

Abstract
The migration of vascular smooth muscle cells (SMCs) and fibroblasts into the intima after vascular injury is a central process in vascular lesion formation. The elevation of transmural interstitial flow is also observed after damage to the vascular endothelium. We have previously shown that interstitial flow upregulates matrix metalloproteinase-1 (MMP-1) expression, which in turn promotes SMC and fibroblast migration in collagen I gels. In this study, we investigated further the mechanism of flow-induced MMP-1 expression. An ERK1/2 inhibitor PD-98059 completely abolished interstitial flow-induced SMC migration and MMP-1 expression. Interstitial flow promoted ERK1/2 phosphorylation, whereas PD-98059 abolished flow-induced activation. Silencing ERK1/2 completely abolished MMP-1 expression and SMC migration. In addition, interstitial flow increased the expression of activator protein-1 transcription factors (c-Jun and c-Fos), whereas PD-98059 attenuated flow-induced expression. Knocking down c-jun completely abolished flow-induced MMP-1 expression, whereas silencing c-fos did not affect MMP-1 expression. Taken together, our data indicate that interstitial flow induces MMP-1 expression and SMC migration in collagen I gels via an ERK1/2-dependent and c-Jun-mediated mechanism and suggest that interstitial flow, ERK1/2 MAPK, c-Jun, and MMP-1 may play important roles in SMC migration and neointima formation after vascular injury.
Keywords: shear stress, matrix metalloproteinase, mitogen-activated protein kinase, activator protein-1, neointima formation, smooth muscle cell, extracellular signal-regulated kinase
vascular smooth muscle cell (SMC) and fibroblast migration and proliferation in the intima play a major role in neointima formation after vascular injury. It is well known that growth factors and inflammatory cytokines can induce SMC migration through three-dimensional (3-D) extracellular matrix (ECM) by stimulating the secretion of matrix metalloproteinases (MMPs), which are capable of digesting ECM (15, 25, 48). For example, the upregulated expression of MMP-1 in the early stages of atherosclerosis is associated with SMC migration (2, 3). Because the major collagen components in the vascular wall are collagen type I and type III and because MMP-1 is responsible for the initial cleavage of collagen I and III, MMP-1 is believed to play an essential role in vascular SMC migration (15).
Transmural interstitial flow driven by the transmural pressure differential is a physiological fluid movement through the vascular vessel interstitium that imposes fluid shear stress on parenchymal cells (41, 44). The biological role of this small flow (shear stress) has not been well recognized (39, 40, 45). However, during the early stages of vascular injury, the interstitial flow is elevated because of a loss of endothelial hydraulic resistance, and we hypothesized that this elevated flow could participate in vascular SMC and fibroblast migration and neointima formation (39). Furthermore, in hypertension, the transmural interstitial flow is also increased because of the elevated transmural differential pressure in the large arteries, which also can lead to neointima formation (20, 26). We have previously shown that interstitial flow can stimulate SMCs and fibroblasts to express MMP-1, which can in turn facilitate cell migration in collagen I gels (39). However, one of the remaining challenges is to determine the exact biomolecular signaling pathway (mechanism) of flow-induced MMP-1 expression.
Therefore, in this study, we investigated the underlying mechanism of interstitial flow-induced MMP-1 expression. The activation of mitogen-activated protein kinases (MAPKs) such as extracellular signal-related kinase-1 and -2 (ERK1/2), the expression of activator protein-1 (AP-1) transcription factors such as c-Jun and c-Fos, and the AP-1 DNA binding activity were examined. Gene silencing was also conducted to confirm the roles of ERK1/2 and AP-1 in the regulation of MMP-1 expression. The results suggest that interstitial flow induces MMP-1 expression and SMC migration via an ERK1/2-dependent AP-1 (c-Jun) activation mechanism.
MATERIALS AND METHODS
Collagen gel preparation and flow experiments.
Rat aorta SMCs were isolated from male Sprague-Dawley rats weighing 150 g (16). The procedure was approved by the City College/City University of New York Medical School Animal Care and Use Committee. As previously described (39), rat aortic SMCs (passages 3–5) were suspended in rat-tail collagen I (BD Science) gels (cell density, 2.5 × 105 cells/ml; and final gel concentration, 4 mg/ml), and pH was adjusted to 7.0 by mixing the appropriate amount of NaOH. For cell migration experiments, 200 μl of gel were loaded into each 12-well cell culture insert with 8-μm pores (BD Science). For RNA extraction and protein extraction experiments, 6-well cell culture inserts with 8-μm pores (BD Science) were used. To keep the same level of shear stress, the same gel thickness was maintained in both the 6- and 12-well experiments, thus 1 ml of gel was used for a 6-well insert. The gels were incubated for 24 h to allow cell spreading. Gels were then subjected to interstitial flow driven by a 1-cmH2O pressure drop (shear stress was ∼0.05 dyn/cm2) for various time periods according to the specific experimental design. This shear stress level elicited the maximum enhancement of migration in our previous study (39). For MAPK inhibition experiments, after 24 h of cell spreading in the collagen gel, the cells were treated with p38 MAPK inhibitor InSolution SB-203580 (Calbiochem) or ERK1/2 inhibitor InSolution PD-98059 (Calbiochem) for 1 h followed by various time periods of interstitial flow with or without inhibitors in the flow media.
Western blot analysis.
