

Abstract
Estrogen, in addition to its genomic effects in brain, causes rapid and reversible changes to synaptic operations. We report here that these acute actions are due to selective activation of an actin-signaling cascade normally used in the production of long-term potentiation (LTP). Estrogen, or a selective agonist of the steroid's β-receptor, caused a modest increase in fast glutamatergic transmission and a pronounced facilitation of LTP in adult hippocampal slices; both effects were completely eliminated by latrunculin, a toxin that prevents actin filament assembly. Estrogen also increased spine concentrations of filamentous actin and strongly enhanced its polymerization in association with LTP. A search for the origins of these effects showed that estrogen activates the small GTPase RhoA and phosphorylates (inactivates) the actin severing protein cofilin, a downstream target of RhoA. Moreover, an antagonist of RhoA kinase (ROCK) blocked estrogen's synaptic effects. Estrogen thus emerges as a positive modulator of a RhoA>ROCK>LIM kinase>cofilin pathway that regulates the subsynaptic cytoskeleton. It does not, however, strongly affect a second LTP-related pathway, involving the GTPases Rac and Cdc42 and their effector p21-activated kinase, which may explain why its acute effects are reversible. Finally, ovariectomy depressed RhoA activity, spine cytoskeletal plasticity, and LTP, whereas brief infusions of estrogen rescued plasticity, suggesting that the deficits in plasticity arise from acute, as well as genomic, consequences of hormone loss.
Introduction
Discussions about how estrogen produces strong, positive effects on memory and cognition have largely focused on genomic actions of the steroid. Circulating levels of estrogen influences the number of dendritic spines in certain hippocampal subfields (Gould et al., 1990) and regulates neurotransmitter levels throughout much of the cortical telencephalon (Fink et al., 1996; Lee and Pfaff, 2008). Estrogen also modulates the production of brain growth factors responsible for the maintenance of neuronal processes (Singh et al., 1995; Sohrabji et al., 1995; Pan et al., 1999). Effects of these types, as discussed in the literature, provide reasonable explanations for the link between estrogen and cognition.
Despite the above, recent work suggests that acute, nontranscriptional effects may also contribute to estrogen's influence over behavior. Short infusions of 17-β-estradiol (hereafter referred to as estradiol or E2), a main form of estrogen, cause small but rapid increases in fast excitatory synaptic responses and markedly facilitate the formation of LTP in hippocampal slices (Foy et al., 1999; Bi et al., 2000; Sharrow et al., 2002; Foy et al., 2008). The plasticity effects are mediated by estradiol β-type receptors (ERβs), which are present in synaptic membranes (Milner et al., 2005), and are accompanied by increased surface expression of AMPA-type glutamate receptors (Liu et al., 2008). These short-latency effects on transmission and plasticity are likely related to the acute, nongenomic influence of estradiol on learning (Packard and Teather, 1997; Luine et al., 2003; Rhodes and Frye, 2006) and could therefore underlie variations in memory performance associated with cyclic or age-related changes in E2 production.
Surprisingly little is known about the steps linking estradiol receptors to the complex machinery that regulates synaptic strength. However, work on non-neuronal tissue indicates that the steroid regulates actin dynamics and thereby modifies the submembrane cytoskeleton (Giretti et al., 2008). This is of potential relevance to its acute actions in adult brain because rapid, activity-driven changes to the spine cytoskeleton play an essential role in the production of LTP (Lynch et al., 2007; Messaoudi et al., 2007). Questions thus arise as to whether estradiol acutely regulates the cytoskeleton at adult synapses and whether such effects are responsible for its actions on baseline transmission, plasticity, and learning. Moreover, a short-latency connection to actin networks would help explain why natural fluctuations in estradiol levels, or acute injections of the steroid, are accompanied by changes in spine morphology and number.
Here we report that estradiol produces its acute synaptic actions by selectively stimulating one of the multiple actin signaling cascades involved in the production of stable LTP. We also found that ovariectomy depresses the same pathway, suggesting that the loss of plasticity associated with this chronic manipulation (Singh et al., 1994; Fink et al., 1996; Smith and McMahon, 2005, 2006; Liu et al., 2008) arises from the continuing absence of estradiol's acute effects on synapses. Initial tests of this idea proved positive. The linkages between estradiol receptors and the spine cytoskeleton described here may thus have significance for ideas about the origins of, and potential treatments for, memory problems that can accompany surgical ovariectomy or menopause.
Materials and Methods
All animal procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with protocols approved by the Institutional Animal Care and Use Committee of the University of California at Irvine. This includes efforts to minimize animal suffering and numbers of rats used in the work described.
Slice preparation and recording.
Studies used young adult (30–42 d old) male Sprague Dawley rats (Charles River) and middle-aged (retired breeders at 9–10 months old) ovariectomized (OVX) Long–Evans rats (Charles River). Methods were slightly modified from those described previously (Kramár et al., 2006). Briefly, acute hippocampal slices were prepared and maintained in an interface recording chamber containing preheated artificial CSF (aCSF) of the following composition (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 1.5 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 10 glucose and maintained at 31 ± 1°C. Slices were continuously perfused with this solution at a rate of 1.75–2 ml/min while the surface of the slices were exposed to warm, humidified 95% O2/5% CO2. Recordings began following at least 1.5 h of incubation.
Field EPSPs (fEPSPs) were recorded from CA1b stratum radiatum using a single glass pipette (2–3 MΩ). Stimulation electrodes (twisted nichrome wires, 65 μm) were positioned at two sites (CA1a and CA1c) in the apical Schaffer collateral–commissural projections to provide convergent activation of CA1b pyramidal cells. Pulses were administered in an alternating manner to the two electrodes at 0.05 Hz using a current that elicited a 50% maximal response. After establishing a 10–20 min stable baseline, test compounds (see below) were introduced into the infusion line by switching from control aCSF to drug-containing aCSF. To determine the LTP threshold and ceiling effect in control and drug-treated slices, the number of “theta” bursts delivered to induce LTP varied from 2 to 10, with each burst consisting of four pulses at 100 Hz and the bursts themselves separated by 200 ms (i.e., theta burst stimulation or TBS). The stimulation intensity was not increased during TBS. Data were collected and digitized by NAC 2.0 Neurodata Acquisition System (Theta Burst) and stored on a disk.
Reagents.
WAY200070 (WAY) and 4,4′,4″-[4-propyl-(1H)-pyrazole-1,3,5-triyl]trisphenol (PPT) were provided by Wyeth Pharmaceuticals. H-1152P dihydrochloride (Tocris Bioscience), d-(−)APV (Tocris Bioscience), DNQX (Tocris Bioscience), picrotoxin (Sigma), and saclofen (Tocris Bioscience) were prepared fresh in aCSF, while PPT, WAY, 17-β estradiol, ICI-182,780 (Tocris Bioscience), and latrunculin A (EMD Chemicals) were dissolved in DMSO and diluted to working concentrations in aCSF (DMSO <0.01%, final).
In situ phalloidin labeling.
Methods were slightly modified from those described previously (Kramár et al., 2006). Rhodamine- (Sigma) or Alexa568 (Invitrogen)-phalloidin (6 μm) was applied topically from a micropipette every 5 min for 20 min either (1) at the end of the experiment in slices that received low-frequency stimulation in the presence or absence of drugs or (2) 10 to 60 min after the delivery of theta bursts. Slices were then collected and prepared for microscopy. Labeling was examined using a Zeiss Axioskop microscope (40×, Apochromat, NA 1.2) and an Axiocam camera. Identification and measurement of labeled spines was performed on a 35,700 μm2 sampling area within the zone of physiological recording as described previously (Lin et al., 2005; Kramár et al., 2006; Rex et al., 2007). To quantify the total number of labeled spines, a fixed-interval intensity threshold series (15–75% of maximum, spaced by 4%) was used to detect boundaries of both faintly and densely labeled spines under conditions of drug application during low-frequency stimulation. Redundant observations from the threshold series were analyzed using Matlab (MathWorks) to produce a best boundary estimate for each spine. Spine intensity was calculated as mean pixel intensity for all pixels assigned to a given spine. In slices receiving TBS, analysis by single threshold was set at 75% of maximum image intensity to detect only densely labeled spines.
Chronic estradiol treatment.
