A for 15 minutes to promote exchange of the bound GDP for GTP. The enzyme was then diluted to 20 μM in buffer A containing charged tRNA (10 μM) and 2 mM GTP, and incubated for another 15 minutes. The mixture was incubated with an equal volume of RNC (final concentration ~ 100 nM). Reaction was stopped by the addition of KOH to a concentration of 100 mM. Reaction products were resolved by electrophoretic TLC as above, and analyzed similarly. Reactions performed with total tRNAs were carried out with final concentrations of EF-Tu and tRNA of 100 μM and 80 μM, respectively. The rate constants for these experiments were determined by fitting curves following the fraction of substrate (e.g. dipeptide) that disappeared as a function of time.
Toe-print assay
Initiated and elongated peptidyl-tRNA complexes were prepared as above, except that the mRNAs used had extra sequence at the 3′-end to allow for an oligonucleotide primer to anneal and be extended by reverse transcriptase. The toe-printing reactions were then carried out essentially as described39. The RNCs were resuspended in buffer A that was supplemented with an additional 10 mM MgCl2. A trace amount of 5′-radio-labeled RT primer (5′ phosphorylated using polynucleotide kinase and [γ-32P]-ATP), and dNTPs (600 μM each) were added. Primer extension was initiated by the addition of AMV reverse transcriptase at a concentration of 1U/μL. The reaction was incubated at 37°C for 10 minutes, followed by the addition of NaOH at 100 mM and incubation at 90°C for 10 minutes to digest the RNA. The reaction was ethanol precipitated before analysis on long format 6% PAGE.
2D TLC separation
For resolution of complex PT reactions incubated with total tRNA mixtures, the PT reactions were performed essentially as described above. At the end of the reaction, peptidyl-tRNA was hydrolyzed with 100 mM KOH before spotting the sample on a 20 × 20 cm cellulose TLC. In the first dimension the mobile phase was composed of ethanol:water:acetic acid at a ratio of 70:20:10. The TLC was then thoroughly dried and run electrophoretically in pyridine-acetate buffer (pH 2.8) for the second dimension.
S100 in vitro translation
For Fig. 5b, purified initiation complexes, instead of the post-translocation complexes used in previous reactions, were prepared as described above, and then incubated (~50 nM final concentration) with an S100 extract containing 120 μM tRNA (pre-charged with a tRNA synthetase mix lacking AsnRS), 2 mM GTP, 6 mM PEP and 0.02 mg/mL pyruvate kinase in buffer A at 37°C for 10 minutes. For Supplementary Fig. 14, post-translocation tripepidyl-RNCs were prepared as above, and reacted in a similar fashion with S100 and aminoacyl-tRNA mixture. The samples were resolved using the electrophoretic TLC system described above. Observed Asn-tRNAAsn activity in Fig. 5b likely derives from contaminating reagent that carries through in the bulk tRNA mixture or through impurities in the individual amino acids, as seen for Leu-tRNALeu activity in Supplemental Fig. 1.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Edelmann P, Gallant J. Mistranslation in E. coli. Cell. 1977;10:131–137. doi: 10.1016/0092-8674(77)90147-7. [DOI] [PubMed] [Google Scholar]
- 2.Bouadloun F, Donner D, Kurland CG. Codon-specific missense errors in vivo. Embo J. 1983;2:1351–1356. doi: 10.1002/j.1460-2075.1983.tb01591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell. 2004;13:191–200. doi: 10.1016/s1097-2765(04)00005-x. [DOI] [PubMed] [Google Scholar]
- 4.Guth EC, Francklyn CS. Kinetic discrimination of tRNA identity by the conserved motif 2 loop of a class II aminoacyl-tRNA synthetase. Mol. Cell. 2007;25:531–542. doi: 10.1016/j.molcel.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt E, Schimmel P. Mutational isolation of a sieve for editing in a transfer RNA synthetase. Science. 1994;264:265–267. doi: 10.1126/science.8146659. [DOI] [PubMed] [Google Scholar]