Collagen gels were washed once with ice-cold PBS, and 2× lysis buffer was then added immediately to the gels followed by a sonication for 30 s on ice. The 2× lysis buffer was composed of 2× radioimmunoprecipitation assay buffer, containing 300 mM NaCl, 2% Nonidet P-40, 100 mM Tris, 0.2% Brij 35, and 2 mM EDTA (pH 7.5), with a supplement of 2× protease inhibitor cocktail (Roche Diagnostics), 2× phosphatase inhibitor cocktail (Roche Diagnostics), 2 mM activated Na3VO4, and 2 mM PMSF. Lysates were centrifuged in a microfuge (14,000 rpm for 1 h at 4°C), and the supernatants were then collected and the remaining gel pellets were discarded as previously described (16). The supernatants were concentrated using a YM-10 Microcon centrifugal filter device (Millipore). Protein concentrations in supernatants were evaluated using a total protein assay (Bio-Rad). The protein samples were then boiled for 5 min after mixing with 4× sample buffer, containing 400 mM Tris·HCl, 8% SDS, 40% glycerol, 0.04% bromphenol blue, and 20% β-mercaptoethanol (pH 6.8), and stored at −80°C. Equal amounts of total protein per well were loaded onto 10% SDS-polyacrylamide gels. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad) and blocked at room temperature with 2% enhanced chemiluminescence (ECL) advance blocking agent (Amersham, GE Healthcare) in TBS plus Tween-20 (16). The membranes were incubated overnight with a 1:1,000 dilution of a specific rabbit primary antibody (monoclonal antibodies: ERK1/2, phospho-ERK1/2, phospho-p38 MAPK, c-Fos, and c-Jun; and polyclonal antibodies: p38 MAPK, α-tubulin, and β-actin; all from Cell Signaling), followed by a 1.5-h room temperature incubation with an ECL horseradish peroxidase-linked anti-rabbit IgG antibody (Amersham, GE Healthcare). The proteins on polyvinylidene difluoride membranes were then detected using an ECL advanced Western blotting detection kit (Amersham, GE Healthcare) and the ChemiDoc XRS system with the Quantity One software (Bio-Rad). Some membranes were stripped using Restore Plus Western blot stripping buffer (Thermo Scientific Pierce) for a subsequent detection.
Nuclear protein extraction and AP-1 DNA binding activity assay.
Cells were released from collagen gels by 0.2% of collagenase I in growth medium and washed with ice-cold PBS as previously described (39). To extract nuclear protein, the cells were treated with NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific Pierce) by following the manufacturer's procedure. Because of the presence of EDTA, the extracts were mixed with CaCl2 to a final concentration of 5 mM to inactivate EDTA. Three micrograms of nuclear extract per well were used for an AP-1 DNA binding activity assay using the AP-1 transcription factor microplate assay (Marligen), following the manufacturer's procedure.
RNA extraction and gene expression analysis.
Total RNA was isolated from cells in collagen matrixes using TRIzol LS Reagent (Invitrogen) with the following modification. During RNA precipitation, high-salt (1.2 M Na-citrate/0.8 M NaCl) was added together with isopropanol. Under these conditions, RNA can be effectively precipitated while contaminating polysaccharides and proteoglycans remain in the soluble form (6). RNA samples were then converted to cDNA by reverse transcription (RT). For analyzing gene expression, the polymerase chain reaction (PCR) was performed using the following protocol: predenaturation at 95 °C for 5 min and then either 40 cycles (for MMP-1) or 30 cycles (for c-jun, c-fos, and GAPDH) of denaturation at 94 °C for 35 s, annealing at 52 °C for 35 s, and extension at 72 °C for 35 s, followed by a final extension at 72 °C for 10 min. The amplified products were separated by electrophoresis in 2.5% agarose gels and photographed under ultraviolet (UV) light in the presence of ethidium bromide. Quantitative real-time PCR (RT-qPCR) was also performed for c-fos, c-jun, and MMP-1 expression on the ABI PRISM 7000 sequence detection system (Applied Biosystems). GAPDH served as an internal control. The reactions were performed in 30-μl reaction mixture volumes containing SYBR Green PCR Master Mix (Applied Biosystems), cDNA, and specific primer pairs. RT-qPCR programs were set to 2 min at 50°C and 10 min at 95°C, followed by 45 cycles of 35 s at 95°C, 35 s at 52°C, and 40 s at 72°C. Following each PCR, a dissociation curve analysis was used to assess the specificity of product amplification. Primer sequences are listed in Table 1.
Table 1.
Primer sequences and product sizes for rat genes
| Target Gene | Primer Sequence | Genbank Locus | Size |
|---|---|---|---|
| GAPDH | NM_017008 | 232 | |
| Forward (372–390) | 5′-TCTTCACCACCATGGAGAA-3′ | ||
| Reverse (603–585) | 5′-ACTGTGGTCATGAGCCCTT-3′ | ||
| MMP-1 | M60616 | 383 | |
| Forward (1016–1036) | 5′-GACCTCATGTTCATCTTTAGA-3′ | ||
| Reverse (1398–1378) | 5′-CACCACAATAAGGAATTCGTT-3′ | ||
| c-fos | X06769 | 239 | |
| Forward (1665–1684) | 5′-GGAATTAACCTGGTGCTGGA-3′ | ||
| Reverse (1903–1884) | 5′-TCAGACCACCTCAACAATGC-3′ | ||
| c-jun | X17163 | 291 | |
| Forward (2592–2611) | 5′-GAAGTAGCCCCCAACCTCTC-3′ | ||
| Reverse (2882–2863) | 5′-ATGGCTCTCAACTCAAGCGT-3′ |
MMP-1, matrix metalloproteinase-1.
RNA interference.
To silence ERK1/2, two ERK1 short hairpin (sh)RNAs and two ERK2 shRNAs, which were subcloned into pSUPER vector (kindly donated by Dr. Michal Hetman), were cotransfected into SMCs. The ERK1/2 shRNA target sequences were as follows: shERK1-1, GACCGGATGTTAACCTTTA; shERK1-2, ATGTCATAGGCATCCGAGA; shERK2-1, GTACAGAGCTCCAGAAATT; and shERK2–2, AGTTCGAGTTGCTATCAAG (21). To silence c-jun and c-fos, two c-jun shRNAs and c-fos shRNAs, which were subcloned into BS/U6 vector (kindly donated by Dr. Mingtao Li), were cotransfected into cells. The c-jun shRNA target sequences were as follows: shc-jun-a, ACAGGTGGCACAGCTTAAA; and shc-jun-b, AGTCATGAACCACGTTAAC. The c-fos shRNA target sequences were as follows: shc-fos-a, GGAGACAGATCAACTTGAA; and shc-fos-b, GCTGAAGGCTGAACCCTTT (27, 47). The transfections were conducted using Lipofectamine LTX and PLUS reagents (Invitrogen).
Data analysis.