Long–Evans rats at middle age (9–10 months old) were ovariectomized and 6 months later randomly divided into two groups: One group received estradiol replacement therapy (control group) and the other remained estradiol-depleted (experimental group). For hormone replacement, the estradiol analog 17-β estradiol benzoate (EB), maintained in a stock solution of 10 μg/0.1 ml in sesame oil, was injected subcutaneously once per day, every other day, for 2 weeks (Woolley and McEwen, 1993). Experimental animals received similar injections of sesame oil. At 24 h after the last injection, rats were decapitated, one side of the hippocampus was used for slice physiology experiments and the other was frozen and used to measure activated RhoA (see below). To verify the ovariectomy and sufficiency of EB replacement, the uterus was severed from the cervix, cleared of fat from the uterine tubes, and blotted dry on tissue paper to remove luminal fluid before being weighed. Uteri from EB-injected animals weighed, on average (±SEM), twice as much as uteri collected from vehicle-injected rats and were highly vascularized and filled with fluid (uterine weight: 0.185 ± 0.02 g, and 0.373 ± 0.02 g for vehicle- and EB-treated rats, respectively; n = 8/group).
Western blot analysis.
Hippocampal slices were pooled (2–3/group), homogenized in radioimmunoprecipitation assay buffer containing Phosphatase Inhibitor Cocktails 1 and 2 and Complete Protease Inhibitor Cocktail (Roche Diagnostics) (Rex et al., 2007), normalized for protein content (Bio-Rad Protein Assay), separated by 12% SDS-PAGE, and processed for Western blot analysis using rabbit antisera to pPAK1/2/3 (44-940G; S141, Biosource) and pCofilin (112866; S3, Abcam) and the ECL Plus chemiluminescent detection system (Amersham). Blots were reblotted (Millipore) and probed for β-tubulin and β-actin (Sigma) as loading controls. ImageJ (NIH) software was used to measure the intensity of immunoreactive bands. Statistical significance was determined by two-tailed Student's t test. N values are presented as number of samples tested.
Pull-down assays for activated Rho GTPases.
Samples (6–10 slices pooled) were homogenized in 1× Mg2+ lysis buffer (MLB) (Millipore) with Complete Protease Inhibitor Cocktail (Roche Diagnostics) and 10% glycerol. Protein levels were measured and normalized. Samples were incubated with Rac/Cdc42 (PAK-1 PBD; 14-325, Millipore) or RhoA (Rhotekin RBD; 14-383, Millipore) assay reagent and gently rocked for 4 h at 4°C. Substrate rhotekin binds strongly to both RhoA and RhoC, but weakly to RhoB (Reid et al., 1996). RhoC is not present in brain, thus RhoA was specifically detected in blots probed with the RhoA antibody. Agarose beads were collected by centrifugation (10 s, 14,000 × g, 4°C) and washed three times with MLB, resuspended and boiled in Laemmli buffer, and then separated by SDS-PAGE. Western blot analysis was performed using mouse anti-RhoA (418; Santa Cruz Biotechnology), mouse anti-Rac (05-389; Millipore), or rabbit anti-Cdc42 (4201; Millipore). For GTP and GDP loading controls, samples were incubated with 100 μm GTPγS or 1 mm GDP (30 min at 30°C with agitation) before assay with reagents.
Data analysis.
All electrophysiological measures are presented in the text as mean ± SD and in figures as mean ± SEM. Data were collected from control slices in parallel with experimental tissue for each treatment tested. The sample size for all experiments represents the number of animals used unless otherwise noted. At the end of each experiment, measurements from control slices were pooled and used as the comparison to drug-treated slices. Student's t tests, using p < 0.05 significance criteria (two-tailed), were used to compare groups for electrophysiology, Western blot analyses and measures of spine labeling. Analysis of mean spine labeling intensity used the nonparametric Kruskal–Wallis one-way ANOVA by ranks test. Pairwise comparisons were evaluated post hoc using the method of Bonferroni. The α level of these comparisons was set at p < 0.01, following a round-robin-style assessment of variance between repeated trials (>100) of the Kruskal–Wallis test with control group data.
Results
Estradiol increases synaptic responses via an action on AMPA receptors
In agreement with earlier reports (Foy et al., 1999; Bi et al., 2000; Sharrow et al., 2002; Foy et al., 2008), infusion of 1 nm estradiol (E2) caused a rapid increase in the synaptic response elicited by stimulation of the Schaffer–commissural projections to field CA1b stratum (str.) radiatum. The effect, which began within 5–10 min after the start of infusion and reversed completely during washout, was highly reproducible and consisted of a 17 ± 3% increase over baseline by 20 min after the start of infusion. The magnitude of the increase was comparable to that reported by other groups. The threshold for the E2-induced changes in baseline transmission fell between 0.01 nm (−1 ± 0.3%) and 0.1 nm (+6 ± 1.6%) (Fig. 1A). The changes to the fEPSP slope produced by 0.1 and 1.0 nm E2 were significantly different from those obtained with 0.01 nm E2 (p < 0.05, ANOVA with post hoc tests). Comparable results were obtained with a 20 min infusion of WAY, a selective agonist of ER-β: 10 and 100 nm treatments increased fEPSP slopes by 5 ± 2% and 17 ± 4% respectively, while 1 nm had no detectable effect (Fig. 1B). PPT, an agonist for estradiol receptor α (ER-α), at 100 nm did not reliably affect synaptic responses (+1 ± 1%). The differences between groups infused with WAY and PPT were highly significant (p < 0.001, ANOVA: F = 56.09) and the increases found with WAY at 10 and 100 nm were reliably larger in post hoc tests than those obtained with 1 nm WAY or PPT.
Figure 1.

Estrogen receptor stimulation reversibly increases synaptic transmission. Field EPSPs (fEPSPs) elicited by stimulation of the Schaffer–commissural projections were recorded in CA1b stratum radiatum of slices prepared from the hippocampus of adult male rats. A, Infusion of estradiol (E2) at concentrations of 0.01 (n = 3), 0.1 (n = 4), and 1 (n = 4) nm caused a rapid, dose-dependent, and reversible increase in fEPSP slope (infusion period indicated by bar). Inset, Traces collected during baseline and at the end of E2 infusion (calibration: 1 mV, 5 ms). B, The effects of E2 were reproduced by 20 min infusions of WAY (1–100 nm, n = 3–5), a selective agonist for estrogen's β receptors. An agonist for the α receptors of estrogen (PPT: 100 nm, n = 3) had no evident effects on synaptic responses. Inset, Traces collected during baseline and at peak response to 100 nm WAY (calibration: 1 mV/5 ms). C, The graph shows the percentage (mean ± SEM) paired-pulse facilitation (ppf) of the initial slope of fEPSPs expressed as a function of the interpulse interval (40, 60, 100, and 200 ms) for responses recorded before (dotted lines) and during (solid lines) infusion of 1 nm estradiol (n = 5) or the selective agonist 100 nm WAY (n = 5). Neither treatment significantly influenced paired-pulse facilitation.
The enhanced responses in the presence of 1 nm E2 and 100 nm WAY were not accompanied by changes in paired-pulse facilitation (p ≥ 0.5 at all intervals tested), and so are not likely due to changes in transmitter release kinetics (Fig. 1C). They were also selective to glutamate receptors because E2 did not affect the size of the “field IPSP” (fIPSP), a picrotoxin-sensitive response recorded in the presence of AMPA and NMDA receptor antagonists. Specifically, the amplitude of the fIPSP recorded 30 min after infusing 1 nm E2 was within 1 ± 11% of the prehormone baseline (Fig. 2A). We also tested whether E2 affects the presynaptic GABAB autoreceptors that shape transmitter release from feedforward GABAergic interneurons with repetitive stimulation (Arai et al., 1995). As shown in Figure 2A, the fIPSP generated by the second of two stimulation pulses under baseline conditions (aCSF) is reduced relative to the first response when the pulses are separated by 200 ms. The paired-pulse depression effect during baseline (−47 ± 11%), which is significantly reduced by the GABAB receptor antagonist saclofen (−36 ± 6.8%, p < 0.002), was unaffected by infusions of 1 nm E2 (−46 ± 6%; aCSF vs E2, p = 0.38).
Figure 2.

