

Abstract
Background
During acute HIV infection, high viral loads and the induction of host immune responses typically coincide with the onset of clinical symptoms. However, clinically severe presentations during acute HIV-1 infection, including AIDS-defining symptoms, are unusual.
Methods
Virus isolates were tested for clade, drug susceptibility, coreceptor usage, and growth rate for two cases of clinically severe sexual transmission. HLA genotype was determined, and HIV-1-specific CTL responses to an overlapping peptide set spanning the entire HIV clade A and clade B proteome were assayed.
Results
The virus isolated from the two unrelated cases of severe primary HIV-1 infection showed R5/X4 dual/mixed tropism, belonged to clade B and CRF02-AG, and were highly replicative in peripheral blood mononuclear cell culture. Impaired humoral responses were paralleled by a profound absence of HIV-1-specific CTL responses to the entire viral proteome in the two study cases. One case for which the virus source was available, showed a remarkable HLA similarity between the transmission pair as all 4 HLA-A and -B alleles were HLA supertype-matched between the subjects involved in the transmission case.
Conclusions
The data suggest that concurrence of viral and host factors contribute to the clinical severity of primary HIV-1 infection and that subjects infected with highly replicative dual tropic viruses are more prone to develop AIDS-defining symptoms during acute infection if they are unable to mount humoral and cellular HIV-1-specific immune responses. Concordant HLA supertypes might facilitate the preferential transmission of HLA-adapted viral variants, further accelerating disease progression.
Keywords: primary HIV-1 infection, HLA supertypes, CTL responses, R5/X4 dual tropism, rapid disease progression
Introduction
Symptoms of acute retroviral syndrome typically coincide with high-level viremia and the induction of the host's initial adaptive immune response [1, 2]. However, clinically severe presentations during acute HIV-1 infection, including AIDS-defining symptoms, are considered to occur infrequently [3]. Furthermore, epidemiological studies have shown that, in the absence of treatment, less than 0.5% of HIV-1-infected individuals progresses to AIDS within a year after primary infection [4]. A complex interplay between multiple viral and host factors is most likely to be involved in accelerating disease progression. Among these myriad factors, CXCR4 tropism has been associated with higher viral virulence [5]. Moreover, HLA class I concordance between individuals and the inability to elicit specific CTL responses have been suggested to increase HIV-1 transmission and disease progression [6, 7].
Here, we investigate the immunological and virological factors that contribute to development of AIDS-defining pathogenesis in two independent cases of unusually severe acute sexually transmitted HIV infection. Their clinical and diagnostic outcome is described in Figure 1.
Figure 1.
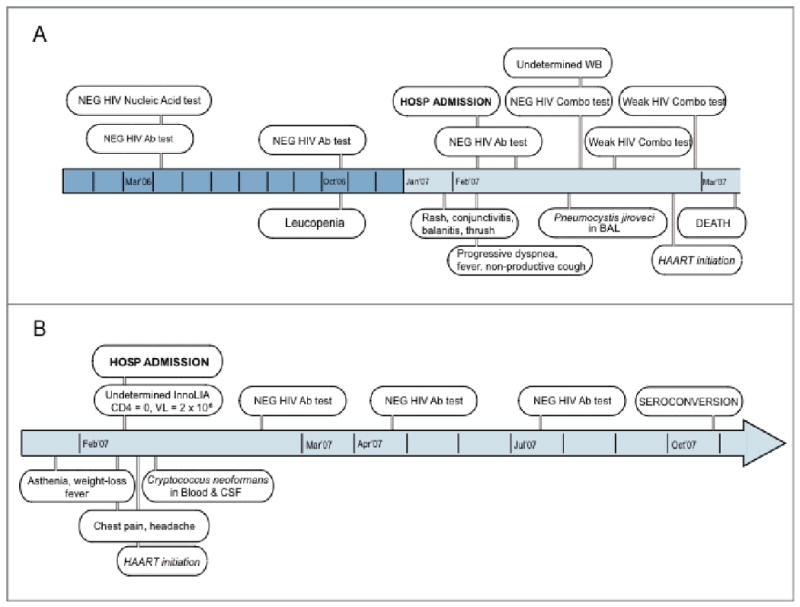
Description and outline of clinical and diagnostic outcome of the two severe primary HIV-1 infection cases in this study.
Material and methods
The study subjects gave written informed consent to participate in this study, which was approved by the institutional review boards of the hospitals where they received medical care.
HIV-1 was isolated from the patients' peripheral blood mononuclear cells (PBMC), and the viral stocks were titrated in TZM-bl cells [8]. Coreceptor usage of primary HIV-1 isolates was assessed by infection of U87.CD4 cells expressing either CCR5 or CXCR4 [9]. Syncytia induction was determined in vitro in MT-2 cells. CCR5 was genotyped in genomic DNA extracted from cryopreserved PBMCs to detect the Δ32 deletion. The growth rate of the viral isolates was determined by infecting phytohemagglutinin (PHA)-stimulated donor PBMCs [10]. To assess the presence of drug resistance-associated mutations, we sequenced the HIV protease region (codons 1 to 99) and reverse transcriptase (RT) region (codons 40 to 247) from a plasma sample drawn prior to initiation of antiretroviral therapy.