Results are presented as means ± SE. Data sets were analyzed for statistical significance using a Student's t-test with a two-tailed distribution, and P < 0.05 was considered statistically significant. For comparison of more than two groups, we used one-way ANOVA followed by the t-test with Bonferroni correction, and P < 0.05/N (N stands for number of comparisons) was considered statistically significant.
RESULTS
Interstitial flow induces SMC migration and MMP-1 expression, whereas PD-98059 abolishes the flow effects.
MAPKs are important mediators that regulate a variety of cellular processes, including gene expression, proliferation, survival, apoptosis, migration, and differentiation (36). Therefore, the roles of ERK1/2 and p38 MAPK were examined in this study. As shown in Fig. 1, interstitial flow significantly induced vascular SMC migration by twofold and ERK1/2 inhibitor (PD-98059) completely abolished flow-induced migration. Surprisingly, p38 MAPK inhibitor SB-203580 markedly enhanced flow-induced migration. In no-flow control cases, PD-98059 did not affect SMC migration, whereas SB-203580 promoted SMC migration.
Fig. 1.
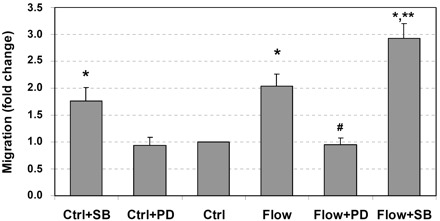
Interstitial flow induces vascular smooth muscle cell (SMC) migration, ERK1/2 inhibitor PD-98059 (PD) suppresses flow-induced SMC migration, and p38 MAPK inhibitor SB-203580 (SB) promotes SMC migration. After 24 h of cell spreading in collagen I gels, media was replaced with either fresh media or 5 μM SB-203580 or 10 μM PD-98059 for 1 h, and some gels were then exposed to 1 cmH2O of interstitial flow (∼0.05 dyn/cm2) either with or without inhibitors for 6 h followed by 48 h migration to 20 ng/ml PDGF-BB. The numbers of migrated cells to the undersides of cell culture inserts were counted. Other gels that were not exposed to flow served as no-flow controls (Ctrl). Data are presented as means ± SE; n = 6–8. *P < 0.05 vs. Ctrl; **P < 0.02 vs. Ctrl + SB; #P < 0.005 vs. flow.
We have previously shown that interstitial flow-induced cell migration in collagen I gels is controlled by MMP-1 (39). Therefore, we examined the effects of interstitial flow and MAPK inhibitors on MMP-1 gene expression. As shown in Fig. 2, interstitial flow significantly induced MMP-1 gene expression in vascular SMCs, and PD-98059 completely abolished flow-induced MMP-1 expression, whereas SB-203580 markedly enhanced flow-induced MMP-1 expression. In no-flow control cases, PD-98059 inhibited MMP-1 expression and SB-203580 slightly promoted MMP-1 gene expression. The combination of SMC migration data and MMP-1 expression data suggests that interstitial flow-induced cell migration and MMP-1 expression are ERK1/2 dependent, whereas p38 MAPK appears to play a downregulatory role.
Fig. 2.
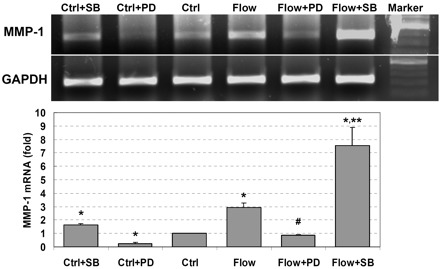
Interstitial flow induces matrix metalloproteinase-1 (MMP-1) mRNA expression, ERK1/2 inhibitor PD-98059 attenuates flow-induced MMP-1 expression, and p38 MAPK inhibitor SB-203580 promotes MMP-1 expression. After 24 h of cell spreading in collagen I gels, media was replaced with either fresh media or 5 μM SB-203580 or 10 μM PD-98059 for 1 h, and some gels were then exposed to 1 cmH2O of interstitial flow either with or without MAPK inhibitors for 6 h. Other gels that were not exposed to flow served as no-flow controls. MMP-1 gene expression was checked by both RT-PCR and RT-quantitative PCR (RT-qPCR), and GAPDH served as an internal control. RT-qPCR data (bottom) are presented as means ± SE; n = 4. *P < 0.05 vs. Ctrl; #P < 0.01 vs. flow; **P < 0.05 vs. flow.
Interstitial flow induces ERK1/2 and p38 MAPK activation.
To further investigate whether the interstitial flow and MAPK inhibitors really affect ERK1/2 activation, Western blot analysis was used to analyze the levels of both phosphorylated and total ERK1/2 (shown in Fig. 3A). During 60 min of exposure to interstitial flow, ERK1/2 was markedly activated as the phosphorylated ERK1/2 level increased greatly. PD-98059 dramatically inhibited ERK1/2 activation for no-flow (0 min) and flow (15, 30, and 60 min) cases compared with the cases without an addition of PD-98059. Interestingly, p38 MAPK inhibitor SB-203580 significantly enhanced ERK1/2 activation for no-flow (0 min) and flow (15, 30, and 60 min) cases. This unexpected stimulatory role of SB-203580 on ERK1/2 activation has been reported elsewhere (14, 24, 35). Sixty minutes of interstitial flow, with or without SB-203580 or PD-98059, had no effect on total ERK1/2.
Fig. 3.
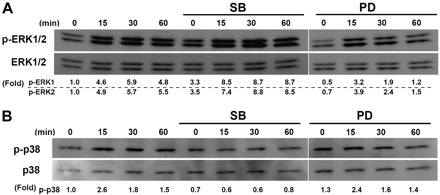
Interstitial flow induces ERK1/2 and p38 MAPK activation. A: PD-98059 markedly inhibited flow-induced ERK1/2 activation, and SB-203580 significantly activated ERK1/2. B: SB-203580 significantly inhibited flow-induced p38 MAPK activation, and PD-98059 did not significantly affect p38 MAPK activation. After 24 h of cell spreading in collagen I gels, media was replaced with either fresh media or 5 μM SB-203580 or 10 μM PD-98059 for 1 h, and the gels were then exposed to 1 cmH2O of interstitial flow either with or without MAPK inhibitors for 0, 15, 30, or 60 min. Total ERK1/2 and phosphorylated ERK1/2 (p-ERK1/2) levels were analyzed by Western blot analysis. Effects of PD-98059 on ERK1/2 and p38 phosphorylation were run in a different SDS-PAGE, and the gel panels were spliced together. The experiments were run three times, and similar results were observed. The gel panels are representative images. Fold change values are the ratios of p-ERK1 and ERK2 over the total ERK1 and ERK2 or p-p38 over p38 and then normalized to no-flow control cases without inhibitors, respectively.