Estradiol selectively facilitates AMPA receptor-mediated components of fast EPSPs. A, Pharmacologically isolated IPSPs (responses recorded in the presence of 20 μm DNQX and 100 μm APV, antagonists for AMPA and NMDA receptors, respectively) were elicited by stimulation of the Schaffer–commissural projections. These responses were completely blocked by the GABAA receptor antagonist picrotoxin (PTX, upper right traces) and were unaffected by infusions of 1 nm E2 (upper left traces; also see text). Paired-pulse depression (200 ms interstimulus interval), a characteristic feature of fast IPSPs (see text), was unaffected by E2 (upper left traces and graph); subsequent infusion of the presynaptic GABAB receptor antagonist saclofen (100 μm) (upper right traces and graph) confirmed that the depression was due to GABA autoreceptors (n = 5). Calibration: 0.1 mV, 50 ms. B, The NMDA receptor component of the fEPSP was isolated by incubating the slices with 20 μm DNQX and 100 μm PTX. The resultant evoked potentials were unaffected by 20 min infusions of 1 nm E2 and were eliminated by APV (n = 4). The traces included in the inset illustrate the slow kinetics of the isolated NMDA response (calibration: 0.1 mV, 20 ms) and the extent to which it is insensitive to E2. C, E2 (1 nm) enhances isolated AMPA-receptor-mediated fEPSPs. The graph summarizes the results for a group of slices (n = 4) tested in the presence of APV and PTX. E2 (1 nm) caused an ∼20% increase in the amplitude of the isolated AMPA receptor-mediated fEPSP, a value comparable to that obtained in untreated slices (see Fig. 1). The traces included in the inset illustrate the E2 effect and the extent to which the responses were eliminated by the AMPA receptor antagonist DNQX.
By exclusion, the above results point to ionotropic glutamate receptors as the likely sites at which E2 acts to increase transmission. Given their preeminent role in mediating fast EPSPs, AMPA receptors emerge as the probable targets. This does not rule out the possibility that the steroid also impacts the NMDA receptors, something that would be of great importance to an analysis of how it influences LTP. Accordingly, we pharmacologically isolated the NMDA receptor-mediated component of the synaptic response by infusing DNQX (20 μm) and picrotoxin (100 μm) to block AMPA and GABAA receptor-mediated response components, and then tested for possible effects of E2. Infusions that reliably enhanced fEPSPs in untreated slices did not measurably change the amplitude of the NMDA receptor-dependent fEPSP 20 min after the start of infusion (aCSF = +1 ± 2.5% vs 1 nm E2 = −1 ± 1.1%, p > 0.60) (Fig. 2B). This result, in addition to ruling out an action on NMDA receptors, reinforces the conclusion that E2 does not enhance release or cause generalized changes to the postsynaptic environment. Finally, we confirmed, as expected from the above, that pharmacologically isolated AMPA receptor-mediated fEPSPs (NMDA and GABAA receptors blocked) are positively modulated by 1 nm E2 (Fig. 2C).
Estradiol lowers the threshold and raises the ceiling for LTP
We tested for LTP in the presence of E2 after adjusting the stimulation pulse strength to produce responses equivalent in size to those recorded during the preinfusion baseline. Past studies have shown that estradiol enhances the potentiation effect (Foy et al., 1999, 2008; Bi et al., 2000); we were interested in whether this reflects a reduction in the LTP threshold or an elevation of its normally present ceiling, two variables that are diagnostic with regard to mechanisms. A single pair of theta bursts did not produce stable potentiation in vehicle-treated slices (30 min after induction = +2 ± 4%), but did cause potentiation (+23 ± 3%) in slices infused with 1 nm E2. The delivery of three bursts had small, variable effects in control slices (+14 ± 6%) but generated robust LTP when delivered in the presence of 1 nm E2 (+44 ± 5%). Five theta bursts produced stable potentiation in the controls (+27 ± 2%) and a supranormal amount of potentiation in slices infused with E2 (+63 ± 6%). Slices perfused with 100 nm WAY reproduced the pattern of results obtained with E2 (2 bursts = +20 ± 4%, 3 bursts = +33 ± 5%, 5 bursts = +73 ± 11%), while responses in slices infused with the ER-α agonist PPT (2 bursts = +2 ± 2%, 3 bursts = +10 ± 3%, 5 bursts = +30 ± 9%) were comparable to controls (Fig. 3A). Past studies have shown that TBS trains with >10 bursts, or with spaced administration of multiple 10 burst trains, do not generate greater responses than are obtained with a single train (Larson et al., 1986; Kramár et al., 2004); both E2 (+80 ± 15%) and WAY (+109 ± 26%) nearly doubled the amount of potentiation normally obtained with a conventional 10-burst train (Veh: +45 ± 6%; PPT: +50 ± 18%). Slices infused with E2 or WAY had significantly greater LTP at 30 min after TBS than control or PPT-treated cases (F = 30.5, p < 0.001). Thus, E2 both lowers the threshold and raises the ceiling for LTP induced by naturalistic patterns of afferent stimulation.
Figure 3.

Effects of estrogen receptor stimulation on the threshold and ceiling for LTP in field CA1. A, Two or three theta bursts (arrows) did not reliably change the fEPSP slope in hippocampal slices (male rats) continuously infused with vehicle (0.01% DMSO) or the ER-α agonist PPT, but produced stable potentiation following pretreatment with estradiol (E2) or the ER-β agonist WAY. Five or ten theta bursts (arrows) generated greater potentiation in E2- or WAY-treated slices than in vehicle- or PPT-infused slices (n = 5/group). The upper dashed line denotes maximum LTP during control (aCSF) conditions. Inset: Traces collected during baseline recording (solid line) and 30 min after delivery of 3, 5 or 10 theta bursts (dotted lines) from slices infused with vehicle, E2 or WAY. Calibration: 1 mV, 5 ms. B, Percentage increase in burst response area across a series of 10 theta bursts in vehicle, PPT, WAY, and E2-treated slices; within-train facilitation was not significantly different between groups. C, Traces show the composite responses generated by a pair of theta bursts (200 ms between-burst interval) recorded in the presence and absence of 100 μm APV; percentage change in the areas of the fourth EPSP (red hash marks) provides a measure of NMDA receptor-mediated depolarization (calibration: 1 mV, 10 ms). As described in text, E2 did not affect this component of burst responses used to induce LTP (n = 4).
In an effort to determine whether the steroid acts on the synaptic events used to induce LTP, we measured the extent to which the burst responses facilitated over the course of a theta train. There were no evident differences between control slices and those treated with estradiol, or the estrogen receptor agonists, on this measure (p = 0.22, one-way ANOVA) (Fig. 3B). The magnitude of the NMDA receptor-mediated component of the burst responses was assessed by comparing the size of the second of the responses to a pair of theta bursts in the presence and absence of the selective NMDA receptor antagonist APV (Fig. 3C). The NMDA receptor contribution to burst size was the same in control (+37 ± 6%) and 1 nm E2-treated slices (+35 ± 4%; n = 4, p > 0.8). We conclude from this set of results that acute estradiol exerts its effects at a stage following the triggering events for LTP.
Acute synaptic effects of estradiol involve actin polymerization within dendritic spines
Infusions of ICI-182,780 (100 μm), which blocks binding to estradiol's nuclear receptors, did not alter baseline synaptic transmission or affect the rapid increases in fEPSP responses induced by 1 nm E2 (Fig. 4A). The response facilitation produced by E2 in the presence of ICI-182,780 (+14 ± 3%) was comparable to that obtained in multiple experiments (e.g., Fig. 1) without the inhibitor (+17 ± 3%; p > 0.3).
Figure 4.

Latrunculin blocks the effects of estradiol on synaptic physiology. Responses to Schaffer–commissural stimulation were recorded from field CA1b of slices (from male rats) treated with reagents during periods indicated by the horizontal bars. A, Treatment with ICI-182,780 (ICI, 100 μm) to block estradiol's nuclear receptor did not reduce E2-induced increases in fEPSP slope (n = 5). Inset: representative traces collected during baseline, ICI-182,780 infusion and at peak 1 nm E2 response. Calibration: 1 mV, 5 ms. B, Pretreatment with 500 nm latrunculin (latrunc.) blocked the effects of E2 on fEPSPs elicited by a collection of Schaffer–commissural projections stimulated at 0.05 Hz stimulation throughout the experiment (open circles). Ten theta bursts delivered to a second population of afferents produced a robust short-term potentiation that rapidly decayed back to baseline (closed circles). A 90 min period of latrunculin washout restored responsiveness of the fEPSP to 1 nm E2. C, Infusions of 500 nm latrunculin blocked, in a fully reversible manner, the increase in baseline responses produced by the ER-β agonist WAY (100 nm; n = 6). Inset, Representative traces collected during periods of treatment with latrunculin, WAY, and both. Calibration 1 mV, 5 ms.
