

Sorting of both soluble and integral membrane proteins is disrupted by loss of ADF/cofilin, suggesting that actin severing controls expansion of a sorting domain within the TGN.
Abstract
Knockdown of the actin-severing protein actin-depolymerizing factor (ADF)/cofilin inhibited export of an exogenously expressed soluble secretory protein from Golgi membranes in Drosophila melanogaster and mammalian tissue culture cells. A stable isotope labeling by amino acids in cell culture mass spectrometry–based protein profiling revealed that a large number of endogenous secretory proteins in mammalian cells were not secreted upon ADF/cofilin knockdown. Although many secretory proteins were retained, a Golgi-resident protein and a lysosomal hydrolase were aberrantly secreted upon ADF/cofilin knockdown. Overall, our findings indicate that inactivation of ADF/cofilin perturbed the sorting of a subset of both soluble and integral membrane proteins at the trans-Golgi network (TGN). We suggest that ADF/cofilin-dependent actin trimming generates a sorting domain at the TGN, which filters secretory cargo for export, and that uncontrolled growth of this domain causes missorting of proteins. This type of actin-dependent compartmentalization and filtering of secretory cargo at the TGN by ADF/cofilin could explain sorting of proteins that are destined to the cell surface.
Introduction
Newly synthesized signal sequence–containing proteins that enter the ER have basically two fates: (1) to stay in the ER or (2) to be exported by COPII-coated transport carriers. Cargo sorting, packing, and export from the ER require a large number of specific receptors, guides, and chaperones (Lee et al., 2004; Saito et al., 2009). The secretory proteins travel via the ER–Golgi intermediate compartment to the Golgi. The mechanism of cargo transport across the Golgi stack remains a controversial issue (Glick and Malhotra, 1998; Matsuura-Tokita et al., 2006; Patterson et al., 2008; Glick and Nakano, 2009). At the TGN, the secretory cargoes are sorted for distribution to their respective destinations (Mellman and Warren, 2000). Compared with export from the ER, sorting of proteins at the TGN is more complicated and less well understood. For a start, there are several exit routes from the TGN but none comparable with the stable exit sites of the ER. The sorting of lysosomal hydrolases is well understood, which is mediated binding to the mannose 6-phosphate receptor and subsequent export by clathrin-coated vesicles (Kornfeld and Mellman, 1989; Ghosh et al., 2003). Integral membrane proteins destined to the cell surface are known to contain export signals in their cytoplasmic tail, but no general rule has emerged thus far for their export from the TGN (Fölsch et al., 1999, 2009; Ang et al., 2003, 2004; Salvarezza et al., 2009). The mechanism by which soluble secretory cargo is sorted and packed for export at the TGN remains largely obscure. The yeast exomer is required for the export of a population of secretory cargo at the TGN (Wang et al., 2006). This complex of proteins does not have a homologue in other eukaryotes. By and large, coats, cargo receptors, and guides for the sorting and export of secretory cargo (other than those used for clathrin-mediated transport to the endosomes) at the TGN in the mammalian cells remain elusive.
We performed a genome-wide screen to identify new components of the secretory pathway in Drosophila melanogaster tissue culture (S2) cells (Bard et al., 2006). This procedure revealed several new components, including twinstar (the Drosophila homologue of cofilin), which regulates actin polymerization (Kueh et al., 2008; Chan et al., 2009; Kardos et al., 2009). Apart from the finding that twinstar knockdown inhibited secretion of the soluble secretory protein HRP, we could not deduce anything else about its role in protein secretion (Bard et al., 2006). Mammalian cells express three different isoforms of twinstar called cofilin1, cofilin2, and actin-depolymerizing factor (ADF; Bamburg, 1999). Phosphorylation of cofilin at serine3 (Ser3) by LIM kinase (LIMK) inactivates cofilin, whereas dephosphorylation reactivates it (Arber et al., 1998). Exogenously expressed LIMK1 in neurons localizes to the Golgi membranes, and cofilin is reported to play a role in the export of p75-GFP to the apical surface of the polarized MDCK cells (Rosso et al., 2004; Salvarezza et al., 2009). These findings prompted us to readdress the role of cofilin in the secretory pathway. Our results reveal a surprising function of cofilin in sorting of proteins at the TGN. The discussion of our findings follows.