In order to determine whether case 2 harbored the same virus as the suspected source patient, viral RNA was extracted from plasma. The pol (protease and first 235 codons of the RT) and env (C2 to V5 regions) genes were sequenced [10, 11]. In addition, a total of 46 molecular clones encompassing the env gene were used to estimate diversity in the plasma viral RNA for the source and index patients [11]. Sequence alignments were obtained using Sequencher v4.6 (Gene Codes Corporation) and ClustalW, and manually edited in the regions of variable length. Genetic distances and evolutionary rates were computed using a Kimura 2-parameter model. Neighbour-joining phylogenetic trees of each subject's pol and env sequences were constructed using MEGA3. The reliability of phylogram clustering was assessed by bootstrapping analyses. Co-receptor usage was inferred from env clonal sequences using phenotype prediction tools (http://coreceptor.bioinf.mpi-inf.mpg.de/).
HLA class I and class II genotypes were identified by high resolution sequencing in an approved clinical laboratory. HLA class I supertype assignment was based on functional classification for the many different four-digit high-resolution HLA alleles that overlap in their peptide-binding specificities [12].
Cellular immunity to HIV and EBV was assessed by IFNγ elispot assays. T cell responses were detected to an overlapping peptide (OLP) set spanning the entire HIV clade A and clade B protein sequence [13]. In addition, optimal epitopes known to be presented by the subjects HLA class I alleles were included in either their clade-specific consensus version or based on sequence variants identified in the index or source subject (Table I suppl.). To assess general immune reactivity, three peptide pools containing a previously described set of EBV-derived optimal CTL epitopes were also tested [14]. Specific cut-offs for positive responses were used as previously defined [15].
Results
Case 1
Laboratory assessment of case 1 indicated a change in his HIV-1 antibody reactivity around the time of presentation. Three previous determinations—HIV-1 antibody-, nucleic acid-, and antigen-based assays within nine months before presentation—were all negative. Antibody/antigen and WB tests became partially reactive, and plasma HIV-1 RNA was positive at time of presentation, suggesting HIV-1 primary infection (Figure 1 and Table 1).
Table 1. Laboratory assessment of the patients involved in 2 case reports of sexual transmission of severe HIV-1 infection.
| Case report 2 | |||
|---|---|---|---|
| Case report 1 | |||
| Variable | Index patient | Source patient | |
| Plasma HIV-1 RNA level, copies/mL | 32 × 104 | 2 × 106 | 4 × 104 |
| CD4+ T cell count, cells/mm3 | 108 | 0 | 33 |
| CD4+ T cell percentage | 27 | 0 | 4 |
| CD8+ T cell count, cells/mm3 | Not determined | 203 | 481 |
| CD8+ T cell percentage | Not determined | 59 | 59 |
| Result of standard HIV Ab test | Negative | Weaka | Positive |
| HIV Western blot findings | Undetermined | Undetermined | Positive |
| Result of nucleic acid and/or viral load test | Positive | Positive | Positive |
| Plasma p24 level, pg/mL | 6.3 | 72.6 | 12.5 |
| Viral subtype | B | AG | AG |
| Drug resistance genotype | |||
| Protease | K20M and M36I | L10V, I13V, G16E, M36I, H69K and L89I | L10V, I13V, G16E, K20I, M36I, H69K and L89I |
| Reverse transcriptase | None | None | None |
| Coreceptor useb | R5/X4 | R5/X4 | R5/X4 |
| CCR5 Δ32 genotype | WT/WT | WT/WT | WT/WT |
| HLA alleles | |||
| Class Ic | A*0201 (A2), A*1101 (A3) | A*0201 (A2), A*0301 (A3) | A*6802 (A2), A*6801 (A3) |
| B*3503 (B7), B*4001 (B44) | B*0702 (B7), B*1801 (B44) | B*5101 (B7), B*5301 (B7) | |
| Cw*0304, Cw*0401 | Cw*0702, Cw*1203 | Cw*0401, Cw*1502 | |
| Class II | DRB1*0701, DRB1*1302 | DRB1*0405, DRB1*1201 | DRB1*0101, DRB1*0701 |
| DQB1*0202, DQB1*0604 | DQB1*0301, DQB1*0302 | DQB1*0202, DQB1*0501 | |
NOTE. WT, wild type.
Different HIV antibody tests provided nonreactive or weakly reactive results.
Viruses from all of the patients were syncytia-inducing viruses. Supertypes are shown in parenthesis.
Supertypes are shown in parenthesis. A*68, B*53 (increased susceptibility), B*51 (increased protection).
The replication-competent virus isolated from PBMCs was able to infect and replicate in both CCR5 and CXCR4-U87.CD4 cells as concluded from the p24 antigen production and the formation of syncytia in the cell cultures (Figures 2A and 2B). The subject did not have a Δ32 genotype in the CCR5 chemokine receptor gene that might have explained an early selection of CXCR4-tropic viruses [16]. The production of p24 antigen in growth kinetics cultures of donor PBMCs was similar to the laboratory-adapted viral strain HIV-1NL4-3 (Figure 2C). Phylogenetic analyses with bootscanning methods for the genetic subtyping of pol indicated the presence of a subtype B virus. The HIV-1 genotype showed no drug resistance-associated mutations. The results of HLA-typing are shown in Table 1.
Figure 2.