We then used latrunculin, a toxin that blocks the addition of actin monomers to growing filaments, to test whether actin polymerization plays a role. The compound had no measurable influence on baseline fEPSP slope, in accord with previous reports (Krucker et al., 2000; Rex et al., 2007), but eliminated the effects of E2 on fEPSPs (Fig. 4B). E2 also failed to promote LTP in latrunculin-treated slices: Attempts to induce potentiation in the experimental pathway resulted in a robust short-term potentiation that gradually decayed back to baseline (60 min after TBS = +4 ± 5% vs baseline). These results confirm that facilitation of LTP by E2 is thoroughly dependent upon the same actin assembly processes that operate in the absence of the steroid.
Latrunculin by itself did not alter the amplitude of the isolated NMDA receptor response (baseline = 0.22 ± 0.03 mV vs 60 min infusion of latrunculin = 0.22 ± 0.03 mV), the composite response (4 overlapping EPSPs) to a single theta burst (areas of burst response 54 ± 11 mV · ms for vehicle-treated slices and 45 ± 8 mV · ms for the latrunculin cases; p = 0.15), or the NMDA receptor-mediated component of burst size (Veh = 33.0 ± 8.9% vs latrunculin = 33.7 ± 11.6% of the total response area). The facilitation of burst responses that occurs within a train was similarly unaffected by the toxin (% facilitation of area from burst 1 to burst 5 was 88 ± 42% and 66 ± 22% for vehicle vs latrunculin slices; p > 0.3). Theta burst responses and their enhancement are markedly affected by manipulations acting on frequency facilitation of transmitter release, NMDA receptors, GABA receptors, and afterhyperpolarizing potentials (Larson et al., 1986; Arai and Lynch, 1992; Sah and Bekkers, 1996). The failure of latrunculin to change burst responses or short-term potentiation thus constitutes strong evidence that acute administration of the toxin does not significantly influence the complex machinery that regulates the magnitude of synaptic responses. More extended treatments, however, would be expected to disrupt ongoing filament assembly/disassembly processes underlying the maintenance of cell junctions including synapses.
E2 produced its normal increase in synaptic responses in both control and experimental pathways when applied after a 90 min washout of latrunculin (control path = +14 ± 4% and experimental path = +20 ± 8%) (Fig. 3B). WAY-induced increases in fEPSPs were also thoroughly and reversibly blocked by latrunculin (latrunculin + WAY = −6 ± 6%; WAY alone = +16 ± 7%, p < 0.001) (Fig. 4C).
The above results suggest that E2 triggers actin filament assembly in the subsynaptic region. Direct tests of this were performed by applying fluorescence- tagged phalloidin, a toxin that selectively binds to filamentous (F-) actin, to slices following a 20 min infusion of E2. The slices were fixed, sectioned, and examined with epifluorescence microscopy. Field CA1 str. radiatum contained a sparse population of small, densely fluorescent structures in slices receiving low-frequency stimulation (LFS) in the absence of estrogen (Fig. 5A, top left); previous work indicates that these profiles correspond to labeled dendritic spines (Lin et al., 2005; Kramár et al., 2006; Rex et al., 2007). It appeared from survey micrographs that E2 increased the number of phalloidin-positive spines throughout field CA1 (Fig. 5A, top right). This effect was altogether absent in E2 slices that had been pretreated for 30 min with latrunculin (Fig. 5A, bottom right). Blind quantitative analyses using an automated system (see Materials and Methods) confirmed that slices receiving a 20 min infusion of E2 had a greater number of spines containing high concentrations of F-actin than did yoked control slices (p = 0.0005) (Fig. 5B). Further analysis of all (light to dense) labeled spines showed that the frequency distribution of labeling intensities was shifted in a latrunculin-sensitive manner toward higher values (Kruskal–Wallis, χ2 = 343.75, p < 0.001) (Fig. 5C). This is as expected if E2 acts uniformly to increase F-actin in a population in which individual elements (spines) differed significantly with regard to starting concentrations of F-actin.
Figure 5.
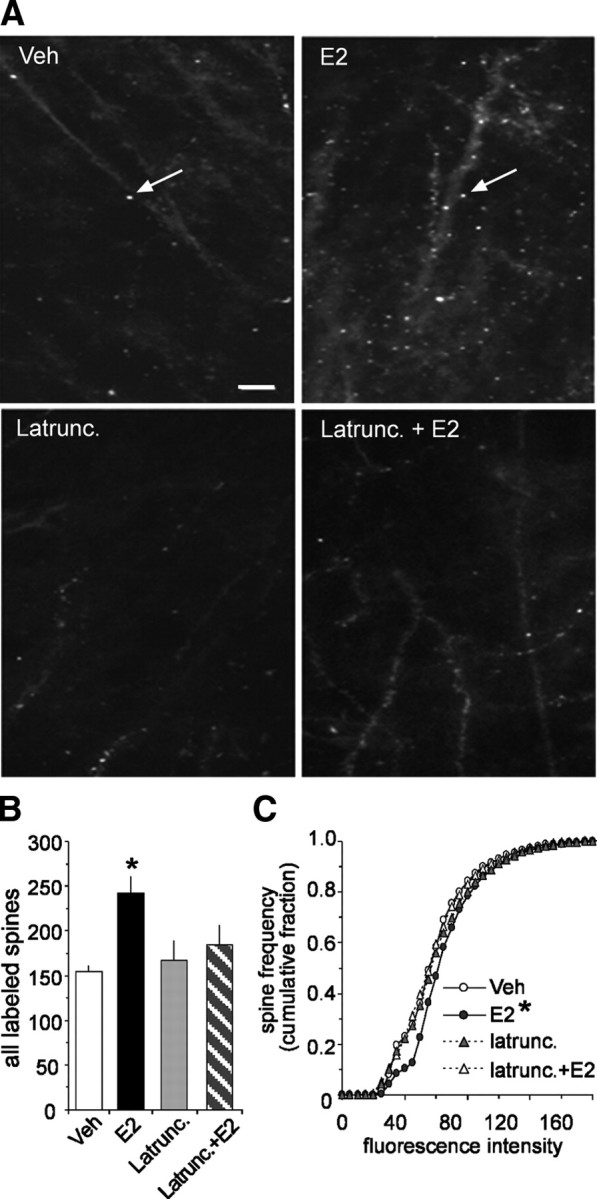
Estradiol promotes actin polymerization in dendritic spines. A, The top two panels compare phalloidin labeling in slices treated with 1 nm E2 or vehicle; the increase in spine-like profiles (arrows) produced by the steroid is typical of what is found in survey micrographs. The bottom two panels illustrate the effects of latrunculin (500 nm) alone on baseline and in combination with E2-induced labeling. Scale bar, 5 μm. B, The group mean (±SEM) number of dendritic spines with phalloidin labeling was significantly greater in E2 slices (n = 21 slices) compared with vehicle controls (n = 14 slices; p = 0.0005). E2 did not produce a significant effect in slices that had been pretreated with latrunculin. C, Cumulative frequency distribution of phalloidin-labeling intensities in CA1b str. radiatum in control versus E2-treated slices; the steroid shifted the distribution significantly to the right (i.e., toward more intense labeling) relative to the control profile (p < 0.001, Kruskal–Wallis test). This effect was blocked by latrunculin.
If estradiol by itself causes a modest increase in actin filaments in spine heads, then it could potentially augment the theta-burst induced polymerization needed for stable LTP. Tests of this were performed using a TBS train that was subthreshold for eliciting a reliable increase in the number of spines containing high concentrations of F-actin. Three theta bursts produced a pronounced short-term potentiation that rapidly decayed back to baseline in vehicle-treated slices (Fig. 6A). Slices collected 60 min after TBS had about the same number of phalloidin-labeled spines within the target dendritic lamina as did control (no TBS) slices (Fig. 6B,C). A 20 min infusion of E2 caused the expected increase in fEPSPs that decayed over a 60 min washout (Fig. 6A); these slices, collected at the end of the washout, had the same number of densely labeled spines as untreated slices (Fig. 6B,C). This result establishes the point that the E2-driven actin polymerization described in Figure 5 is fully reversible. The three-burst theta train that failed to produce stable LTP under control conditions caused a robust, nondecremental potentiation effect when administered at the end of a 20 min E2 infusion (Fig. 6A). It also produced a lasting increase in phalloidin-labeled spines as seen in slices collected after a 60 min washout of E2 and, accordingly, 60 min after TBS (Fig. 6B,C). In all, a brief exposure to E2 markedly enhances the effects of TBS on both actin polymerization and LTP.