Results
HRP secretion requires twinstar in Drosophila S2 cells
A genome-wide screen revealed that twinstar was required for the secretion of signal sequence HRP (ss-HRP) in Drosophila S2 cells (Bard et al., 2006). However, the specificity of twinstar in this process and the site of its action along the secretory pathway were not reported. We first reconfirmed the requirement of twinstar in protein secretion from S2 cells with a double-stranded RNA (dsRNA) different (dstsr-2) from that reported previously (dstsr-1). The efficient knockdown of twinstar by dstsr-1 and dstsr-2 in S2 cells was confirmed by RT-PCR (Fig. 1 A). Secretion of HRP from S2 cells was monitored as described previously (Bard et al., 2006). In the presence of dstsr-2, HRP secretion was inhibited by 75%, which is similar to that observed previously (Fig. 1 B). To examine the effect of twinstar knockdown on the organization of the actin cytoskeleton, Drosophila S2 cells treated with control dsRNA or dstsr-2 were stained with fluorescently labeled phalloidin and visualized by fluorescence microscopy. Inhibition of twinstar by dstsr-2 revealed accumulation of actin filaments, which were removed by treatment of the cells with the actin-depolymerizing agent latrunculin A (LatA; Fig. 1 C). Importantly, LatA treatment of twinstar knockdown cells restored secretion of HRP (Fig. 1 D).
Figure 1.

Twinstar is required for secretion of HRP in Drosophila S2 cells. (A) Drosophila S2 cells stably transfected with a plasmid containing ss-HRP were incubated with two different dsRNAs (ds-R1 and ds-R2) specific for twinstar (tsr). The knockdown efficiency was monitored by RT-PCR. (B) S2 cells stably expressing ss-HRP were incubated with dsRNA specific for twinstar and syntaxin 5 (Syx5; the Golgi-specific t-SNARE) and tested for secretion of HRP. (C) S2 cells, control, and twinstar knockdown were treated with DMSO or LatA stained with phalloidin to monitor actin organization. Bar, 5 µm. (D) S2 cells stably expressing ss-HRP, control, and twinstar knockdown were treated with DMSO or LatA, and HRP secretion was measured. (E) S2 cells stably expressing ss-HRP were incubated with two different dsRNAs specific for dLIMK (dsLIM-R1 and dsLIM-R2) and dsSSH (dsSSH-R1 and dsSSH-R2). The effects of dLIMK and dSSH knockdown on HRP secretion were monitored. Error bars indicate mean ± SD of HRP activity in the medium normalized by HRP activity in cell lysates of triplicate measurements from representative experiments. Compared datasets were termed as statistically significant when P < 0.01 (**).
Twinstar is phosphorylated by LIMK (Ohashi et al., 2000) and dephosphorylated by the protein phosphatase slingshot (SSH). The phosphorylated form of twinstar is inactive in actin severing (Niwa et al., 2002). To further ascertain the role of twinstar in ss-HRP secretion, S2 cells were incubated with specific dsRNAs to knockdown Drosophila LIMK (dLIMK) and Drosophila SSH (dSSH). We verified the efficacy of dLIMK and dSSH knockdown by RT-PCR (unpublished data). Although knockdown of dLIMK had no effect on HRP secretion, dSSH knockdown was very effective (Fig. 1 E). This fits well with the known inhibitory effect of LIMK-mediated phosphorylation and inactivation of cofilin's actin-severing activity. Collectively, these results confirm the scope of our genome-wide screen and strengthen our proposal that twinstar was required for secretion of HRP.
HRP export from the Golgi requires ADF and cofilin1 in mammalian cells
The mammalian homologue of twinstar called cofilin has three isoforms: cofilin1, cofilin2, and ADF (Bamburg, 1999). To determine which of the three actin-severing proteins are present in HeLa cells, equal amounts of lysates were analyzed by Western blotting with specific antibodies to cofilin1, cofilin2, and ADF. Both cofilin1 and ADF were detected in HeLa cells (Fig. 2 A).
Figure 2.

Expression of cofilin isoforms in HeLa cells and their knockdown by siRNA. (A) Equal amount of HeLa cell lysates were analyzed by Western blotting using antibodies recognizing cofilin1 (CFL-1), cofilin2 (CFL-2), and ADF. Positions of cofilin1, cofilin2, and ADF are indicated by arrowheads. (B) Lysates of HeLa cells transfected with control or ADF + cofilin1 siRNA were analyzed by Western blotting using anti-cofilin1 and anti-ADF antibodies, respectively. The lysates were also blotted with anti–β-actin antibody to monitor and normalize protein concentration of samples analyzed by Western blotting. (C) The effect of ADF + cofilin1 siRNA was quantified by densitometry using the ImageJ software and normalized to the expression of ADF and cofilin1, respectively, compared with cells transfected with control siRNA. Error bars represent the SD of triplicate experiments.