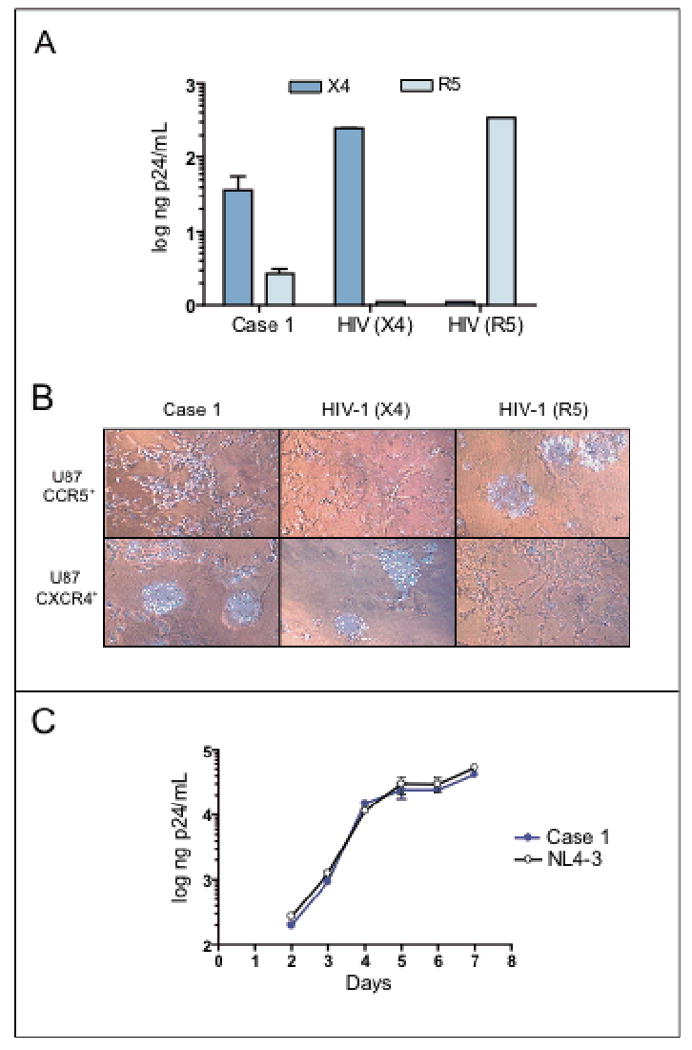
Case 1 virological data. Viral coreceptor usage based on p24 production (A) and syncytia formation (B) in U87.CD4 cells expressing either CXCR4 or CCR5. Control viral strains HIV-1NL4-3 (CXCR4-tropic, syncytia inducer) and HIV-1NFN-SX (CCR5-tropic, non-syncytia inducer) were included in both assays. Viral replication growth rates in PHA-stimulated primary donor PBMC infected with the patient's viral isolate (C). The laboratory-adapted HIV-1NL4-3 reference strain was grown in parallel. One representative experiment out of two with PBMCs from two different donors is shown.
Case 2
Clinical symptoms and analytical results in the index patient were consistent with a diagnosis of advanced HIV-1 infection and AIDS. However, the patient denied other HIV risks than sexual contact with her partner for the past two years. Her mother tested negative for HIV-1 infection, thus excluding a potential vertical transmission. Moreover, the viruses isolated from the source and index patients were similar both phenotypically and genotypically (Figure 3 and Table 1).
Figure 3.
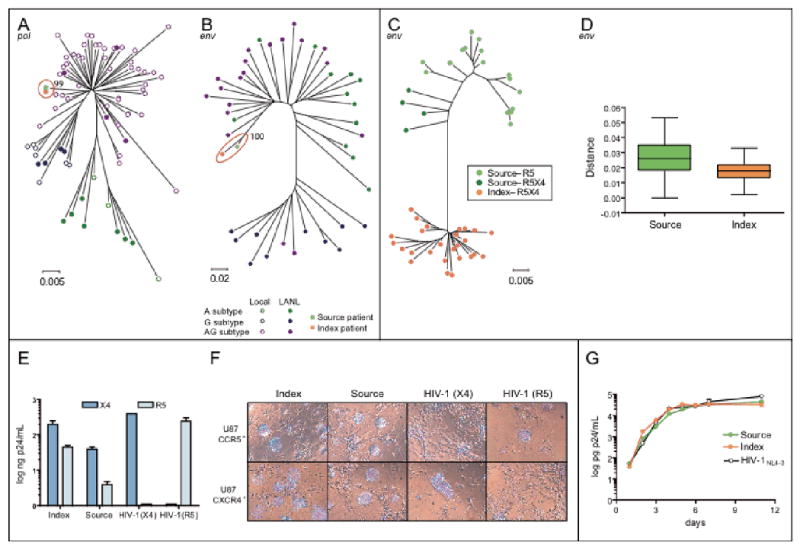
Case 2 virological data, including source patient and index patient samples. Neighbour-joining phylograms of pol (A) and env (B) sequences derived from viral RNA in plasma. Case sequences are represented by an orange square (index) or a green square (source). The scale for the genetic distance, based on the Kimura 2-parameter method, is indicated below each phylogram. Phylogenetic analysis of the env C2V3 clonal sequences derived from the index and source viral isolates with different colour patterns represent the virtual tropism of each clone (C). Intra-patient diversity is shown (D). The scale for the genetic distance, based on the Kimura 2-parameter method, is indicated below the phylogram. Viral coreceptor usage based on p24 production (E) and syncytia formation (F) in U87.CD4 cells expressing either CXCR4 or CCR5. Control viral strains HIV-1NL4-3 (CXCR4-tropic, syncytia inducer) and HIV-1NFN-SX (CCR5-tropic, non-syncytia inducer) were included in both assays. Viral replication growth rates in PHA-stimulated primary donor PBMCs infected with the patient's viral isolate (G). The laboratory-adapted HIV-1NL4-3 reference strain was grown in parallel. One representative experiment out of three with different PBMC donors is shown.
Laboratory assessment of the index patient's original sample provided clear reactivity data on the presence of HIV-1 antigens, but, despite high levels of immunoglobulins (Igs), antibody-based systems provided partial reactivity, indicating a lack of HIV-1-specific antibodies (Figure 1 and Table 1).