Figure 6.

E2 enhances theta burst-induced LTP and actin polymerization. A, E2 (1 nm) or vehicle was infused for 20 min. and then washed out for 60 min. The steroid produced its typical ∼20%, reversible increase in the slope of the fEPSPs elicited in field CA1 by single pulse stimulation of the Schaffer–commissural fibers. A single train of three theta bursts delivered at the end of vehicle infusion caused a robust short-term potentiation that decayed back to baseline over the following 60 min. The same train applied at the end of E2 infusion generated robust and stable LTP (33 ± 9% at 60 min after infusion; p = 0.005 vs TBS in vehicle-treated slices). B, Rhodamine-labeled phalloidin was applied at the end of the 60 min washout period and the slices processed for epifluorescence microscopy. Survey micrographs indicated that slices receiving LFS during E2 treatment (“E2-LFS”) did not increase the number of labeled spines above levels seen in vehicle-treated cases (“Veh-LFS”). Three theta bursts also appeared to be without effect when applied in the presence of vehicle (“Veh+TBS”) but caused an evident increase in the numbers of labeled spines when delivered at the end of E2 infusion (“E2+TBS”). Scale bar, 5 μm. C, An automated system was used to count the number of spines associated with dense phalloidin labeling in slices collected 60 min after the various treatments. Slices that received only LFS had the same, low number of labeled spines whether treated with E2 or vehicle. Three theta bursts did not affect the frequency of phalloidin-positive spines when delivered to vehicle-treated slices but caused a pronounced increase when administered 60 min earlier in the presence of E2 (*p < 0.0001 vs E2 without TBS) (n = 8 slices/group).
Estradiol selectively facilitates a signaling pathway that controls actin polymerization
How does E2 promote actin polymerization in dendritic spines? We investigated this beginning with cofilin, a constitutively active protein that severs dynamic actin polymers. Phosphorylation inactivates cofilin and accordingly clears the way for the elongation, branching, and stabilization of filaments. Western blot assays showed that E2, infused for 20 min as in the electrophysiology experiments, caused an ∼50% increase in the concentration of phosphorylated (p)-cofilin (n = 6/group; p = 0.04), whereas total cofilin levels were unaffected by treatment (Fig. 7A, left).
Figure 7.

Estradiol selectively activates a RhoA-to-actin signaling pathway. Hippocampal slices (male rats) were treated with vehicle (Veh) or 1 nm estradiol (E2) for 20 min. A, Representative Western blots show at left levels of phosphorylated (p) cofilin and total cofilin and at right levels of pPAK and total PAK1 in hippocampal slices treated with vehicle or E2. Quantitative analyses (n = 6 slices/group) confirmed that E2 selectively increased levels of pCofilin relative to total cofilin (bar graph; p < 0.05 for E2 vs Veh), whereas E2 modestly but significantly reduced pPAK relative to measures from vehicle controls (n = 8/group; p < 0.01). Total PAK levels were unaffected. B, Total and active (GTP-bound) levels of RhoA, Cdc42, and Rac in vehicle- and E2-treated slices as seen in Western blots of material collected with the pull-down assay. Plots of band densities (bottom panel) indicate that E2 increased slice concentrations of activated RhoA (n = 3–5/group; p < 0.05 vs Veh control) while having no effect on total RhoA. Results from similar experiments detected no effect of E2 on active or total levels of the related GTPases Rac and Cdc42. Following a 60 min washout of E2, activated RhoA levels were not significantly different from those in vehicle-treated slices (n = 3/group). C, The ROCK inhibitor H-1152P (250 nm; open horizontal bar) reversibly blocked the increase in synaptic responses induced by E2 (n = 4). Inset, Representative traces collected during treatment with H-1152P, and E2 alone and in combination. Calibration: 1 mV, 5 ms.
LIM kinases (LIMK), the primary up-stream regulators of cofilin, are targets of two signaling cascades initiated by Rho family GTPases: (1) Rac/Cdc42>p21-activated kinase (PAK) and (2) RhoA>RhoA kinase (ROCK) (Edwards et al., 1999; Maekawa et al., 1999). We used pull-down assays to test whether either of these pathways is used by E2 and found that the steroid causes a profound increase in activated (GTP-bound) RhoA (n = 5/group; p = 0.04) with little if any change in Rac and Cdc42 activity (n = 3–5/group; p = 0.08) (Fig. 7B). The E2-induced increase in RhoA activity returned to control levels following a 60 min washout (Fig. 7B, right); this supports the hypothesis that acute applications of E2 cause transient and reversible forms of plasticity. Total concentrations of the GTPases (active and inactive) were not affected by 1 nm E2. In agreement with its lack of effect on Rac/Cdc42, E2 caused a small decrease in the phosphorylation (activation) of PAK, a primary effector for these two GTPases (n = 8/group; p = 0.008) (Fig. 7A, right).
Collectively, the results just described point to the conclusion that E2 phosphorylates cofilin by activating RhoA- rather than Rac/Cdc42-initiated signaling. If RhoA initiates the pathway primarily responsible for E2's effects on synaptic physiology, then selective inhibitors of RhoA's effector ROCK should block E2-induced changes in fEPSPs. In accord with this, 30 min pretreatment of hippocampal slices with the selective ROCK inhibitor H1152P (Sasaki et al., 2002), at concentrations (250 nm) shown in other studies to block TBS-induced phosphorylation of cofilin (Rex et al., 2009) eliminated E2's effects on fEPSPs (Fig. 7C). A 20 min infusion of H1152P alone did not alter baseline fEPSP slope (+1 ± 2%) but completely blocked E2-induced increases in synaptic transmission (−2 ± 3%). Following a 60 min washout period, reintroduction of 1 nm E2 alone caused significant increases in fEPSP slope (+17 ± 2%, p = 0.003 vs E2 + H1152P) that returned to baseline during washout.
Actin signaling after prolonged loss of estradiol
Finally, the question arises as to whether effects of the type described above contribute to the behavioral changes associated with extended periods of low estradiol. We explored this using long-term (6 month) ovariectomized rats given estrogen replacement (OVX + EB) or vehicle injections (OVX + Veh) for 2 weeks before testing. Input/output curves (Fig. 8A) and paired-pulse facilitation (Fig. 8B) were comparable for the two groups, and not noticeably different from results collected from young adult rats (see Fig. 1C). However, LTP (fEPSP slope 60 min after TBS = +17 ± 15%) was clearly impaired in the OVX + Veh slices relative to what is found at the same set of synapses in young adult or middle-aged male rats (Fig. 8C) (Rex et al., 2005). OVX rats given estrogen replacement for 2 weeks before slice preparation expressed normal appearing LTP (fEPSP slope 60 min after TBS = +54 ± 10%; p < 0.0001 vs OVX + Veh), as previously reported for mice (Gureviciene et al., 2003) and for short-term OVX rat models (Smith and McMahon, 2005, 2006). The cause of the impairment in the OVX + Veh group appears to reside in the processes underlying the stabilization, as opposed to the induction and expression, of LTP. The size (area) of the four EPSP responses to a single theta burst was not detectably different in slices from OVX + EB versus OVX + Veh rats (data not shown). The burst facilitation that occurs within the train was also comparable for the two groups: The mean percentage enhancement in area for bursts 2–10 relative to the first burst was 30 ± 16% and 33 ± 20% in slices from OVX rats with and without EB replacement, respectively (Fig. 8D). Beyond this, LTP expression for the first 2 min after TBS was 84 ± 45% above baseline in slices prepared from OVX + Veh and 94 ± 17% from OVX + EB rats (p > 0.5). However, despite this initial robust potentiation, LTP decayed rapidly and failed to establish a stable plateau in the slices from vehicle-injected OVX rats.
Figure 8.