To knockdown ADF and cofilin1, HeLa cells were transfected with ADF- and cofilin1-specific siRNAs. After 48 h, cells lysates were probed with specific antibodies to quantify knockdown efficacy (Fig. 2 B). Compared with control siRNA, the level of ADF and cofilin1 was reduced by 75% with our procedure (Fig. 2 C).
To test the role of ADF and cofilin1 in protein secretion, HeLa cells were transfected with control, ADF, cofilin1, or ADF and cofilin1-specific siRNA. After 24 h, cells were transfected with ss-HRP plasmid for an additional 20 h. The culture medium from these cells was analyzed for HRP secretion as described previously (Bossard et al., 2007). Although knockdown of either ADF or cofilin1 had no significant effect on HRP secretion, simultaneous knockdown of both inhibited HRP secretion by 75% compared with control cells (Fig. 3 A). This is not surprising because ADF and cofilin1 have overlapping functions in actin severing in mammalian cells (Hotulainen et al., 2005). Therefore, all of the following experiments described were performed with HeLa cells in which both ADF and cofilin1 were knocked using this protocol.
Figure 3.

Depletion or inactivation of ADF and cofilin1 inhibits secretion of HRP in HeLa cells. (A) HeLa cells were transfected with control or ADF, cofilin1 (CFL-1), or ADF + cofilin1 siRNA. After 24 h, cells were transfected with ss-HRP–Flag. The medium from these cells was analyzed for HRP secretion. (B) HeLa cells were transfected with control or ADF and cofilin1 siRNA. In parallel, ADF and cofilin1 knockdown HeLa cells were transfected with a rat HA-tagged cofilin1 plasmid. The cells were transfected with ss-HRP–Flag. The medium from these cells was analyzed for HRP secretion. LatA and Jasp were added 4 h before collection of the medium. (A and B) Error bars indicate the mean SD of HRP activity in the medium normalized by HRP activity in cell lysates. (C) Control and ADF + cofilin1 knockdown cells were transfected with a control or HA rat cofilin1 plasmid, lysed, and subsequently analyzed by Western blotting using an anti-cofilin1 antibody. (D) HeLa cells were transfected with control (left) or ADF + cofilin1 siRNA (right) followed by transfection with ss-HRP–Flag. HRP was accumulated in the TGN at 20°C for 2 h in the presence of 100 µM CHX. The cells were shifted to 32°C for the indicated time, and the location of HRP was detected with an anti-HRP antibody (red). Cells were also stained with an anti-TGN46 (green) antibody to visualize the TGN. (E) 100 cells from each sample (control and ADF + cofilin1 siRNA–treated cells) in three different experiments were randomly counted to visualize intracellular HRP location compared with TGN46 after a 0- and 60-min shift to 32°C. Error bars represent the mean ± SD of 100 counted cells from three independent experiments. Compared datasets were termed as statistically significant when P < 0.01 (**). Bars, 5 µm.
To test whether the defect in HRP secretion was ADF and cofilin1 specific and whether it was a result of the reorganization of the actin cytoskeleton caused by ADF and cofilin1 depletion, various control experiments were performed. HeLa cells were transfected with control or ADF- and cofilin1-specific siRNA. After 24 h, cells were transfected with a rat HA-tagged cofilin1 or a control plasmid and an ss-HRP plasmid for an additional 20 h. ADF and cofilin1 knockdown cells were also treated with the actin-depolymerizing agent LatA or actin-stabilizing reagent Jasplakinolide (Jasp). The medium from these cells was analyzed for HRP secretion. Control or ADF and cofilin1 knockdown cells expressing a control or HA rat cofilin1 plasmid as described for the HRP secretion assay were lysed and analyzed by Western blotting with an anti-cofilin1 antibody. The antibody recognized endogenous cofilin1 (Fig. 3 C, lanes 1 and 3) and HA-tagged rat cofilin1 (Fig. 3 C, lanes 3 and 4). Importantly, HA-tagged rat cofilin1 was not silenced by ADF and cofilin1 siRNA, and it rescued inhibition of HRP secretion (Fig. 3, B and C [lane 4]). HRP secretion was also rescued by treatment of ADF and cofilin1 knockdown cells with LatA but not with Jasp (Fig. 3 B). Collectively, these findings demonstrate that knockdown of ADF and cofilin1 inhibits secretion of HRP in HeLa cells. The fact that treatment of ADF and cofilin1 knockdown cells with LatA rescued HRP secretion demonstrates that the defect was caused by hyperpolymerization of actin. Knockdown of ADF and cofilin1 was also found to inhibit secretion of the endogenous interleukin-8 from MDA-MB cells (unpublished data).