Bootstrap analysis of pol and env sequences from the index and the source patients revealed values of ≥99% in 1000 replicates (Figure 3A and 3B), indicating that sequence clustering was unlikely to have occurred by chance. The genetic distance of the pol sequences between the index patient and the source patient was <0.1%, whereas the mean genetic diversity between randomly selected sequences from local, epidemiologically unrelated HIV-infected individuals with the CRF02-AG subtype was 3.0%. The high degree of similarity between viral sequences indicates a likely viral transmission from one patient to the other. Clonal analysis of env sequences indicated that all sequences from the index patient were closely related, with a mean diversity of 1.8%, while the source patient's sequences had a mean diversity of 2.4% (Figure 3C and 3D).
After five days of culture, the viruses isolated from the source and the index patient were able to infect and replicate in both CCR5- and CXCR4-U87.CD4 cells as indicated by the p24 antigen production and the formation of syncytia in the cell cultures (Figure 3E and 3F). None of the individuals showed a Δ32 genotype in the CCR5 chemokine receptor gene. Phenotypic inference of the V3 amino acid sequence in multiple clones from each individual suggests that all clones from the index patient could use CXCR4 (R5X4 tropism) for viral entry, whereas the source patient contained clones which could only use CCR5 and clones that could use CXCR4 (R5X4 tropism) (Figure 3C). The production of p24 antigen in growth kinetics cultures in PBMCs was equal in the two viral isolates and comparable with the laboratory-adapted viral strain HIV-1NL4-3 (Figure 3G).
Genetic subtyping of the gag, pol, and env genes in the patients' virus indicated that both subjects contained the AG circulating recombinant form 02 (CRF02-AG). Drug-resistance genotyping showed no resistance-associated mutations in the RT. Several polymorphisms were detected in the protease gene, which might have been associated with possible tipranavir resistance in non-subtype B viruses (Table 1).
Cellular immune responses
Case 1 showed a single weak response against one OLP, which was not subsequently observed in the reconfirmation test, and only a borderline response to one EBV-peptide pool. This atypical lack of EBV-specific CTL responses suggests a widespread impairment of the ability to mount adequate CTL responses [15].
The index and source patient of case 2 expressed HLA class I alleles that were highly related. In fact, 3 out of 4 HLA-A and -B alleles were HLA supertype-matched between the two subjects (Table 1). As the transmission of escape mutants arising in the source may have prevented the induction of an effective T cell response in the index patient, the cellular immune response to the entire viral proteome was assessed in both subjects at 1 and 9 months after the transmission event. The analyses included two comprehensive sets of OLP spanning HIV clade A and clade B consensus sequences as well as autologous (index and source patient) peptide variants of optimally defined epitopes presented by the subjects' HLA types (Table I suppl.). The source subject showed weak responses to three different regions of the virus (Table 2), representing an overall weak response rate in comparison to more than 300 previously tested chronically HIV-infected subjects with a median of 17 responses [13, unpublished data]. The index case showed an even weaker HIV-specific T cell response to only a single peptide, which was detected before the subject was treated but was subsequently lost 9 months after infection. Importantly, the index subject was able to mount a T cell response to a peptide pool containing EBV-derived CTL epitopes, indicating that the absence of HIV-specific T cells was not due to poor cell viability or a general immune incompetence in this individual.
Table 2. Optimal epitopes showing sequence diversity between viruses from the source and index patients on case report 2.
| Epitope | Protein | HXB2 position | Reference | Sequence |
|---|---|---|---|---|
| A02 AL9 | Vpr | 59-67 | con B | AIIRILQQL |
| source/index | --------- | |||
| ConAG | --------- | |||
| A02 FK10 | Gag | 70-79 | con B | FLGKIWPSYK |
| source/index | -------- H- | |||
| ConAG | ---------- | |||
| A02 GT9 | Vpr | 41-49 | con B | GLGQHIYET |
| source | ------- N- | |||
| index | ------- D- | |||
| ConAG | --------- | |||
| A02 SL9 | Gag | 77-85 | con B | SLYNTVATL |
| source/index | -- F-- I--- | |||
| ConAG | -- F-- I--- | |||
| A02 RI9 | Vpr | 62-70 | con B | RILQQLLFI |
| source/index | -------- T | |||
| ConAG | -------- V | |||
| A02 RI10 | Env | 311-320 | con B | RGPGRAFVTI |
| source/index | I--- QT- Y-- | |||
| ConAG | I--- QT- YAT | |||
| A02 YV9 | Pol | 127-135 | con B | YTAFTIPSV |
| source/index | -------- L | |||
| ConAG | --------- | |||
| A03 AK9 | Pol | 158-166 | con B | AIFQSSMTK |
| source/index | ---- A---- | |||
| ConAG | ---- A---- | |||
| A03 RK9 | Gag | 20-28 | con B | RLRPGGKKK |
| source/index | -------- Q | |||
| ConAG | --------- | |||
| A03 RY10 | Gag | 20-29 | con B | RLRPGGKKKY |
| source/index | --------- Q- | |||
| ConAG | ---------- | |||
| A03 TN10 | Vpr | 19-28 | con B | TLELLEELKN |
| source/index | ---------H | |||
| ConAG | ---------H | |||
| A68 DL9 | Pol | 30-38 | con B | DTVLEEWNL |
| source/index | ------ I-- | |||
| ConAG | ----- I- L- | |||
| A68 IV9 | Pol | 3-11 | con B | ITLWQRPLV |
| source/index | -------- V- | |||
| ConAG | --------- | |||
| A*6801 DR11 | Vpr | 52-62 | con B | DTWAGVEAIIR |
| source | --- E-- M---- | |||
| index | --- E-- V---- | |||