- 6.Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. USA. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ninio J. Kinetic amplification of enzyme discrimination. Biochimie. 1975;57:587–595. doi: 10.1016/s0300-9084(75)80139-8. [DOI] [PubMed] [Google Scholar]
- 8.Soll D. The accuracy of aminoacylation--ensuring the fidelity of the genetic code. Experientia. 1990;46:1089–1096. doi: 10.1007/BF01936918. [DOI] [PubMed] [Google Scholar]
- 9.Szaflarski W, et al. New features of the ribosome and ribosomal inhibitors: non-enzymatic recycling, misreading and back-translocation. J. Mol. Biol. 2008;380:193–205. doi: 10.1016/j.jmb.2008.04.060. [DOI] [PubMed] [Google Scholar]
- 10.Jelenc PC, Kurland CG. Nucleoside triphosphate regeneration decreases the frequency of translation errors. Proc. Natl. Acad. Sci. USA. 1979;76:3174–3178. doi: 10.1073/pnas.76.7.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brutlag D, Kornberg A. Enzymatic synthesis of deoxyribonucleic acid. 36. A proofreading function for the 3′ leads to 5′ exonuclease activity in deoxyribonucleic acid polymerases. J. Biol. Chem. 1972;247:241–248. [PubMed] [Google Scholar]
- 12.Precup J, Parker J. Missense misreading of asparagine codons as a function of codon identity and context. J. Biol. Chem. 1987;262:11351–11355. [PubMed] [Google Scholar]
- 13.Brunelle JL, Shaw JJ, Youngman EM, Green R. Peptide release on the ribosome depends critically on the 2′ OH of the peptidyl-tRNA substrate. RNA. 2008;14:1526–31. doi: 10.1261/rna.1057908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youngman EM, He SL, Nikstad LJ, Green R. Stop codon recognition by release factors induces structural rearrangement of the ribosomal decoding center that is productive for peptide release. Mol. Cell. 2007;28:533–543. doi: 10.1016/j.molcel.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Dincbas-Renqvist V, et al. A post-translational modification in the GGQ motif of RF2 from Escherichia coli stimulates termination of translation. Embo J. 2000;19:6900–6907. doi: 10.1093/emboj/19.24.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freistroffer DV, Kwiatkowski M, Buckingham RH, Ehrenberg M. The accuracy of codon recognition by polypeptide release factors. Proc. Natl. Acad. Sci. USA. 2000;97:2046–2051. doi: 10.1073/pnas.030541097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baranov PV, Gesteland RF, Atkins JF. Recoding: translational bifurcations in gene expression. Gene. 2002;286:187–201. doi: 10.1016/s0378-1119(02)00423-7. [DOI] [PubMed] [Google Scholar]
- 18.Hartz D, McPheeters DS, Traut R, Gold L. Extension inhibition analysis of translation initiation complexes. Methods Enzymol. 1988;164:419–425. doi: 10.1016/s0076-6879(88)64058-4. [DOI] [PubMed] [Google Scholar]
- 19.Brown CM, McCaughan KK, Tate WP. Two regions of the Escherichia coli 16S ribosomal RNA are important for decoding stop signals in polypeptide chain termination. Nucleic Acids Res. 1993;21:2109–2115. doi: 10.1093/nar/21.9.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katunin VI, Muth GW, Strobel SA, Wintermeyer W, Rodnina MV. Important contribution to catalysis of peptide bond formation by a single ionizing group within the ribosome. Mol. Cell. 2002;10:339–346. doi: 10.1016/s1097-2765(02)00566-x. [DOI] [PubMed] [Google Scholar]
- 21.Youngman EM, Brunelle JL, Kochaniak AB, Green R. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004;117:589–599. doi: 10.1016/s0092-8674(04)00411-8. [DOI] [PubMed] [Google Scholar]