Overexpression of LIMK–wild type (LIMK-wt; to inactivate ADF and cofilin1) and cofilin S3E (phosphomimetic and therefore inactive) but not of LIMK–kinase dead (LIMK-kd) or cofilin S3A (not phosphorylatable and therefore active) inhibited export of HRP from the Golgi (Fig. S1).
To determine the site of HRP accumulation in the secretory pathway as a result ADF and cofilin1 knockdown, HeLa cells (control or ADF and cofilin1 siRNA treated) were transfected with ss-HRP and incubated at 20°C in the presence of cycloheximide (CHX) to arrest newly synthesized secretory proteins in the TGN (Fig. 3 D) followed by their shift to the permissive temperature of 32°C to release the cargo for 60 min. The cells were stained with anti-HRP and anti-TGN46 antibodies. HRP was found to colocalize with TGN46-containing Golgi membranes in both control (Fig. 3 D, left) and ADF/cofilin1 knockdown cells (Fig. 3 D, right) at 20°C. Within the subsequent 60 min at 32°C, HRP in control cells was depleted from the Golgi apparatus in >90% of cells (Fig. 3, D and E). However, HRP was arrested in the TGN in ADF and cofilin1 knockdown cells at this time point (Fig. 3, D [right] and E), indicating their requirement in the TGN to cell surface transport.
The same procedure was repeated to monitor trafficking of signal sequence RFP (ss-RFP) in ADF and cofilin1 knockdown cells. In ADF and cofilin1 knockdown HeLa cells, RFP accumulated in the TGN (Fig. S2).
Transport of vesicular stomatitis virus G (VSV-G) protein, GP75, and GPI-GFP is unaffected in ADF and cofilin1 knockdown cells
To assess the role of ADF and cofilin1 in transport of membrane-associated cargoes, HeLa cells stably expressing a temperature-sensitive mutant of the VSV-G protein (GFP-ts045–VSV-G) were either transfected with ADF and cofilin1 or control siRNAs as described in Fig. 3. This mutant VSV-G protein harbors a thermosensitive-folding defect and accumulates in the ER at the nonpermissive temperature of 40°C. Upon shifting cells to 20°C, the ts045–VSV-G protein folds, exits the ER, and is accumulated at the TGN. Shifting cells to 32°C allows the export of VSV-G from the TGN to the cell surface. The cells in this experiment were fixed without permeabilization and incubated with an antibody that recognizes the exoplasmic domain of VSV-G at indicated time points after the shift to 32°C. Confocal microscopy revealed that ADF and cofilin1 knockdown did not affect transport of VSV-G from the TGN to the cell surface (Fig. 4 A). To quantify this result, cells from this experiment were analyzed by FACS, which confirmed that ADF and cofilin1 knockdown did not affect traffic of VSV-G to the cell surface (Fig. 4 B). Overexpression of LIMK-wt, LIMK-kd, cofilin S3E, and cofilin S3A did not affect transport of VSV-G protein to the cell surface in HeLa cells (Fig. S3).
Figure 4.

Knockdown of ADF and cofilin1 does not affect transport of VSV-G and p75. (A) HeLa cells stably expressing ts045–VSV-G–GFP proteins were transfected with control or ADF + cofilin1 (CFL-1) siRNA. After 24 h incubation at 37°C, cells were shifted to 40°C for 30 h (to promote clearance of the cell surface VSV-G by endocytosis and degradation in the lysosomes). To accumulate VSV-G in the TGN, cells were incubated for 2 h at 20°C in the presence of 100 µM CHX. Finally, cells were shifted 32°C to allow progression of newly synthesized VSV-G–GFP from the TGN to the cell surface. Samples were taken at the indicated time points, and VSV-G–GFP was localized using an antibody against the extracellular domain of VSV-G. (B) HeLa cells were transfected with control or ADF + cofilin1 siRNA. VSV-G–GFP was transfected, and VSV-G transport assay was performed as described in A for 90 min. The levels of VSV-G at the cell surface and inside the cells were determined by FACS analysis. Error bar indicates the relative ratio of VSV-G–GFP at the surface to the total in cells. (C, top) Schematic outline of the experimental setup for iFRAP-based analysis of GFP-p75 transport as described previously by Lázaro-Diéguez et al. (2007). (bottom left) The GFP-p75 intensity was plotted over time by fitting the data points (dotted lines) to a one-phase exponential decay equation (solid lines). The graph represents iFRAP decay curves of GFP-p75 intensities in control or ADF/cofilin1 siRNA–treated cells. (bottom right) Quantification of data shown in C (right). MF, mobile fraction of GFp-p75; t1/2, loss of 50% mobile GFP-p75 pool. Error bars represent the mean ± SD of the relative ratio of VSV-G–GFP at the surface to the total in cells of three independent experiments. Bars, 5 µm.