| ConAG | --- E------- | |||
| A*6802 EV10 | Vpr | 48-57 | con B | ETYGDTWAGV |
| source | N------- E-- | |||
| Index | D------ E-- | |||
| ConAG | ------- E-- | |||
| B07 RI10 | Env | 298-307 | con B | RPNNNTRKSI |
| source | -- S----- N- | |||
| Index | --------- GV | |||
| ConAG | --------- V | |||
| B07 TL9 | Gag | 48-56 | con B | TPQDLNTML |
| source/index | ------ M-- | |||
| ConAG | --------- | |||
| B18 FK10 | Gag | 161-170 | con B | FRDYVDRFYK |
| source/index | -------- F- | |||
| ConAG | --------F- | |||
| B51 LI9 | Env | 416-424 | con B | LPCRIKQII |
| source/index | - Q------- | |||
| ConAG | --------- | |||
| B51 TI8 | Pol | 128-135 | con B | TAFTIPSI |
| source/index | ------- L | |||
| ConAG | -------- | |||
| B53 QW9 | Gag | 176-184 | con B | QASQEVKNW |
| source | -- T---- H- | |||
| index | -- T------ | |||
| ConAG | -- T------ | |||
| B53 TL9 | Gag | 48-56 | con B | TPYDINQML |
| source/index | -- Q- L- T-- | |||
| ConAG | --------- | |||
| X-GL12 | Vpr | 9-20 | con B | GPQREPHNEWTL |
| source/index | ----- F----- | |||
| ConAG | ----- F----- |
NOTE. Changes in the sequences are represented by boldface type.
Although some responses to autologous sequence variants that were not tested may exist, the data are in line with a remarkable absence of HIV-specific T cell immunity in both case 1 and the index patient in case 2, which may be related to the extraordinarily fast disease progression in these individuals.
Discussion
The interplay between the viral and host factors influencing accelerated disease progression is complex and poorly understood. The two temporarily coincident cases reported here suggest immediate progression to AIDS from primary HIV-1 infection after sexual transmission. In both cases, the diagnosis of primary HIV-1 infection was supported by nucleic acid- and antigen-based screening tests, with an evolving antibody pattern. Case 1 tested negative for HIV several times before presentation and subsequent HIV-1 WB was only partially reactive. The index patient in case 2 lacked previous negative test results, but presented with very high plasma HIV RNA level, which is consistent with acute HIV infection [17]. In addition, HIV-1-specific antibody tests were only partially reactive and did not become positive until nine months after antiretroviral treatment. Although detection of HIV-specific antibodies after the onset of primary infection symptoms can take between 5 and 15 days [18], complete seroconversion may occasionally be delayed until 12 months after identification of infection by antigen, once virological control has been achieved with effective antiretroviral therapy [18, 19]. The fact that serum levels of IgG, IgM, and IgA (in the index patient in case 2) were within the reference range or higher suggests polyclonal B cell activation [20]. Moreover, positive IgG responses to cytomegalovirus, Toxoplasma gondii, and hepatitis A indicate the ability of antibodies to maintain an appropriate response against microorganisms that cause persistent latent infection. Although plasma viremia had greatly decreased upon treatment, CD4+ T cell recovery increased slowly from total absence at presentation, which could have delayed HIV seroconversion.
The diversity of the viral population in HIV-1 env increases in parallel with divergence at a rate of 1% per year for a few years after seroconversion, before reaching a peak and then levelling off or decreasing [21]. Nevertheless, the rates of diversity are higher among patients with a sharp decline in the number of CD4 T cells [22]. In case 2, mean HIV-1 diversity was lower in the index patient than in the source patient, indicating that viral evolution was longer in the latter. Although viral diversity tends to decrease in the later stages of infection, most of the genetic distances would remain above 2% [21], thus supporting the direction of transmission in this pair and the theory of a very early presentation after viral transmission in the index patient.
The development of acute retroviral syndrome typically coincides with high-level viremia and the host's initial immune response. However, these two cases illustrate primary HIV-1 infection with unusually severe clinical symptoms. Other reports have described severe presentations during primary HIV-1 infection, including acute myopericarditis, renal failure, acute liver failure, and opportunistic infections [23-26], but viral and host factors have not been addressed in detail.
In both cases, the virus isolated from the patients' PBMCs was able to use CCR5 and/or CXCR4 as entry co-receptors and replicate very efficiently in PHA-stimulated donor PBMCs. These data indicate that both viral isolates are either dual-tropic viruses or a mixed population of CCR5-tropic and CXCR4-tropic viruses with high replication capacity. This observation would suggest that the transmitted virus had the ability to deplete CCR5+ as well as CXCR4+/CD4+ T lymphocytes, which may help to explain the total loss of the CD4+ T cell population and rapid clinical progression observed in the index patient upon transmission. Infection with dual-tropic HIV-1 variants in injecting drug users has been associated with immediate and rapid total T cell decline and progression to AIDS within four years of the estimated time of infection [27]. Furthermore, CCR5-Δ32/Δ32 seroconverters who showed the uncommon pattern of early syncytia-inducing virus and rapid CD4 decline had a uniformly high viral load and dual-tropic coreceptor usage [28]. A link between the detection of syncytia-inducing variants and a rapid CD4+ T cell decline in vivo has already been established [29].