Ovariectomy disrupts actin signaling and synaptic plasticity in CA1. Hippocampal slices were prepared from middle-aged (9–10 months old) rats that had been ovariectomized (OVX) for 6 months before receiving either estrogen replacement (17β-estradiol benzoate or EB; 10 μg/0.1 ml, n = 8) or vehicle injections (Veh, sesame oil; n = 8) every other day for 2 weeks. A, Input/output curves compare amplitudes of the presynaptic fiber volley to the fEPSP. Linear regression slopes were not detectably different at any current tested (10, 20, 30, 40, and 50 μA) for slices prepared from OVX + Veh versus OVX + EB rats (n = 8 slices/group). Representative traces are shown at right (calibration: 1 mV, 5 ms). B, Paired-pulse facilitation of the initial slope of the synaptic response was comparable at all interpulse intervals (40, 60, 100, and 200 ms) in the two groups of slices. C, A train of 10 theta bursts produced stable potentiation in slices from OVX rats given EB but not from those given vehicle. Insets show fEPSP traces collected during baseline recording (solid line) and 30 min after TBS (dotted line). Calibration: 1 mV, 5 ms. D, The facilitation of theta burst responses within a train was comparable in slices prepared from OVX rats given EB or vehicle. The data are expressed as the percentage increase in the areas of the numbered burst response over the area of the first burst response (n = 8/group). E, Survey micrographs of phalloidin labeling in slices prepared from OVX rats. Slices from OVX + Veh or OVX + EB rats had few labeled spines following LFS. TBS (10 bursts) produced what appeared to be a modest increase in labeling in OVX + Veh slices and a pronounced increase in OVX + EB slices. Scale bar, 5 μm. F, Numbers of phalloidin-labeled profiles measured with an automated counting program. Estimates for the LFS condition were comparable in slices prepared from OVX + Veh and OVX + EB rats (3 ± 2 and 2 ± 3 spines per 550 μm2, respectively) and the groups are combined in the graph (OVX + LFS, n = 10 slices). The TBS-induced increase in numbers of labeled puncta was significantly greater in slices from OVX + EB (p = 0.0007; n = 9 slices) than in OVX + Veh (n = 7 slices) rats. G, Representative Western blots of activated and total RhoA (as determined in pull-down assays) and bar graph showing quantification of active and total RhoA bands from four experiments. Levels of active RhoA were significantly lower in hippocampal tissue collected from OVX + Veh rats than in OVX + EB rats (p = 0.03).
Since actin dynamics play a central role in LTP stabilization (Okamoto et al., 2004; Kramár et al., 2006; Chen et al., 2007; Rex et al., 2007), we used the in situ phalloidin labeling technique to test for changes in TBS-induced actin polymerization. A high detection threshold was used so that the results could be directly compared with those from prior work on slices from young adult rats (Lin et al., 2005; Kramár et al., 2006). The two groups of OVX slices (with and without prior EB replacement) had the same number of labeled spines under low-frequency stimulation conditions but differed markedly in their responses to TBS. A 10-burst train produced an ∼11-fold increase in the number of densely labeled spines in slices from OVX + EB rats (Fig. 8E,F); this value is comparable to the 10- to 14-fold effects reported for slices from 2- to 3-month-old male rats (Lin et al., 2005; Kramár et al., 2006). A substantially smaller increase (less than fivefold) was obtained in slices from the OVX + Veh group; the difference between the two OVX groups was highly significant (p = 0.0007; n = 8 slices/group).
It appears that chronic reduction of E2 depresses one or more steps in the sequences driving actin filament assembly in spines. Given that E2 causes a substantial increase in RhoA activity (see Fig. 7B), we asked whether baseline levels of the small GTPase differ between OVX rats with and without hormone replacement. Pull-down assays showed that concentrations of activated GTP-bound RhoA were substantially lower in hippocampal homogenates prepared from OVX + Veh versus OVX + EB rats (n = 4/group; p = 0.03) (Fig. 8G).
The above findings raise the broad and therapeutically important question of whether ovariectomy, without hormone replacement, blocks the acute effects of E2 on actin signaling and synaptic plasticity. Experiments directed at this issue showed that a 30 min infusion of 1 nm E2 produces the same rapid and modest increase in baseline synaptic responses (13 ± 4%, p < 0.001 vs baseline) in slices from OVX + Veh rats as in slices from young adult male rats (Fig. 9A). Acute infusions of E2 also significantly increased numbers of spines with dense concentrations of F-actin [1 nm E2 (15 slices) = 35 ± 19 vs controls (12 slices) = 18 ± 5; p = 0.008], caused a significant rightward shift in the phalloidin-label intensity frequency distribution (p < 0.001) (Fig. 9B,C), and rescued TBS-driven increases in actin polymerization (data not shown). As anticipated from these effects, 20 min pretreatment with the steroid fully restored stable LTP in slices prepared from middle-aged, OVX rats (60 min after TBS: +49 ± 12% vs +19 ± 8% for untreated slices; p = 0.002) (Fig. 9D).
Figure 9.

Acute infusions of estradiol offset the effects of ovariectomy on actin dynamics and synaptic plasticity. A, E2 (1 nm) infusions produced a rapid and reversible increase in synaptic responses collected from OVX slices (n = 6) comparable in magnitude to that found in slices from young adult rats (see Fig. 1). Inset: traces collected during baseline (solid line) and at peak response to infusion of E2 (dotted line). Calibration: 0.5 mV, 5 ms. B, Twenty minute infusions of 1 nm E2 increased phalloidin labeling of spine heads in OVX slices relative to vehicle-treated cases. Scale bar, 5 μm. C, E2 caused a significant rightward shift in the intensity versus frequency distribution for phalloidin labeling (p < 0.001, Kruskal–Wallis test). D, The impairment in LTP (10 theta bursts) found in slices from OVX rats was offset by infusion of 1 nm E2 (horizontal bar); percentage LTP at 60 min after TBS was 49 ± 12% for E2-treated (n = 5) and 19 ± 8% for vehicle (Veh)-treated (n = 5; p = 0.002) slices. Inset: fEPSP traces collected during baseline (solid line) and at 60 min after TBS (dotted line). Calibration: 1 mV, 5 ms.
Discussion
The acute effects of estradiol on LTP and memory encoding have important implications for arguments about why cognitive performance fluctuates with the estrous cycle and declines with age (Markowska, 1999). They are also suggestive of therapeutic applications, particularly in light of evidence that highly selective agonists for ER-β enhance plasticity and memory in the absence of many of estradiol's peripheral actions (Liu et al., 2008). Further progress in these areas will require information about how estradiol produces its rapid actions. The present results point to a specific conclusion regarding this question; namely, that estradiol affects synaptic physiology by partially activating one, but not all, of the actin signaling pathways used in the generation of stable LTP. The hormone thus produces a weak and transient form of LTP; this has the effect of modestly increasing baseline responses and markedly enhancing the LTP produced by theta activity.
The first suggestion of a previously unsuspected influence of estradiol on actin dynamics came with the observation that latrunculin, a selective toxin that blocks the addition of actin monomers to growing filaments, eliminated the hormone's influence on baseline synaptic responses. We then found, as anticipated from the latrunculin results, that estradiol caused a modest but reliable increase in spine concentrations of filamentous actin. Also in agreement with the idea that estradiol acts on LTP-related actin signaling pathways, it markedly amplified theta burst-induced actin polymerization in spines. Given the now extensive evidence that actin polymerization is essential for the formation of stable LTP (Krucker et al., 2000; Fukazawa et al., 2003; Okamoto et al., 2004; Kramár et al., 2006; Rex et al., 2007, 2009), the observed interaction between estradiol and TBS provides a likely explanation for why the steroid so potently enhances the potentiation effect. A selective agonist of ER-β produced effects comparable to those obtained with estradiol and these were also blocked by latrunculin.
An analysis of the signaling cascades upstream from actin provided further support for estradiol's links to the substrates of LTP. Theta bursts cause a rapid but transient (∼15 min) increase in cofilin phosphorylation (Chen et al., 2007) and we found that estradiol produces a similar effect. Cofilin is a constitutively active, actin severing protein; phosphorylation inactivates cofilin and facilitates actin filament assembly (Gungabissoon and Bamburg, 2003; Thirone et al., 2009). Estradiol therefore promotes changes in actin management that enable actin polymerization and support the production of LTP. However, while the steroid engages certain actin-related effects that follow LTP induction, it leaves others relatively unaffected. In addition to driving cofilin phosphorylation, theta bursts cause a rapid and transient increase in the number of synapses in the activated dendritic zone that are associated with dense concentrations of phosphorylated (activated) PAK. It was originally thought that this PAK response to TBS leads to increases in cofilin phosphorylation via PAK>LIMK>cofilin signaling (Chen et al., 2007) but recent results have called this idea into question. Specifically, adenosine, a negative modulator of LTP, blocks TBS-induced cofilin phosphorylation and actin polymerization without depressing TBS effects on PAK phosphorylation (Rex et al., 2009). Estradiol, which promotes LTP, produces an inverse pattern of results in that it facilitates cofilin phosphorylation and actin assembly while mildly depressing PAK activation. The steroid did not measurably affect baseline activation of PAK's upstream regulators Rac/Cdc42 (Fig. 7B), so its effect on p-PAK could reflect an action on the multiple protein phosphatases that target the kinase (Zhan et al., 2003).