HeLa cells contain the machinery necessary for sorting apical and basolateral proteins in the TGN (Yeaman et al., 2004). Therefore, we tested whether ADF and cofilin1 were required for trafficking of the apically targeted protein p75 in HeLa cells. To visualize GFP-p75 trafficking along the secretory pathway, HeLa cells treated with ADF and cofilin1 or control siRNA were transfected with the apically targeted GFP-p75. Subsequently, the cells were incubated at 20°C to arrest proteins in the TGN followed by a release to 32°C for synchronous export to the cell surface. GFP-p75 was detected at the cell surface 30 min after transfer to 32°C in both control and ADF/cofilin1 knockdown cells (unpublished data). To obtain more quantitative data on the effect of ADF and cofilin1 knockdown on p75 export from the TGN, we used inverse FRAP (iFRAP) as described previously (Lázaro-Diéguez et al., 2007). The experimental procedure is shown schematically in Fig. 4 C (top). In brief, control and ADF and cofilin1 knockdown HeLa cells were transfected with a plasmid encoding GFP-p75 for 12 h. The cells were shifted to 20°C in the presence of CHX for 2 h to arrest GFP-p75 in the TGN and to prevent new protein synthesis. All GFP-p75 except the Golgi pool was photobleached, and the decrease in fluorescence at the TGN was monitored by live cell imaging (Fig. 4 C, top right). The GFP-p75 intensity was plotted over time (Fig. 4 C, bottom left), and the mobile fraction, i.e., the amount of cargo exported from the Golgi after 15 min at 32°C, was calculated by fitting the data points to a one-phase exponential decay equation using Prism software as described previously by Lázaro-Diéguez et al. (2007; Fig. 4 C, bottom right). The quantitation of t1/2 (loss of 50% mobile GFP-p75 pool) revealed no obvious delay in the export of p75 from TGN in ADF and cofilin1 knockdown HeLa cells compared with control cells (Fig. 4 C, bottom right). Similar fluorescence microscopy–based analysis revealed that knockdown of ADF and cofilin1 in HeLa cells had no effect on the trafficking of glycosylphosphatidylinositol (GPI)-GFP to the cell surface (unpublished data).
ADF and cofilin1 knockdown causes missorting of a subset of soluble proteins in the TGN
Our results present a striking situation: ADF and cofilin1 are required for the secretion of HRP, RFP, and interleukin-8 but not the transport of integral membrane proteins such as the VSV-G, p75, or GPI-anchored GFP from TGN to the cell surface. To gain further insight into the involvement of ADF and cofilin1 in TGN to cell surface transport, we monitored secretion of endogenous soluble secretory proteins.
HeLa cells were transfected with ADF- and cofilin1-specific or control siRNAs. Subsequently, cells were pulsed with medium containing 35S-labeled methionine for 15 min and chased for 2 h in the complete medium. Total medium from the cells was collected to precipitate secreted proteins and analyzed by SDS-PAGE and autoradiography (Fig. 5 A, left). Our findings revealed that secretion of several proteins was inhibited or reduced (Fig. 5 A, asterisks) upon knockdown of ADF and cofilin1. Interestingly, a large number of new proteins or proteins secreted in higher amounts (Fig. 5 A, arrowheads) were evident in the medium from ADF and cofilin1 knockdown cells. As expected, brefeldin A (BFA; blocks conventional protein secretion) treatment of control and ADF and cofilin1 knockdown cells efficiently inhibited the amount of total protein secretion from HeLa cells (Fig. 5 A, right). There was no difference in the levels of β-actin in the medium, which rules out the possibility that new proteins released into the medium were a result of cell death and lysis (Fig. 5 B).
Figure 5.

ADF and cofilin1 knockdown changes the composition of proteins secreted by HeLa cells. (A) HeLa cells were transfected with control or ADF + cofilin1 (CFL-1) siRNA. After 48 h, the cells were pulsed with [35S]methionine for 15 min and chased with standard medium containing unlabeled L-methionine. 2 h after the chase, newly synthesized proteins secreted in the medium were precipitated using TCA and analyzed by SDS-PAGE/autoradiography. BFA was added to one half of control or siRNA-treated cells 60 min before the pulse and kept throughout the chase. Secretion inhibited (asterisks) and increased secretion (arrowheads) in ADF + cofilin1 knockdown cells are shown compared with control cells. (B) Cells described in A were lysed, and aliquots of the lysates and medium of control and ADF + cofilin1 knockdown cells were analyzed by Western blotting with a β-actin antibody. The position of actin is indicated by an arrowhead.