To investigate whether interstitial flow and MAPK inhibitors affect p38 MAPK activation, both phosphorylated and total p38 MAPKs were detected by Western blot analysis. As shown in Fig. 3B, interstitial flow markedly activated p38 MAPK. SB-203580 clearly inhibited flow-induced p38 MAPK activation, whereas PD-98059 did not affect p38 MAPK activation. Sixty minutes of interstitial flow, with or without SB-203580 or PD-98059, had no effect on total p38 MAPK.
Interstitial flow induces SMC migration and MMP-1 expression via ERK1/2.
Since the pharmacological inhibitors may also influence other protein kinases (10), to further confirm the predominant regulatory role of ERK1/2 in MMP-1 expression and cell migration, ERK1/2 shRNAs were transfected into cells. After the knocking down of ERK1/2, both interstitial flow-induced MMP-1 expression (Fig. 4A) and cell migration (Fig. 4B) were completely abolished. In addition, the migration in no-flow control case (vector) was barely dependent on ERK1/2 (Figs. 1 and 4B), which is consistent with our previous observation that the migration of SMCs in the no-flow control case is independent of MMP-1 (39).
Fig. 4.
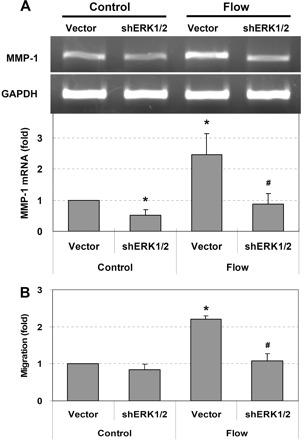
Interstitial flow induces MMP-1 expression and SMC migration via ERK1/2. Silencing ERK1/2 abolished flow-induced MMP-1 expression (A) and cell migration (B). RT-PCR (A, top) and RT-qPCR (A, bottom) were used to measure MMP-1 gene expression. To silence both ERK1 and ERK2 at the same time, the same amount of each ERK1 and ERK2 short hairpin (sh)RNAs was cotransfected into SMCs at 10 μg of total plasmid DNA/2.5×106 cells (i.e., 2.5 μg shERK1-1, 2.5 μg shERK1-2, 2.5 μg shERK2-1, and 2.5 μg shERK2-2 were mixed together). pSUPER vector (10 μg) was used as vector control. Transfection was conducted using Lipofectamine LTX and PLUS reagents (Invitrogen). After 24 h of transfection, cells were suspended into collagen gels and allowed to spread for 24 h before flow experiments. Gels were then exposed to 1 cmH2O of interstitial flow for 3 h, either followed by 48 h migration to 20 ng/ml PDGF-BB or directly subjected to gene analysis. Data are presented as means ± SE. A: *P < 0.05 vs. vector control; #P < 0.01 vs. vector flow; n = 3. B: *P < 0.01 vs. vector control; #P < 0.01 vs. vector flow; n = 4.
Taken together, these data (cell migration, MMP-1 expression, MAPK activation and inhibition, and ERK1/2 gene knockdown) suggest that interstitial flow-induced cell migration and MMP-1 expression proceed through an ERK1/2-dependent pathway. ERK1/2 activation plays a much more direct and critical role in regulating MMP-1 expression than p38 MAPK does, although the exact role of p38 MAPK is not clear.
Interstitial flow induces c-jun and c-fos expression through ERK1/2 activation.
Transcription factors c-jun and c-fos are members of the AP-1 family, and these immediate-early genes are targets of the ERK1/2 MAPK pathway (11). To investigate whether interstitial flow-induced ERK1/2 phosphorylation affects AP-1 transcription factor (c-Jun and c-Fos) expression, we determined c-jun and c-fos gene expression by RT-qPCR and c-Jun and c-Fos protein expression by Western blot analysis. Interstitial flow dramatically induced c-jun and c-fos transient gene expression (Fig. 5A) within 15 min, and the elevated expression then returned to the control level by 60 min. PD-98059 attenuated both c-jun and c-fos transient gene expression to their control levels. In the presence of SB-203580, interstitial flow significantly induced the c-jun and c-fos gene expression that remained at a relatively high level at 60 min although the peak level was not as high as the induction by interstitial flow alone (Fig. 5A). The change of c-fos expression was more pronounced than that of c-jun.
Fig. 5.
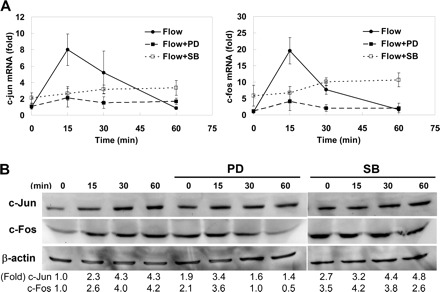
Interstitial flow induces activator protein-1 (AP-1) transcription factor gene (A) and protein (B) expression through ERK1/2 activation. PD-98059 markedly inhibited flow-induced c-Jun and c-Fos expression, and SB-203580 promoted c-Jun and c-Fos expression at longer times (60 min). After 24 h of cell spreading in collagen I gels, media was replaced with either fresh media or 5 μM SB-203580 or 10 μM PD-98059 for 1 h, and the gels were then exposed to 1 cmH2O of interstitial flow either with or without inhibitors for 0, 15, 30, or 60 min. β-Actin served as internal control. A: mRNA expression of c-fos and c-jun were detected by real-time PCR (means ± SE; n = 3–5). B: a representative Western blot result for AP-1 protein expression. Effects of PD-98059 on c-Jun and c-Fos expression were run in a different SDS-PAGE, and the gel panels were spliced together. The experiments were run 3 times, and similar results were observed. The quantifications for protein expression are shown as fold change.