Despite the fact that the characterized viral subtype CRF02-AG in case 2 is rather unusual in our area (1.1% of pol sequences tested for antiretroviral resistance between 1999 and 2007), it is still the second most common non-B subtype. CRF02-AG is the predominant and most rapidly spreading HIV strain in West and West Central Africa [30, 31], thus raising concerns about its superior replication fitness and/or transmission efficiency. In fact, primary HIV-1 CRF02-AG isolates from Cameroon exhibited higher ex vivo replicative fitness than subtype A and G viruses from the same geographic region [32, 33]. These observations are consistent with the high replication rate we observed in primary PHA-stimulated PBMCs, although we compared them to the laboratory-adapted B-subtype HIV-1NL4-3 strain.
In our cases, concurrent host factors may have also contributed to higher susceptibility to HIV-1 infection or disease progression. For example, specific HLA haplotypes have been proposed as an important risk factor in this context [34]. Among these, HLA-B*35, which is in high linkage disequilibrium with HLA-Cw*04, has been consistently associated with rapid progression to AIDS [35-37]. Specifically, the allele HLA*B3503, present in case 1, has been reported to increase the risk of progression to AIDS 2.7-fold (95% CI: 1.7–4.3, P<.001) [38]. In case 2, the source patient expressed the HLA A*68, B*53, and Cw04 alleles, which have been associated with rapid disease progression [34]. Although, none of the alleles in the index patient have been associated with accelerated disease progression, the donor and recipient expressed 3 out of 4 HLA-A and –B alleles that fell in the same HLA supertypes (i.e. clusters of functionally related four-digit high-resolution HLA class I alleles) [12]. This may have facilitated the transmission of viruses with cytotoxic T lymphocyte escape mutations, thus diminishing the number of epitopes recognized in the newly infected individual [6, 7, 39, 40]. Remarkably, optimal epitope variants representing autologous sequence diversity did not elicit a response, suggesting effective CTL escape (Table I suppl.). This hypothesis would fit with the fact that the index patient in case 2 showed almost complete absence of HIV-1-specific CTL responses, something rather unusual during primary HIV-1 infection. In a previous study, only 1 out of 5 patients presenting with primary HIV-1-infection, showed absence of precursor CTL specific for cells expressing viral proteins [1]. Another study in acute and early infected subjects reported a slightly higher breadth and magnitude of HIV-1-specific CTL responses [41]. However, this study used a less comprehensive pool of overlapping peptides and, contrarily to our case which showed a persistent absence of responses at month 9, CTL responses increased after 6 to 12 months of treatment. Although we could not identify the source patient for case 1, the lack of HIV-1-specific CTL responses also in this case might let us to speculate with a potential HLA class I concordance at transmission. In any case, the inability to elicit HIV-1-specific CTL responses at the time of primary infection was paralleled in these patients with AIDS-defining pathogenesis and severe clinical presentation. Although HIV-1-specific CTL responses have been considered a crucial factor in HIV disease progression, we had limited experimental and clinical evidences of the detrimental effect that the inability to elicit these responses might have in symptomatic primary HIV-1 infection [1, 42]. Moreover, the coincident inability in these 2 cases to mount an effective adaptive immune responses against HIV-1, albeit not to other pathogens, might be a consequence of a potential defect in the innate immunity. Clearly, further studies in these and other subjects with accelerated disease progression will be needed to address these factors.
In conclusion, we describe two cases of sexual transmission of a highly replicative dual-tropic HIV-1 of subtypes B and CRF02-AG that resulted in an aggressive clinical progression to severe symptomatic AIDS in young patients. Adaptive cellular and humoral immune responses in the host might have simultaneously failed to control the virus.
Table 3. Cellular immune responses in patients involved in 2 case reports of sexual transmission of severe primary HIV-1 infection.
| Patient, peptide | Protein | HXB2 position | Sequence | SFC per 1 × 106 PBMCs |
|---|---|---|---|---|
| Case report 1: OLP 83 con B | Nef | 118-135 | tQGYFPDWQNYTPGPGIRY | 47 |
| Case report 2 | ||||
| Source patient | ||||
| OLP 42 con A | Gag | 172-189 | LRAEQATQEVKGWMTETL | 41 |
| OLP 42 con B | LRAEQASQEVKNWMTETL | 72 | ||
| B53 QW9 con B | Gag | 176-184 | QASQEVKNW | 97 |
| B53 QW9 index/con A | QATQEVKNW | 97 | ||
| OLP 84 con A | Nef | 126-143 | NYTPGPGIRYPLCFGWCF | 10 |
| OLP 84 con B | NYTPGPGIRYPLTFGWCF | 103 | ||
| OLP 223 con A | Pol | 583-600 | QLEKDPIAGAETFYVDGA | 52 |
| OLP 223 con B | QLEKEPIVGAETFYVDGA | 21 | ||
| OLP 224 con A&B | GAETFYVDGAANRETKL | 72 | ||
| Index patient | ||||
| OLP 296 con A&B | Env | 52-69 | LFCASDAKAYDTEVHNVW | 18 |
| A24 LY10 con B&A/index | Env | 52-61 | LFCASDAKAY | 24 |
NOTE. Sequence changes between peptides are shown in italics, and optimal peptides are shown in boldface type. OLP, overlapping peptide; SFC, spot-forming cells; con, consensus.
Acknowledgments
We thank T. Puig, E. Grau and R. Ayen (irsiCaixa), J.R. Santos (Lluita), V. González (Microbiology Unit, HUGTiP), M. Juan and E. Palou (BST), and B. Ortiga (CESSCAT) for expert medical and technical assistance in this study, A. Gladden (Partners, MGH) for sequencing analysis, and R. Haubrich (UCSD) for helpful discussion and critical manuscript review.