To identify proteins secreted upon ADF and cofilin1 knockdown, we used quantitative stable isotope labeling by amino acids in cell culture (SILAC) mass spectrometry (MS). The procedure for this experiment is shown schematically in Fig. 6. In brief, one pool of HeLa cells was cultured in medium containing normal arginine and lysine, and another was grown in heavy arginine- and lysine-containing medium (Fig. 6). After normalization of growth conditions, cells were transfected with control or ADF- and cofilin1-specific siRNA. 48 h after transfection, the secreted proteins were collected and processed as described in experimental procedures. Based on the ratio between heavy and light peptides secreted into the medium, the proteins were classified as class A (ratio of 1 and no defect in secretion), class B (ratio > 1 and inhibition in secretion), and class C (ratio < 1 and secretion accelerated).
Figure 6.

ADF and cofilin1 knockdown causes missorting of soluble proteins. A schematic presentation of the SILAC experiment. HeLa cells were incubated in medium containing normal arginine and lysine or with the stable isotope arginine (13C6) and lysine (13C6/15N2). After five passages, heavy-labeled cells were transfected with control siRNA and unlabeled cells with ADF + cofilin1 siRNA. 48 h after transfection, cells were washed and incubated in medium without FCS and cultured for 2 h. Media were collected and mixed in 1:1 ratio according to cell number and SILAC-based analysis of the cell pellet. The mixed proteins were hereafter separated by 1D gel electrophoresis, and excised bands were trypsinized. Extracted peptides were analyzed by reverse-phase HPLC coupled to a mass spectrometer. Dotted lines together with arrow lines are used to indicate mass differences between SILAC peptide pairs. Based on the relative abundance ratio of specific peptides (Ong and Mann, 2006; Harsha et al., 2008), the proteins were classified as class A (ratio between heavy [H] and light [L] peptides = 1; secretion unchanged), class B (ratio between H/L > 1; secretion inhibited in ADF/cofilin-depleted cells), and class C (ratio between H/L < 1; secretion accelerated in ADF/cofilin-depleted cells).
Although several proteins were secreted at normal levels from ADF and cofilin1 knockdown cells (class A; Table I), secretion of secretogranin-2 (90% inhibition) and several collagens (40–80% inhibition, depending on the type of collagen) was inhibited under such conditions (class B; Table II). A large number of secretory proteins was secreted in higher amounts (class C; Table III), which included galactosidase A (lysosomal hydrolase) and Cab45 (a soluble resident protein of the Golgi apparatus; Fig. S4).
Table I.
Secretion of cargoes unchanged upon ADF and cofilin1 knockdown (class A)
| Protein name | Unique peptides | Ratio (H/L) control/RNAi |
| Matrix-remodeling–associated protein 5 | 14 | 1 |
| Fibulin 1 | 1 | 1 |
| Procollagen C-endopeptidase enhancer 1 | 5 | 1 |
| Carboxypeptidase A4 | 47 | 1 |
| Follistatin-related protein 1 | 35 | 1 |
| Laminin subunit α5 | 30 | 0.9 |
| Fibrillin-1 | 4 | 0.9 |
| Fibronectin | 12 | 0.8 |
Secretion of proteins into cell culture media from control and ADF and cofilin1 siRNA–treated HeLa cells was analyzed by SILAC-based MS. There was no change in secretion of cargoes classified as class A upon ADF and cofilin1 knockdown (ratio between heavy and light peptides [H/L] = 1).
Table II.
Cargoes inhibited in secretion upon ADF and cofilin1 knockdown (class B)
| Protein name | Unique peptides | Ratio (H/L) control/RNAi |
| Secretogranin-2 | 2 | 11 |
| Collagen α1 (III) chain | 3 | 4.6 |
| Lumican | 3 | 3 |
| Collagen α1 (I) chain | 4 | 2.8 |
| Collagen α1(XVIII) chain | 4 | 2 |
| Agrin (secreted form) | 16 | 1.9 |
| Collagen α1(VII) chain | 42 | 1.9 |
| Collagen α1(XV) chain | 6 | 1.9 |
| Immunoglobulin superfamily member 10 | 7 | 1.8 |
| Perlecan | 51 | 1.6 |
Secretion of proteins into cell culture media from control and ADF and cofilin1 siRNA–treated HeLa cells was analyzed by SILAC-based MS. There was an inhibition of secretion of cargoes classified as class B upon ADF and cofilin1 knockdown (ratio between heavy and light peptides [H/L] > 1).