Similar trends were observed for AP-1 protein expression (Fig. 5B). Interstitial flow markedly induced c-Jun and c-Fos protein expression within 60 min. PD-98059 significantly reduced flow-induced c-Jun and c-Fos expression. SB-203580 raised c-Jun and c-Fos expression for both no-flow (0 min) and flow cases, but there appeared to be little flow induction.
Interstitial flow induces AP-1 DNA binding activity depending on ERK1/2 activation.
To regulate target gene transcription, AP-1 transcription factors have to translocate from the cytoplasm into the nucleus and bind to specific sites on DNA (AP-1 site). As shown in Fig. 6, interstitial flow significantly increased AP-1 DNA binding activity by twofold, and PD-98059 completely abolished the flow-increased DNA binding activity, suggesting that the increased DNA binding activity is ERK1/2 dependent. After treatment with SB-203580, the flow-induced AP-1 DNA binding activity was only slightly, but not significantly, higher than the flow-only case (Fig. 6). This is not consistent with the fact that the MMP-1 gene expression was much higher in the case of flow with SB-203580 than in the flow-only case (Fig. 2). However, it is possible that the sustained expression of AP-1 transcription factor stimulated by SB-203580 (Fig. 5) provided a sustained high level of DNA binding activity which maintained the MMP-1 expression at a high level.
Fig. 6.
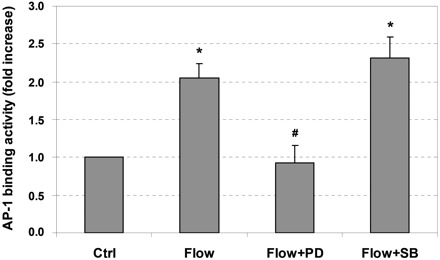
Interstitial flow induces AP-1 DNA binding activity via ERK1/2 activation. ERK1/2 inhibitor PD-98059 abolished flow-induced augmentation of AP-1 binding activity, whereas SB-203580 slightly enhanced flow-induced augmentation of AP-1 binding activity. After 24 h of cell spreading in collagen I gels, media was replaced with either fresh media or 5 μM SB-203580 or 10 μM PD-98059 for 1 h, and the gels were then exposed to 1 cmH2O of interstitial flow either with or without inhibitors for 3 h. After flow, cells were released from collagen gels by incubating with 0.2% collagenase I for 40 min. Nuclear proteins were then extracted followed by AP-1 DNA binding activity assay. Data are presented as means ± SE; n = 3. *P < 0.05 vs. Ctrl; #P < 0.02 vs. flow.
Interstitial flow-induced MMP-1 expression is mediated by c-jun, but not c-fos.
To further investigate whether ERK1/2 regulated MMP-1 expression directly through AP-1, c-jun and c-fos genes were silenced by their shRNAs. As shown in Fig. 7, after knocking down c-jun, the flow-induced MMP-1 expression was completely abolished. Although interstitial flow significantly induced c-fos expression (Fig. 5), surprisingly, the silencing of c-fos did not affect MMP-1 expression (Fig. 8). These data suggest that it was c-jun but not c-fos that mediated flow-induced MMP-1 expression through an ERK1/2-dependent pathway.
Fig. 7.
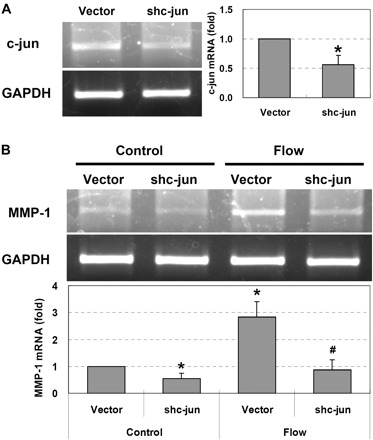
Interstitial flow induces MMP-1 expression depending on c-jun. A: cells that were transfected with c-jun shRNAs expressed less c-jun [RT-PCR (left), and RT-qPCR (right)]. B: silencing c-jun completely abolished flow-induced MMP-1 expression [RT-PCR (top), and RT-qPCR (bottom)]. To silence c-jun, the same amounts of shc-jun-a and shc-jun-b shRNAs were cotransfected into SMCs at 10 μg of total plasmid DNA/2.5×106 cells (i.e., 5 μg of shc-jun-a and 5 μg of shc-jun-b were mixed together). BS/U6 vector (10 μg) was used as vector control. Transfection was conducted using Lipofectamine LTX and PLUS reagents (Invitrogen). After 24 h of transfection, cells were suspended into collagen gels and allowed to spread for 24 h before flow experiments. Gels were then exposed to 1 cmH2O of interstitial flow for 3 h. Data are presented as means ± SE. A: *P < 0.005 vs. vector, n = 3. B: *P < 0.02 vs. vector control; #P < 0.01 vs. vector flow; n = 3.
Fig. 8.
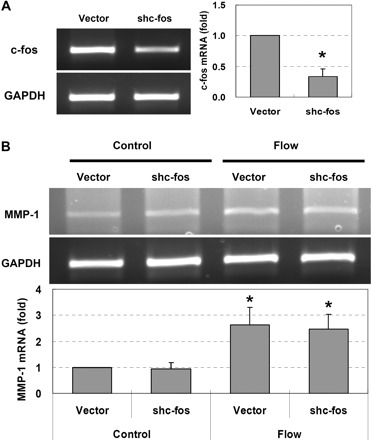
Interstitial flow-induced MMP-1 expression is independent of c-fos. A: cells that were transfected with c-fos shRNAs expressed much less c-fos [RT-PCR (left), and RT-qPCR (right)]. B: silencing c-fos barely affected flow-induced MMP-1 expression [RT-PCR (top), and RT-qPCR (bottom)]. To silence c-fos, the same amounts of shc-fos-a and shc-fos-b shRNAs were cotransfected into SMCs at 10 μg of total plasmid DNA/2.5×106 cells (i.e., 5 μg of shc-fos-a and 5 μg of shc-fos-b were mixed together). BS/U6 vector (10 μg) was used as vector control. Transfection was conducted using Lipofectamine LTX and PLUS reagents (Invitrogen). After 24 h of transfection, cells were suspended into collagen gels and allowed to spread for 24 h before flow experiments. Gels were then exposed to 1 cmH2O of interstitial flow for 3 h. Data are presented as means ± SE. A: *P < 0.001 vs. vector; n = 3. B: *P < 0.02 vs. vector control; n = 3.