- 22.Freistroffer DV, Pavlov MY, MacDougall J, Buckingham RH, Ehrenberg M. Release factor RF3 in E.coli accelerates the dissociation of release factors RF1 and RF2 from the ribosome in a GTP-dependent manner. Embo J. 1997;16:4126–4133. doi: 10.1093/emboj/16.13.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marquez V, Wilson DN, Tate WP, Triana-Alonso F, Nierhaus KH. Maintaining the ribosomal reading frame: the influence of the E site during translational regulation of release factor 2. Cell. 2004;118:45–55. doi: 10.1016/j.cell.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Geigenmuller U, Nierhaus KH. Significance of the third tRNA binding site, the E site, on E. coli ribosomes for the accuracy of translation: an occupied E site prevents the binding of non-cognate aminoacyl-tRNA to the A site. Embo J. 1990;9:4527–4533. doi: 10.1002/j.1460-2075.1990.tb07904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundararajan A, Michaud WA, Qian Q, Stahl G, Farabaugh PJ. Near-cognate peptidyl-tRNAs promote +1 programmed translational frameshifting in yeast. Mol. Cell. 1999;4:1005–1015. doi: 10.1016/s1097-2765(00)80229-4. [DOI] [PubMed] [Google Scholar]
- 26.Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 27.Manley JL. Synthesis and degradation of termination and premature-termination fragments of beta-galactosidase in vitro and in vivo. J. Mol. Biol. 1978;125:407–432. doi: 10.1016/0022-2836(78)90308-x. [DOI] [PubMed] [Google Scholar]
- 28.Dong H, Kurland CG. Ribosome mutants with altered accuracy translate with reduced processivity. J. Mol. Biol. 1995;248:551–561. doi: 10.1006/jmbi.1995.0242. [DOI] [PubMed] [Google Scholar]
- 29.Craigen WJ, Caskey CT. Expression of peptide chain release factor 2 requires high-efficiency frameshift. Nature. 1986;322:273–275. doi: 10.1038/322273a0. [DOI] [PubMed] [Google Scholar]
- 30.Jorgensen F, Adamski FM, Tate WP, Kurland CG. Release factor-dependent false stops are infrequent in Escherichia coli. J. Mol. Biol. 1993;230:41–50. doi: 10.1006/jmbi.1993.1124. [DOI] [PubMed] [Google Scholar]
- 31.Pape T, Wintermeyer W, Rodnina M. Induced fit in initial selection and proofreading of aminoacyl-tRNA on the ribosome. Embo J. 1999;18:3800–3807. doi: 10.1093/emboj/18.13.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartetzko A, Nierhaus KH. Mg2+/NH4+/polyamine system for polyuridine-dependent polyphenylalanine synthesis with near in vivo characteristics. Methods Enzymol. 1988;164:650–658. doi: 10.1016/s0076-6879(88)64075-4. [DOI] [PubMed] [Google Scholar]
- 33.Shaw JJ, Green R. Two distinct components of release factor function uncovered by nucleophile partitioning analysis. Mol. Cell. 2007;28:458–467. doi: 10.1016/j.molcel.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunelle JL, Youngman EM, Sharma D, Green R. The interaction between C75 of tRNA and the A loop of the ribosome stimulates peptidyl transferase activity. RNA. 2006;12:33–39. doi: 10.1261/rna.2256706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu Y, et al. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 36.Blanchard SC, Kim HD, Gonzalez RL, Jr., Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc. Natl. Acad. Sci. USA. 2004;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaher HS, Unrau PJ. T7 RNA polymerase mediates fast promoter-independent extension of unstable nucleic acid complexes. Biochemistry. 2004;43:7873–7880. doi: 10.1021/bi0497300. [DOI] [PubMed] [Google Scholar]
- 38.Moazed D, Noller HF. Sites of interaction of the CCA end of peptidyl-tRNA with 23S rRNA. Proc. Natl. Acad. Sci. USA. 1991;88:3725–3728. doi: 10.1073/pnas.88.9.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorner S, Brunelle JL, Sharma D, Green R. The hybrid state of tRNA binding is an authentic translation elongation intermediate. Nat. Struct. Mol. Biol. 2006;13:234–241. doi: 10.1038/nsmb1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.