Table III.
Cargo secretion accelerated upon ADF and cofilin1 knockdown (class C)
| Protein name | Unique peptides | Ratio (H/L) control/RNAi |
| Thrombospondin 3 | 3 | 0.3 |
| Follistatin-related protein 4 | 10 | 0.3 |
| Thrombospondin 1 | 12 | 0.4 |
| Pappalysin-1 | 4 | 0.4 |
| Serine protease HTRA1 | 6 | 0.4 |
| Laminin subunit β1 | 20 | 0.5 |
| Laminin subunit α4 | 16 | 0.5 |
| Proprotein convertase subtilisin/kexin type 9 | 8 | 0.5 |
| Metalloproteinase inhibitor 1 (TIMP1) | 2 | 0.5 |
| Nidogen-1 | 6 | 0.5 |
| SPARC-related modular calcium-binding protein 1 | 3 | 0.4 |
| Laminin subunit γ1 | 18 | 0.6 |
| Laminin subunit β1 | 20 | 0.5 |
| Galectin-3–binding protein | 3 | 0.6 |
| 72 kD type IV collagenase | 2 | 0.6 |
| Stanniocalcin-2 | 1 | 0.6 |
| Cystatin-C | 1 | 0.6 |
| Cab45 (Golgi) | 2 | 0.6 |
| α-Galactosidase A (lysosome) | 2 | 0.6 |
Secretion of proteins into cell culture media from control and ADF and cofilin1 siRNA–treated HeLa cells was analyzed by SILAC-based MS. There was an accelerated secretion of cargoes classified as class C upon ADF and cofilin1 knockdown (ratio between heavy and light peptides [H/L] < 1). Cab45, a luminal Golgi-resident protein, and α-galactosidase A (lysosomal hydrolase) were secreted upon ADF and cofilin1 knockdown.
To further investigate the effect of ADF and cofilin1 knockdown on secretion of Cab45, we analyzed the medium and HeLa cell lysates (transfected with control and ADF and cofilin1 siRNA) by Western blotting with specific antibodies. The medium was also analyzed with an anti–β-actin antibody to test for cell lysis. Cab45 was detected in the lysates of control and ADF and cofilin1 knockdown cells (Fig. 7 A), and it was secreted upon ADF and cofilin1 knockdown but not by transfection of HeLa cells with control siRNA. There was no release of β-actin from cells under our experimental conditions (unpublished data).
Figure 7.

ADF and cofilin1 knockdown affects secretion of secretory cargo. (A) HeLa cells were transfected with control and ADF + cofilin1 (CFL-1) siRNA. 48 h after transfection, cells were washed in medium without FCS and cultured for 2 h. Equal amounts of cell lysates and media were analyzed by Western blotting using an anti-Cab45 antibody. (B) Equal amounts of the cell lysates and medium, as described in A, were analyzed by Western blotting with an anti-TIMP1 antibody. Arrowheads in A and B indicate the position of Cab45 and α-TIMP1, respectively. (C) HeLa cells were transfected with control (left) or ADF + cofilin1 siRNA (right) for 40 h. Endogenous TIMP1 was accumulated in the TGN at 20°C for 2 h in the presence of 100 µM CHX. The cells were shifted to 32°C, fixed, and stained with an anti-TIMP1 antibody (green) at the indicated times. Cells were also stained with an anti-TGN46 (red) antibody to visualize the TGN. (D) 100 cells from each sample (control and ADF + cofilin1 siRNA–treated cells) in three different experiments were randomly counted to visualize intracellular TIMP1 location compared with TGN46 after a 0- and 60-min shift to 32°C. Compared datasets were termed as statistically significant when P < 0.01 (**). (E) Equal amounts of the medium and the cell lysates as described in A were Western blotted with anti–cathepsin D (left) and anti–β-actin antibody (right). Error bars represent the mean ± SD of 100 counted cells from three independent experiments. Bars, 5 µm.