DISCUSSION
In the present study, we observed that interstitial flow activated both ERK1/2 and p38 MAPK, PD-98059 abolished flow-induced ERK1/2 activation, and in contrast, SB-203580 enhanced flow-induced ERK1/2 activation and abolished flow-induced p38 MAPK activation (Fig. 3). Interstitial flow also induced MMP-1 expression and SMC migration, and PD-98059 inhibited flow-induced MMP-1 expression and cell migration, whereas SB-203580 enhanced MMP-1 expression and cell migration although the activation of p38 MAPK was inhibited (Figs. 1 and 2). Silencing ERK1/2 completely abolished flow-induced MMP-1 expression and cell migration (Fig. 4). These findings indicate that interstitial flow-induced MMP-1 expression and cell migration proceed through an ERK1/2-dependent pathway. ERK1/2 activation plays a much more critical role in regulating flow-induced MMP-1 expression than p38 MAPK although the exact role of p38 MAPK remains unclear.
There is abundant evidence in the literature that the induction of MMP-1 expression is related to the activation of MAPKs such as ERK1/2 and/or p38 MAPK (5, 28, 33). The activation of ERK1/2 by chemical and physical factors such as mitogenic growth factors, proinflammatory cytokines, heat shock, UV light, and other factors often induces MMP-1 expression in many cell types (5, 7, 12, 15, 28, 31). In contrast, the activation of p38 MAPK has opposing effects on MMP-1 gene expression depending on the mode of induction (13). For example, MMP-1 expression in human skin fibroblasts induced by tumor necrosis factor-α is through AP-1-dependent transcriptional activation via the ERK1/2 pathway and AP-1-independent enhancement via p38α MAPK by mRNA stabilization (33); lipopolysaccharide-induced monocyte MMP-1 expression is regulated by both ERK1/2 and p38 MAPK (23), whereas PDGF-BB and arsenite suppress MMP-1 expression via p38 MAPK activation (13, 46). One study has demonstrated that p38α, one isoform of p38 MAPK, inhibits the ERK1/2 pathway (46). Thus, in our study, the cross talk between ERK1/2 and p38 MAPK might play a role in interstitial flow-induced MMP-1 expression. However, although the role of p38 MAPK remains unclear, our data strongly suggest that ERK1/2 plays a predominant regulatory role in flow-induced MMP-1 expression.
In cardiovascular health and disease, it has been shown that the ERK1/2 MAPK plays an important role (30). Increased ERK1/2 activation contributed to augmented vascular SMC proliferation and neointima formation with aging in a rabbit study (17). The rapid activation of ERK1/2 after balloon injury of the rat carotid artery may be associated with vascular SMC migration and proliferation in vivo (19, 22). Completely blocking ERK1/2 activation in balloon-injured carotid arteries can inhibit carotid neointima formation by suppressing SMC migration and proliferation after 2 wk (18). The evidence from in vitro studies has also shown that ERK1/2 activation plays a crucial role in cardiovascular cell proliferation, cell migration, and MMP-1 expression (7–9, 15, 28, 29, 32).
The activation of ERK1/2 MAPK can induce the expression and activation of many transcription factors, such as AP-1. AP-1 transcription factors have a variety of physiological and pathophysiological consequences through their regulation of target gene expression. The AP-1 DNA binding element plays a pivotal role in the regulation of MMP expression since AP-1 sites are present throughout the MMP promoters (4, 43). Jun proteins can form stable dimers that bind AP-1 DNA recognition elements (5′-TGAG/CTCA-3′). Fos proteins can bind DNA by forming heterodimers with Jun proteins (Jun:Fos) that are more stable than Jun:Jun dimers (37). The ERK MAPK cascade is likely involved in the induction of the c-fos gene and in forming the Jun:Fos heterodimer (37). In the present study, we observed that interstitial flow significantly promoted AP-1 (c-Jun and c-Fos) expression (Fig. 5). The increased expression and activation of c-Jun and c-Fos led to an elevated AP-1 DNA binding activity (Fig. 6), which could promote MMP-1 expression. Using an ERK1/2 specific inhibitor PD-98059, we further demonstrated that interstitial flow-induced AP-1 DNA binding activity is ERK1/2 dependent (Fig. 6). However, interestingly, RNA interference results suggest that it was c-jun but not c-fos that regulated MMP-1 expression (Figs. 7 and 8), although c-fos was definitely induced by flow.
Taken together, we described for the first time a mechanism of interstitial flow-induced MMP-1 expression in vascular SMCs in 3-D collagen I gels (shown in Fig. 9). Interstitial flow, by an as-yet to-be-determined mechanotransduction mechanism, can induce ERK1/2 MAPK activation, which promotes AP-1 transcription factor expression and AP-1 DNA binding activity, resulting in an elevated MMP-1 expression. Proteolysis of collagen by upregulated MMP-1 then facilitates SMC migration through collagen I. However, the mechanism by which vascular cells “sense” interstitial flow and then trigger ERK1/2 signaling pathway requires further investigation. With the consideration that cells in 3-D exhibit matrix adhesions all over their surface, cell-matrix adhesion and tethering through integrin-rich focal adhesions would be a potential mechanosensor (38, 39). Another candidate would be the cell surface glycocalyx, which has been shown to mediate fluid shear stress-regulated vascular SMC contraction (1) and various endothelial cell functions (42).
Fig. 9.