How, then, do theta bursts and estradiol inactivate cofilin? Beyond its involvement in the Rac/Cdc42>PAK>LIMK cascade, there is substantial evidence that LIMK is also a target for RhoA's effector ROCK (Maekawa et al., 1999; Ohashi et al., 2000; Thirone et al., 2009). Our results indicate that the latter pathway (i.e., RhoA>ROCK>LIMK>cofilin) is used by estradiol to modulate actin polymerization and synaptic physiology. The hormone activated RhoA while its actions on synaptic responses were completely blocked by a selective inhibitor of ROCK. This again links E2 to an LTP pathway because ROCK inhibitors completely disrupt consolidation of the potentiation effect (Rex et al., 2009).
The above results raise questions about the functional consequences of estradiol's failure to initiate the cascades involving Rac/Cdc42>PAK signaling. Possibly related to this, multiple experiments have shown that the actin filaments that quickly form after TBS are, like LTP itself, unstable for a period of several minutes and then become resistant to disruption (Krucker et al., 2000; Kramár et al., 2006; Rex et al., 2007, 2009). Recent work showed that selective inhibitors of Rac or PAK have no effect on either TBS-induced actin polymerization, but block the stabilization of both the newly formed filaments and LTP that normally occurs in the 15 min following TBS (Rex et al., 2009). These observations help explain why estradiol's acute effects reverse so readily; that is, the failure of the steroid to engage Rac/Cdc42> PAK signaling could mean that the actin networks it causes to assemble remain dynamic and inherently unstable. In accord with this, we found that a 60 min washout eliminates the increase in phalloidin labeling of F-actin produced by E2.
The idea that estradiol-induced increases in synaptic responses represent a type of weak and transient LTP necessarily predicts that the hormone's actions on the cytoskeleton ultimately activate the same synaptic processes that express the potentiation effect. It is now generally agreed that the latter events involve expansion of the pool of AMPA-type glutamate receptors that mediate fast EPSPs in hippocampus (Newpher and Ehlers, 2008). Consonant with this is recent evidence that TBS-induced phosphorylation of synaptic cofilin is associated with substantially enlarged postsynaptic densities (PSDs) (Chen et al., 2007) and therefore with a greater territory for docking AMPA receptors (Matsuzaki et al., 2004; Okamoto et al., 2004). Comparable studies have not been conducted after acute estradiol treatment but it is known that estradiol increases surface expression of the GluR1 subunit of the AMPA receptor (Liu et al., 2008). It will be of considerable interest to test whether the hormone causes a modest enlargement of PSDs (commensurate with its influence on EPSPs) and if this effect, along with estradiol's actions on AMPA receptor surface expression, requires actin filament assembly.
The actions of estradiol on the spine cytoskeleton, as described here, also provide a plausible explanation for fluctuations in spine numbers and LTP in hippocampus across phases of the estrous cycle (Woolley et al., 1990; Warren et al., 1995). Spine growth and retraction in dissociated neurons are driven by changes in the underlying cytoskeleton (Honkura et al., 2008) while the production of LTP, as noted, depends on actin filament assembly and stabilization. Baseline concentrations of F-actin vary considerably across spines (see Fig. 5C) and it is conceivable that some cytoskeletal configurations are more dependent upon estradiol than others. In any event, an across days analysis of spine pCofilin and F-actin concentrations would constitute a first step in determining whether the mechanisms described here are involved in spine changes associated with the different stages of the estrous cycle.
Finally, and of considerable clinical significance, is the issue of whether the absence of estradiol's short-latency actions on the cytoskeleton and plasticity are important contributors to behavioral changes arising from surgical ovariectomy or menopause. We found that RhoA activity, a key element in the path leading to stable LTP, was significantly depressed in OVX rats relative to the levels recorded for hormone replacement animals. Moreover, spine levels of activated LIMK, a target of RhoA and a key upstream regulator of cofilin, are depressed in OVX rats without hormone replacement (Yildirim et al., 2008). Baseline activity levels of the small GTPases are not reliably reflected in downstream effectors (Thaxton et al., 2007; Rex et al., 2009) but depressed levels of RhoA would place the OVX hippocampus at a disadvantage with regard to activity-driven cytoskeletal reorganization and, indeed, we found major deficits in TBS-induced actin polymerization and LTP consolidation.
Ovariectomy thus appears to impair the same signaling cascade used by acute estradiol treatments to produce a mild version of LTP. In accord with these arguments, estradiol infusion quickly restored TBS-induced actin polymerization and the capacity for stable LTP in slices from long-term OVX rats. This is not to say that ovariectomy disrupts plasticity simply by reducing moment-to-moment levels of estradiol available at synapses. Prior studies showed that production of NMDA receptor subunits is disturbed in OVX rats, resulting in an imbalance between the two categories of ionotropic glutamate receptors at synapses (Smith and McMahon, 2005, 2006). There are also a variety of well established genomic effects associated with ovariectomy that are likely to affect synaptic mechanisms (Spencer et al., 2008). Additional studies can thus be expected to reveal defects in the OVX slice that are not reversed by acute estradiol. Integrating this information with the findings described here will provide a framework for addressing the cognitive issues sometimes reported to accompany surgical ovariectomy (Sherwin, 1988) or menopause (Devi et al., 2005; Wegesin and Stern, 2007).
Footnotes
These studies were supported by National Institute of Neurological Disorders and Stroke Grants NS045260 and NS051923 to G.L. and C.M.G. C.S.R. was supported by NS045540 and L.Y.C. by National Institute of Mental Health Fellowship MH083396.