All the authors declare that there are no conflicts of interest. Our work was supported by the Spanish Ministry of Education and Science through grants SAF2004-06991 and SAF2007-64696, the Spanish AIDS network “Red Temática Cooperativa de Investigación en SIDA” (RD06/0006), the “Ciber en Epidemiología y Salud Pública”, the HIVACAT program, and the Fundación para la Investigación y Prevención del Sida en España (FIPSE) through grants 36523/05, 36356/05, and 36621/06. N.I.-U. is supported by DURSI from the Generalitat de Catalunya.
U87.CD4 cells transfected with CCR5 or CXCR4 were obtained from H. Deng and D. Littman through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
References
- 1.Koup RA, Safrit JT, Cao Y, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–5. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schacker T, Collier AC, Hughes J, Shea T, Corey L. Clinical and epidemiologic features of primary HIV infection. Ann Intern Med. 1996;125:257–64. doi: 10.7326/0003-4819-125-4-199608150-00001. [DOI] [PubMed] [Google Scholar]
- 3.Kassutto S, Rosenberg ES. Primary HIV type 1 infection. Clin Infect Dis. 2004;38:1447–53. doi: 10.1086/420745. [DOI] [PubMed] [Google Scholar]
- 4.Munoz A, Wang MC, Bass S, et al. Acquired immunodeficiency syndrome (AIDS)-free time after human immunodeficiency virus type 1 (HIV-1) seroconversion in homosexual men. Multicenter AIDS Cohort Study Group Am J Epidemiol. 1989;130:530–9. doi: 10.1093/oxfordjournals.aje.a115367. [DOI] [PubMed] [Google Scholar]
- 5.Tersmette M, Lange JM, de Goede RE, et al. Association between biological properties of human immunodeficiency virus variants and risk for AIDS and AIDS mortality. Lancet. 1989;1:983–5. doi: 10.1016/s0140-6736(89)92628-7. [DOI] [PubMed] [Google Scholar]
- 6.Dorak MT, Tang J, Penman-Aguilar A, et al. Transmission of HIV-1 and HLA-B allele-sharing within serodiscordant heterosexual Zambian couples. Lancet. 2004;363:2137–9. doi: 10.1016/S0140-6736(04)16505-7. [DOI] [PubMed] [Google Scholar]
- 7.Goulder PJ, Brander C, Tang Y, et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature. 2001;412:334–8. doi: 10.1038/35085576. [DOI] [PubMed] [Google Scholar]
- 8.Wei X, Decker JM, Liu H, et al. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bjorndal A, Deng H, Jansson M, et al. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol. 1997;71:7478–87. doi: 10.1128/jvi.71.10.7478-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villena C, Prado JG, Puertas MC, et al. Relative fitness and replication capacity of a multinucleoside analogue-resistant clinical human immunodeficiency virus type 1 isolate with a deletion of codon 69 in the reverse transcriptase coding region. J Virol. 2007;81:4713–21. doi: 10.1128/JVI.02135-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Picado J, Frost SD, Izquierdo N, et al. Viral evolution during structured treatment interruptions in chronically human immunodeficiency virus-infected individuals. J Virol. 2002;76:12344–8. doi: 10.1128/JVI.76.23.12344-12348.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol. 2008;9:1. doi: 10.1186/1471-2172-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frahm N, Korber BT, Adams CM, et al. Consistent cytotoxic-T-lymphocyte targeting of immunodominant regions in human immunodeficiency virus across multiple ethnicities. J Virol. 2004;78:2187–200. doi: 10.1128/JVI.78.5.2187-2200.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woodberry T, Suscovich TJ, Henry LM, et al. Differential targeting and shifts in the immunodominance of Epstein-Barr virus--specific CD8 and CD4 T cell responses during acute and persistent infection. J Infect Dis. 2005;192:1513–24. doi: 10.1086/491741. [DOI] [PubMed] [Google Scholar]
- 15.Frahm N, Yusim K, Suscovich TJ, et al. Extensive HLA class I allele promiscuity among viral CTL epitopes. Eur J Immunol. 2007;37:2419–33. doi: 10.1002/eji.200737365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balotta C, Bagnarelli P, Violin M, et al. Homozygous delta 32 deletion of the CCR-5 chemokine receptor gene in an HIV-1-infected patient. AIDS. 1997;11:F67–71. doi: 10.1097/00002030-199710000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg ES, Altfeld M, Poon SH, et al. Immune control of HIV-1 after early treatment of acute infection. Nature. 2000;407:523–6. doi: 10.1038/35035103. [DOI] [PubMed] [Google Scholar]
- 18.Morpeth S, Thielman N, Giner J, et al. Time to HIV-1 seroconversion is similar among patients with acute HIV-1 infection, but there are exceptions (abstract 389). 13th Conference on Retroviruses and Opportunistic Infections; Denver, CO, USA. 2006. [Google Scholar]
- 19.Chin BS, Lee SH, Kim GJ, Kee MK, Suh SD, Kim SS. Early identification of seronegative human immunodeficiency virus type 1 infection with severe presentation. J Clin Microbiol. 2007;45:1659–62. doi: 10.1128/JCM.00166-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abelian A, Burling K, Easterbrook P, Winter G. Hyperimmunoglobulinemia and rate of HIV type 1 infection progression. AIDS Res Hum Retroviruses. 2004;20:127–8. doi: 10.1089/088922204322749576. [DOI] [PubMed] [Google Scholar]