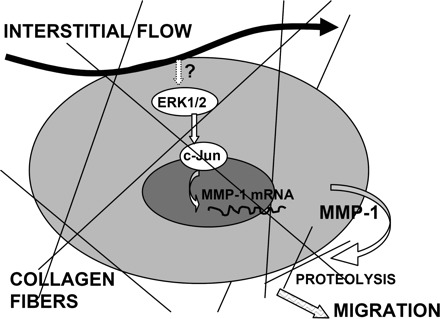
A schematic diagram of the proposed mechanism by which interstitial flow induces MMP-1 expression and SMC migration in collagen I gels via an ERK1/2-dependent and c-Jun-mediated mechanism. Interstitial flow stimulates the cell, inducing ERK1/2 phosphorylation (mechanotransduction), which enhances AP-1 expression and AP-1 DNA binding activity, leading to an increased expression of MMP-1. Upregulated MMP-1 digests collagen fibers and facilitates cell migration. One of the remaining questions is how cells “sense” interstitial flow and then activate the ERK1/2 pathway.
Transmural interstitial flow is elevated when the endothelium is injured because of chemical or mechanical stimuli or when the vascular hydraulic conductivity is enhanced by inflammation and hypertension. Thus elevated interstitial flow may be associated with vessel remodeling and neointima formation (34, 39). The present study reveals a mechanism whereby altered interstitial flow during the early stages of vascular injury stimulates MMP expression and vascular SMC and fibroblast migration, which can lead to neointima formation. These findings suggest that methods to control interstitial flow or directly inhibit MMP expression or suppress ERK1/2 MAPK activation may be able to limit neointima formation clinically after vascular injury.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants RO1-HL-35549 and RO1-HL-57093.
DISCLOSURES
No conflicts of interest are declared by the author(s).
ACKNOWLEDGMENTS
We thank Dr. Yongzhong Hou for valuable discussions and Hui Wang for cell migration assay.
REFERENCES
- 1.Ainslie KM, Garanich JS, Dull RO, Tarbell JM Vascular smooth muscle cell glycocalyx influences shear stress-mediated contractile response. J Appl Physiol 98: 242–249, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Aoyagi M, Yamamoto M, Azuma H, Nagashima G, Niimi Y, Tamaki M, Hirakawa K, Yamamoto K Immunolocalization of matrix metalloproteinases in rabbit carotid arteries after balloon denudation. Histochem Cell Biol 109: 97–102, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Bendeck MP, Irvin C, Reidy M, Smith L, Mulholland D, Horton M, Giachelli CM Smooth muscle cell matrix metalloproteinase production is stimulated via alpha(v)beta(3) integrin. Arterioscler Thromb Vasc Biol 20: 1467–1472, 2000 [DOI] [PubMed] [Google Scholar]
- 4.Borden P, Heller RA Transcriptional control of matrix metalloproteinases and the tissue inhibitors of matrix metalloproteinases. Crit Rev Eukaryot Gene Expr 7: 159–178, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Brauchle M, Gluck D, Di Padova F, Han J, Gram H Independent role of p38 and ERK1/2 mitogen-activated kinases in the upregulation of matrix metalloproteinase-1. Exp Cell Res 258: 135–144, 2000 [DOI] [PubMed] [Google Scholar]
- 6.Chomczynski P, Mackey K Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques 19: 942–945, 1995 [PubMed] [Google Scholar]
- 7.Cortez DM, Feldman MD, Mummidi S, Valente AJ, Steffensen B, Vincenti M, Barnes JL, Chandrasekar B IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol 293: H3356–H3365, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Cospedal R, Abedi H, Zachary I Platelet-derived growth factor-BB (PDGF-BB) regulation of migration and focal adhesion kinase phosphorylation in rabbit aortic vascular smooth muscle cells: roles of phosphatidylinositol 3-kinase and mitogen-activated protein kinases. Cardiovasc Res 41: 708–721, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Cospedal R, Lobo M, Zachary I Differential regulation of extracellular signal-regulated protein kinases (ERKs) 1 and 2 by cAMP and dissociation of ERK inhibition from anti-mitogenic effects in rabbit vascular smooth muscle cells. Biochem J 342: 407–414, 1999 [PMC free article] [PubMed] [Google Scholar]
- 10.Davies SP, Reddy H, Caivano M, Cohen P Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delaney J, Chiarello R, Villar D, Kandalam U, Castejon AM, Clark MA Regulation of c-fos, c-jun and c-myc gene expression by angiotensin II in primary cultured rat astrocytes: role of ERK1/2 MAP kinases. Neurochem Res 33: 545–550, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Di Girolamo N, Coroneo MT, Wakefield D UVB-elicited induction of MMP-1 expression in human ocular surface epithelial cells is mediated through the ERK1/2 MAPK-dependent pathway. Invest Ophthalmol Vis Sci 44: 4705–4714, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Endo H, Utani A, Shinkai H Activation of p38 MAPK suppresses matrix metalloproteinase-1 gene expression induced by platelet-derived growth factor. Arch Dermatol Res 294: 552–558, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Fatima S, Khandekar Z, Parmentier JH, Malik KU Cytosolic phospholipase A2 activation by the p38 kinase inhibitor SB203580 in rabbit aortic smooth muscle cells. J Pharmacol Exp Ther 298: 331–338, 2001 [PubMed] [Google Scholar]
- 15.Game BA, Maldonado A, He L, Huang Y Pioglitazone inhibits MMP-1 expression in vascular smooth muscle cells through a mitogen-activated protein kinase-independent mechanism. Atherosclerosis 178: 249–256, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Garanich JS, Mathura RA, Shi ZD, Tarbell JM Effects of fluid shear stress on adventitial fibroblast migration: implications for flow-mediated mechanisms of arterialization and intimal hyperplasia. Am J Physiol Heart Circ Physiol 292: H3128–H3135, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Gennaro G, Menard C, Giasson E, Michaud SE, Palasis M, Meloche S, Rivard A Role of p44/p42 MAP kinase in the age-dependent increase in vascular smooth muscle cell proliferation and neointimal formation. Arterioscler Thromb Vasc Biol 23: 204–210, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Gennaro G, Menard C, Michaud SE, Deblois D, Rivard A Inhibition of vascular smooth muscle cell proliferation and neointimal formation in injured arteries by a novel, oral mitogen-activated protein kinase/extracellular signal-regulated kinase inhibitor. Circulation 110: 3367–3371, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Cheng L, Hochleitner BW, Xu Q Activation of mitogen-activated protein kinases (ERK/JNK) and AP-1 transcription factor in rat carotid arteries after balloon injury. Arterioscler Thromb Vasc Biol 17: 2808–2816, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 43: 13S–24S, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Kharebava G, Makonchuk D, Kalita KB, Zheng JJ, Hetman M Requirement of 3-phosphoinositide-dependent protein kinase-1 for BDNF-mediated neuronal survival. J Neurosci 28: 11409–11420, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai K, Wang H, Lee WS, Jain MK, Lee ME, Haber E Mitogen-activated protein kinase phosphatase-1 in rat arterial smooth muscle cell proliferation. J Clin Invest 98: 1560–1567, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai WC, Zhou M, Shankavaram U, Peng G, Wahl LM Differential regulation of lipopolysaccharide-induced monocyte matrix metalloproteinase (MMP)-1 and MMP-9 by p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Immunol 170: 6244–6249, 2003 [DOI] [PubMed] [Google Scholar]