References
- Arai A, Lynch G. Factors regulating the magnitude of long-term potentiation induced by theta pattern stimulation. Brain Res. 1992;598:173–184. doi: 10.1016/0006-8993(92)90181-8. [DOI] [PubMed] [Google Scholar]
- Arai A, Silberg J, Lynch G. Differences in the refractory properties of two distinct inhibitory circuitries in field CA1 of the hippocampus. Brain Res. 1995;704:298–306. doi: 10.1016/0006-8993(95)01137-4. [DOI] [PubMed] [Google Scholar]
- Bi R, Broutman G, Foy MR, Thompson RF, Baudry M. The tyrosine kinase and mitogen-activated protein kinase pathways mediate multiple effects of estrogen in hippocampus. Proc Natl Acad Sci U S A. 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi G, Hahn K, Massimi S, Zhivotovskaya E. Prevalence of memory loss complaints and other symptoms associated with the menopause transition: a community survey. Gend Med. 2005;2:255–264. doi: 10.1016/s1550-8579(05)80055-5. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Fink G, Sumner BE, Rosie R, Grace O, Quinn JP. Estrogen control of central neurotransmission: effect on mood, mental state, and memory. Cell Mol Neurobiol. 1996;16:325–344. doi: 10.1007/BF02088099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- Foy MR, Baudry M, Foy JG, Thompson RF. 17beta-estradiol modifies stress-induced and age-related changes in hippocampal synaptic plasticity. Behav Neurosci. 2008;122:301–309. doi: 10.1037/0735-7044.122.2.301. [DOI] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Giretti MS, Fu XD, De Rosa G, Sarotto I, Baldacci C, Garibaldi S, Mannella P, Biglia N, Sismondi P, Genazzani AR, Simoncini T. Extra-nuclear signalling of estrogen receptor to breast cancer cytoskeletal remodelling, migration and invasion. PLoS ONE. 2008;3:e2238. doi: 10.1371/journal.pone.0002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gungabissoon RA, Bamburg JR. Regulation of growth cone actin dynamics by ADF/cofilin. J Histochem Cytochem. 2003;51:411–420. doi: 10.1177/002215540305100402. [DOI] [PubMed] [Google Scholar]
- Gureviciene I, Puoliväli J, Pussinen R, Wang J, Tanila H, Ylinen A. Estrogen treatment alleviates NMDA-antagonist induced hippocampal LTP blockade and cognitive deficits in ovariectomized mice. Neurobiol Learn Mem. 2003;79:72–80. doi: 10.1016/s1074-7427(02)00012-6. [DOI] [PubMed] [Google Scholar]
- Honkura N, Matsuzaki M, Noguchi J, Ellis-Davies GC, Kasai H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron. 2008;57:719–729. doi: 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24:5151–5161. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci U S A. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci U S A. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Wong D, Lynch G. Patterned stimulation at the theta frequency is optimal for the induction of hippocampal long-term potentiation. Brain Res. 1986;368:347–350. doi: 10.1016/0006-8993(86)90579-2. [DOI] [PubMed] [Google Scholar]
- Lee AW, Pfaff DW. Hormone effects on specific and global brain functions. J Physiol Sci. 2008;58:213–220. doi: 10.2170/physiolsci.RV007008. [DOI] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Day M, Muñiz LC, Bitran D, Arias R, Revilla-Sanchez R, Grauer S, Zhang G, Kelley C, Pulito V, Sung A, Mervis RF, Navarra R, Hirst WD, Reinhart PH, Marquis KL, Moss SJ, Pangalos MN, Brandon NJ. Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nat Neurosci. 2008;11:334–343. doi: 10.1038/nn2057. [DOI] [PubMed] [Google Scholar]
- Luine VN, Jacome LF, Maclusky NJ. Rapid enhancement of visual and place memory by estrogens in rats. Endocrinology. 2003;144:2836–2844. doi: 10.1210/en.2003-0004. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM. LTP consolidation: substrates, explanatory power, and functional significance. Neuropharmacology. 2007;52:12–23. doi: 10.1016/j.neuropharm.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Markowska AL. Sex dimorphisms in the rate of age-related decline in spatial memory: relevance to alterations in the estrous cycle. J Neurosci. 1999;19:8122–8133. doi: 10.1523/JNEUROSCI.19-18-08122.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Kanhema T, Soulé J, Tiron A, Dagyte G, da Silva B, Bramham CR. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci. 2007;27:10445–10455. doi: 10.1523/JNEUROSCI.2883-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner TA, Ayoola K, Drake CT, Herrick SP, Tabori NE, McEwen BS, Warrier S, Alves SE. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J Comp Neurol. 2005;491:81–95. doi: 10.1002/cne.20724. [DOI] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD. Glutamate receptor dynamics in dendritic microdomains. Neuron. 2008;58:472–497. doi: 10.1016/j.neuron.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S, Mizuno K. Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem. 2000;275:3577–3582. doi: 10.1074/jbc.275.5.3577. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Packard MG, Teather LA. Posttraining estradiol injections enhance memory in ovariectomized rats: cholinergic blockade and synergism. Neurobiol Learn Mem. 1997;68:172–188. doi: 10.1006/nlme.1997.3785. [DOI] [PubMed] [Google Scholar]
- Pan Y, Anthony M, Clarkson TB. Effect of estradiol and soy phytoestrogens on choline acetyltransferase and nerve growth factor mRNAs in the frontal cortex and hippocampus of female rats. Proc Soc Exp Biol Med. 1999;221:118–125. doi: 10.1046/j.1525-1373.1999.d01-64.x. [DOI] [PubMed] [Google Scholar]
- Reid T, Furuyashiki T, Ishizaki T, Watanabe G, Watanabe N, Fujisawa K, Morii N, Madaule P, Narumiya S. Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J Biol Chem. 1996;271:13556–13560. doi: 10.1074/jbc.271.23.13556. [DOI] [PubMed] [Google Scholar]
- Rex CS, Kramár EA, Colgin LL, Lin B, Gall CM, Lynch G. Long-term potentiation is impaired in middle-aged rats: regional specificity and reversal by adenosine receptor antagonists. J Neurosci. 2005;25:5956–5966. doi: 10.1523/JNEUROSCI.0880-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lin CY, Kramár EA, Chen LY, Gall CM, Lynch G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–3029. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho-GTPase signaling pathways initiate sequential steps in LTP consolidation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes ME, Frye CA. ERbeta-selective SERMs produce mnemonic-enhancing effects in the inhibitory avoidance and water maze tasks. Neurobiol Learn Mem. 2006;85:183–191. doi: 10.1016/j.nlm.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Suzuki M, Hidaka H. The novel and specific Rho-kinase inhibitor (S)-(+)-2-methyl-1-[(4-methyl-5-isoquinoline)sulfonyl]-homopiperazine as a probing molecule for Rho-kinase-involved pathway. Pharmacol Ther. 2002;93:225–232. doi: 10.1016/s0163-7258(02)00191-2. [DOI] [PubMed] [Google Scholar]
- Sharrow KM, Kumar A, Foster TC. Calcineurin as a potential contributor in estradiol regulation of hippocampal synaptic function. Neuroscience. 2002;113:89–97. doi: 10.1016/s0306-4522(02)00151-3. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. Estrogen and/or androgen replacement therapy and cognitive functioning in surgically menopausal women. Psychoneuroendocrinology. 1988;13:345–357. doi: 10.1016/0306-4530(88)90060-1. [DOI] [PubMed] [Google Scholar]
- Singh M, Meyer EM, Millard WJ, Simpkins JW. Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female Sprague-Dawley rats. Brain Res. 1994;644:305–312. doi: 10.1016/0006-8993(94)91694-2. [DOI] [PubMed] [Google Scholar]
- Singh M, Meyer EM, Simpkins JW. The effect of ovariectomy and estradiol replacement on brain-derived neurotrophic factor messenger ribonucleic acid expression in cortical and hippocampal brain regions of female Sprague-Dawley rats. Endocrinology. 1995;136:2320–2324. doi: 10.1210/endo.136.5.7720680. [DOI] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estrogen-induced increase in the magnitude of long-term potentiation occurs only when the ratio of NMDA transmission to AMPA transmission is increased. J Neurosci. 2005;25:7780–7791. doi: 10.1523/JNEUROSCI.0762-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, McMahon LL. Estradiol-induced increase in the magnitude of long-term potentiation is prevented by blocking NR2B-containing receptors. J Neurosci. 2006;26:8517–8522. doi: 10.1523/JNEUROSCI.5279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrabji F, Miranda RC, Toran-Allerand CD. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:11110–11114. doi: 10.1073/pnas.92.24.11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer JL, Waters EM, Romeo RD, Wood GE, Milner TA, McEwen BS. Uncovering the mechanisms of estrogen effects on hippocampal function. Front Neuroendocrinol. 2008;29:219–237. doi: 10.1016/j.yfrne.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaxton C, Lopera J, Bott M, Baldwin ME, Kalidas P, Fernandez-Valle C. Phosphorylation of the NF2 tumor suppressor in Schwann cells is mediated by Cdc42-Pak and requires paxillin binding. Mol Cell Neurosci. 2007;34:231–242. doi: 10.1016/j.mcn.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Thirone AC, Speight P, Zulys M, Rotstein OD, Szászi K, Pedersen SF, Kapus A. Hyperosmotic stress induces Rho/Rho kinase/LIM kinase-mediated cofilin phosphorylation in tubular cells: key role in the osmotically triggered F-actin response. Am J Physiol Cell Physiol. 2009;296:C463–475. doi: 10.1152/ajpcell.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren SG, Humphreys AG, Juraska JM, Greenough WT. LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res. 1995;703:26–30. doi: 10.1016/0006-8993(95)01059-9. [DOI] [PubMed] [Google Scholar]
- Wegesin DJ, Stern Y. Effects of hormone replacement therapy and aging on cognition: evidence for executive dysfunction. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2007;14:301–328. doi: 10.1080/13825580600802893. [DOI] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Roles of estradiol and progesterone in regulation of hippocampal dendritic spine density during the estrous cycle in the rat. J Comp Neurol. 1993;336:293–306. doi: 10.1002/cne.903360210. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim M, Janssen WG, Tabori NE, Adams MM, Yuen GS, Akama KT, McEwen BS, Milner TA, Morrison JH. Estrogen and aging affect synaptic distribution of phosphorylated LIM kinase (pLIMK) in CA1 region of female rat hippocampus. Neuroscience. 2008;152:360–370. doi: 10.1016/j.neuroscience.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Q, Ge Q, Ohira T, Van Dyke T, Badwey JA. p21-activated kinase 2 in neutrophils can be regulated by phosphorylation at multiple sites and by a variety of protein phosphatases. J Immunol. 2003;171:3785–3793. doi: 10.4049/jimmunol.171.7.3785. [DOI] [PubMed] [Google Scholar]