- 21.Shankarappa R, Margolick JB, Gange SJ, et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999;73:10489–502. doi: 10.1128/jvi.73.12.10489-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Markham RB, Wang WC, Weisstein AE, et al. Patterns of HIV-1 evolution in individuals with differing rates of CD4 T cell decline. Proc Natl Acad Sci U S A. 1998;95:12568–73. doi: 10.1073/pnas.95.21.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demarest JF, Jack N, Cleghorn FR, et al. Immunologic and virologic analyses of an acutely HIV type 1-infected patient with extremely rapid disease progression. AIDS Res Hum Retroviruses. 2001;17:1333–44. doi: 10.1089/08892220152596597. [DOI] [PubMed] [Google Scholar]
- 24.Markowitz M, Mohri H, Mehandru S, et al. Infection with multidrug resistant, dual-tropic HIV-1 and rapid progression to AIDS: a case report. Lancet. 2005;365:1031–8. doi: 10.1016/S0140-6736(05)71139-9. [DOI] [PubMed] [Google Scholar]
- 25.Isaksson B, Albert J, Chiodi F, Furucrona A, Krook A, Putkonen P. AIDS two months after primary human immunodeficiency virus infection. J Infect Dis. 1988;158:866–8. doi: 10.1093/infdis/158.4.866. [DOI] [PubMed] [Google Scholar]
- 26.Michael NL, Brown AE, Voigt RF, et al. Rapid disease progression without seroconversion following primary human immunodeficiency virus type 1 infection--evidence for highly susceptible human hosts. J Infect Dis. 1997;175:1352–9. doi: 10.1086/516467. [DOI] [PubMed] [Google Scholar]
- 27.Yu XF, Wang Z, Vlahov D, Markham RB, Farzadegan H, Margolick JB. Infection with dual-tropic human immunodeficiency virus type 1 variants associated with rapid total T cell decline and disease progression in injection drug users. J Infect Dis. 1998;178:388–96. doi: 10.1086/515646. [DOI] [PubMed] [Google Scholar]
- 28.Sheppard HW, Celum C, Michael NL, et al. HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion. J Acquir Immune Defic Syndr. 2002;29:307–13. doi: 10.1097/00126334-200203010-00013. [DOI] [PubMed] [Google Scholar]
- 29.Fauci AS. Host factors and the pathogenesis of HIV-induced disease. Nature. 1996;384:529–34. doi: 10.1038/384529a0. [DOI] [PubMed] [Google Scholar]
- 30.Morison L, Buve A, Zekeng L, et al. HIV-1 subtypes and the HIV epidemics in four cities in sub-Saharan Africa. AIDS. 2001;15(Suppl 4):S109–16. doi: 10.1097/00002030-200108004-00012. [DOI] [PubMed] [Google Scholar]
- 31.Nyambi P, Heyndrickx L, Vereecken K, et al. Predominance of infection with HIV-1 circulating recombinant form CRF02_AG in major Cameroonian cities and towns. AIDS. 2002;16:295–6. doi: 10.1097/00002030-200201250-00022. [DOI] [PubMed] [Google Scholar]
- 32.Konings FA, Burda ST, Urbanski MM, Zhong P, Nadas A, Nyambi PN. Human immunodeficiency virus type 1 (HIV-1) circulating recombinant form 02_AG (CRF02_AG) has a higher in vitro replicative capacity than its parental subtypes A and G. J Med Virol. 2006;78:523–34. doi: 10.1002/jmv.20572. [DOI] [PubMed] [Google Scholar]
- 33.Njai HF, Gali Y, Vanham G, et al. The predominance of human immunodeficiency virus type 1 (HIV-1) circulating recombinant form 02 (CRF02_AG) in West Central Africa may be related to its replicative fitness. Retrovirology. 2006;3:40. doi: 10.1186/1742-4690-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Brien SJ, Gao X, Carrington M. HLA and AIDS: a cautionary tale. Trends Mol Med. 2001;7:379–81. doi: 10.1016/s1471-4914(01)02131-1. [DOI] [PubMed] [Google Scholar]
- 35.Carrington M, Nelson GW, Martin MP, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–52. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 36.Itescu S, Mathur-Wagh U, Skovron ML, et al. HLA-B35 is associated with accelerated progression to AIDS. J Acquir Immune Defic Syndr. 1992;5:37–45. [PubMed] [Google Scholar]
- 37.Sahmoud T, Laurian Y, Gazengel C, Sultan Y, Gautreau C, Costagliola D. Progression to AIDS in French haemophiliacs: association with HLA-B35. AIDS. 1993;7:497–500. doi: 10.1097/00002030-199304000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Gao X, Nelson GW, Karacki P, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med. 2001;344:1668–75. doi: 10.1056/NEJM200105313442203. [DOI] [PubMed] [Google Scholar]
- 39.Bienzle D, MacDonald KS, Smaill FM, et al. Factors contributing to the lack of human immunodeficiency virus type 1 (HIV-1) transmission in HIV-1-discordant partners. J Infect Dis. 2000;182:123–32. doi: 10.1086/315670. [DOI] [PubMed] [Google Scholar]
- 40.Trachtenberg E, Korber B, Sollars C, et al. Advantage of rare HLA supertype in HIV disease progression. Nat Med. 2003;9:928–35. doi: 10.1038/nm893. [DOI] [PubMed] [Google Scholar]
- 41.Altfeld M, Rosenberg ES, Shankarappa R, et al. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J Exp Med. 2001;193:169–80. doi: 10.1084/jem.193.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–10. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]