- 24.Li J, Gorospe M, Barnes J, Liu Y Tumor promoter arsenite stimulates histone H3 phosphoacetylation of proto-oncogenes c-fos and c-jun chromatin in human diploid fibroblasts. J Biol Chem 278: 13183–13191, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Lovdahl C, Thyberg J, Hultgardh-Nilsson A The synthetic metalloproteinase inhibitor batimastat suppresses injury-induced phosphorylation of MAP kinase ERK1/ERK2 and phenotypic modification of arterial smooth muscle cells in vitro. J Vasc Res 37: 345–354, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res 68: 75–103, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Mei Y, Yuan Z, Song B, Li D, Ma C, Hu C, Ching YP, Li M Activating transcription factor 3 up-regulated by c-Jun NH(2)-terminal kinase/c-Jun contributes to apoptosis induced by potassium deprivation in cerebellar granule neurons. Neuroscience 151: 771–779, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Mu H, Wang X, Lin PH, Yao Q, Chen C Chlorotyrosine promotes human aortic smooth muscle cell migration through increasing superoxide anion production and ERK1/2 activation. Atherosclerosis 201: 67–75, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mu H, Wang X, Wang H, Lin P, Yao Q, Chen C Lactosylceramide promotes cell migration and proliferation through activation of ERK1/2 in human aortic smooth muscle cells. Am J Physiol Heart Circ Physiol 297: H400–H408, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Muslin AJ MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci (Lond) 115: 203–218, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park CH, Lee MJ, Ahn J, Kim S, Kim HH, Kim KH, Eun HC, Chung JH Heat shock-induced matrix metalloproteinase (MMP)-1 and MMP-3 are mediated through ERK and JNK activation and via an autocrine interleukin-6 loop. J Invest Dermatol 123: 1012–1019, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Rao GN, Alexander RW, Runge MS Linoleic acid and its metabolites, hydroperoxyoctadecadienoic acids, stimulate c-Fos, c-Jun, and c-Myc mRNA expression, mitogen-activated protein kinase activation, and growth in rat aortic smooth muscle cells. J Clin Invest 96: 842–847, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reunanen N, Li SP, Ahonen M, Foschi M, Han J, Kahari VM Activation of p38 alpha MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J Biol Chem 277: 32360–32368, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Rizzo V Enhanced interstitial flow as a contributing factor in neointima formation: (shear) stressing vascular wall cell types other than the endothelium. Am J Physiol Heart Circ Physiol 297: H1196–H1197, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenberger SF, Gupta A, Bowden GT Inhibition of p38 MAP kinase increases okadaic acid mediated AP-1 expression and DNA binding but has no effect on TRE dependent transcription. Oncogene 18: 3626–3632, 1999 [DOI] [PubMed] [Google Scholar]
- 36.Roux PP, Blenis J ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320–344, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaulian E, Karin M AP-1 in cell proliferation and survival. Oncogene 20: 2390–2400, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Shemesh T, Geiger B, Bershadsky AD, Kozlov MM Focal adhesions as mechanosensors: a physical mechanism. Proc Natl Acad Sci USA 102: 12383–12388, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi ZD, Ji XY, Qazi H, Tarbell JM Interstitial flow promotes vascular fibroblast, myofibroblast, and smooth muscle cell motility in 3-D collagen I via upregulation of MMP-1. Am J Physiol Heart Circ Physiol 297: H1225–H1234, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swartz MA, Fleury ME Interstitial flow and its effects in soft tissues. Annu Rev Biomed Eng 9: 229–256, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Tada S, Tarbell JM Flow through internal elastic lamina affects shear stress on smooth muscle cells (3D simulations). Am J Physiol Heart Circ Physiol 282: H576–H584, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Tarbell JM, Pahakis MY Mechanotransduction and the glycocalyx. J Intern Med 259: 339–350, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Vincenti MP, Brinckerhoff CE Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res 4: 157–164, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang DM, Tarbell JM Modeling interstitial flow in an artery wall allows estimation of wall shear stress on smooth muscle cells. J Biomech Eng 117: 358–363, 1995 [DOI] [PubMed] [Google Scholar]
- 45.Wang S, Tarbell JM Effect of fluid flow on smooth muscle cells in a 3-dimensional collagen gel model. Arterioscler Thromb Vasc Biol 20: 2220–2225, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Westermarck J, Li SP, Kallunki T, Han J, Kahari VM p38 mitogen-activated protein kinase-dependent activation of protein phosphatases 1 and 2A inhibits MEK1 and MEK2 activity and collagenase 1 (MMP-1) gene expression. Mol Cell Biol 21: 2373–2383, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan Z, Gong S, Luo J, Zheng Z, Song B, Ma S, Guo J, Hu C, Thiel G, Vinson C, Hu CD, Wang Y, Li M Opposing roles for ATF2 and c-Fos in c-Jun-mediated neuronal apoptosis. Mol Cell Biol 29: 2431–2442, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zempo N, Koyama N, Kenagy RD, Lea HJ, Clowes AW Regulation of vascular smooth muscle cell migration and proliferation in vitro and in injured rat arteries by a synthetic matrix metalloproteinase inhibitor. Arterioscler Thromb Vasc Biol 16: 28–33, 1996 [DOI] [PubMed] [Google Scholar]