We also tested the effect of ADF and cofilin1 knockdown on the secretion of TIMP1 (tissue inhibitor of metalloproteinases-1) from HeLa cells. The medium and the cell lysates from control siRNA–treated and ADF and cofilin1 knockdown HeLa cells were analyzed by Western blotting with an anti-TIMP1 antibody. There was a marked increase of TIMP1 in the medium of ADF and cofilin1 knockdown HeLa cells compared with control siRNA–transfected cells (Fig. 7 B). To further examine the site of TIMP1 transport acceleration in ADF and cofilin1 knockdown cells, we monitored its export from the TGN by fluorescence microscopy. To visualize TIMP1 export from the TGN under both conditions, control siRNA or ADF- and cofilin1-treated HeLa cells were incubated at 20°C in the presence of CHX to arrest TIMP1 in the TGN. The cells were shifted to 32°C to revive protein export from the TGN. The cells were fixed and stained at the onset and 15 min after the shift to 32°C. At 20°C, TIMP1 was arrested in the TGN in both conditions (Fig. 7 C). In control cells, after 15 min at 32°C, TIMP1 was still present in the TGN (Fig. 7 C, left). Interestingly, in ADF and cofilin1 knockdown cells, TIMP1 was not detectable in the TGN at this time point (Fig. 7 C, right). 100 cells from this experiment were counted for the presence of TIMP1 in the TGN, and the data support our findings that ADF and cofilin1 knockdown accelerated the export of TIMP1 from the TGN (Fig. 7 D). The localization of Golgi-resident enzymes GFP-conjugated sialyltransferase (TGN), galactosyltransferases (medial/trans-Golgi), and M2 (mannosidase II; cis-/medial Golgi) was not affected upon knockdown of ADF and cofilin1 in HeLa cells (unpublished data).
The lysosomal hydrolase cathepsin D is synthesized in the ER as a precursor polypeptide of 53 kD, which is transported to the Golgi. The precursor is subsequently sorted to lysosomes and further converted to a 47-kD intermediate and a 31-kD mature form (Gieselmann et al., 1985; Zaidi et al., 2008). Compared with control siRNA–treated cells, cells depleted of ADF and cofilin1 secreted large amounts of cathepsin D precursor (53 kD; Fig. 7 E). β-Actin was not detected in the medium upon ADF and cofilin1 knockdown, confirming that the release of Cab45 and cathepsin D was not a result of cell lysis (Fig. 7 E). There was no difference in the localization of the lysosomal integral membrane protein LAMP1 (unpublished data). Early endosomal protein, EEA, transferrin, and mannose 6-phosphate receptor were also unaffected upon ADF/cofilin1 knockdown (unpublished data). Therefore, the secretion of lysosomal enzymes is not the result of global disruption of the endosomal–lysosomal compartments.
To discard the possibility that the ADF/cofilin1 knockdown–dependent sorting defect was a result of effects on protein synthesis, we performed the following experiment. Control and ADF/cofilin1 knockdown HeLa cells were pulsed with medium containing 35S-labeled methionine for 15 min and chased with unlabeled methionine on 20°C for 4 h to prevent export of cargo from the TGN. Subsequently, the cells were lysed, and equal amounts of cell lysates were used to immunoprecipitate Cab45 with anti-Cab45 antibody. The immunoprecipitates were analyzed by SDS-PAGE/autoradiography, and the data revealed no difference in the intracellular levels of Cab45 between control and ADF/cofilin knockdown HeLa cells (Fig. S5). The effects on protein secretion are not the result of effects on protein synthesis upon ADF/cofilin1 knockdown (Fig. S5).
ADF and cofilin1 are required for sorting of a subset of integral membrane proteins
To understand the generality of ADF and cofilin1 in transport of membrane-associated proteins, we monitored the transport of all newly synthesized endogenous membrane proteins to the cell surface. HeLa cells were transfected with control or ADF- and cofilin1-specific siRNA. The cells were pulsed with 35S-labeled methionine for 15 min followed by incubation with unlabeled methionine for 2 h with or without BFA. Intact cells were surface biotinylated with NHS-biotin by the established procedures (Nishimura et al., 2002). The cells were subsequently lysed, and the same amount of lysate from control or ADF and cofilin1 siRNA–transfected cells (based on [35S]methionine content) was incubated with avidin-conjugated beads. The proteins bound to the beads were analyzed by SDS-PAGE/autoradiography. Our results revealed that ADF and cofilin1 knockdown cells contained several new cell surface proteins (Fig. 8, new surface proteins [N]). There were also several proteins missing from the cell surface or present in lower quantity under such conditions (Fig. 8, less or missing [L]). A large number of proteins showed no difference in their levels at the cell surface (Fig. 8, asterisks). In cells treated with BFA to block protein secretion, the appearance of methionine-labeled proteins at the cell surface was highly diminished, confirming that polypeptides identified by this procedure were newly synthesized proteins. Although the identity of these proteins is not known (it is technically difficult to obtain their sequence at present even by MS), these results suggest that knockdown of ADF and cofilin1 affects sorting of a subset of integral membrane proteins as well.
Figure 8.
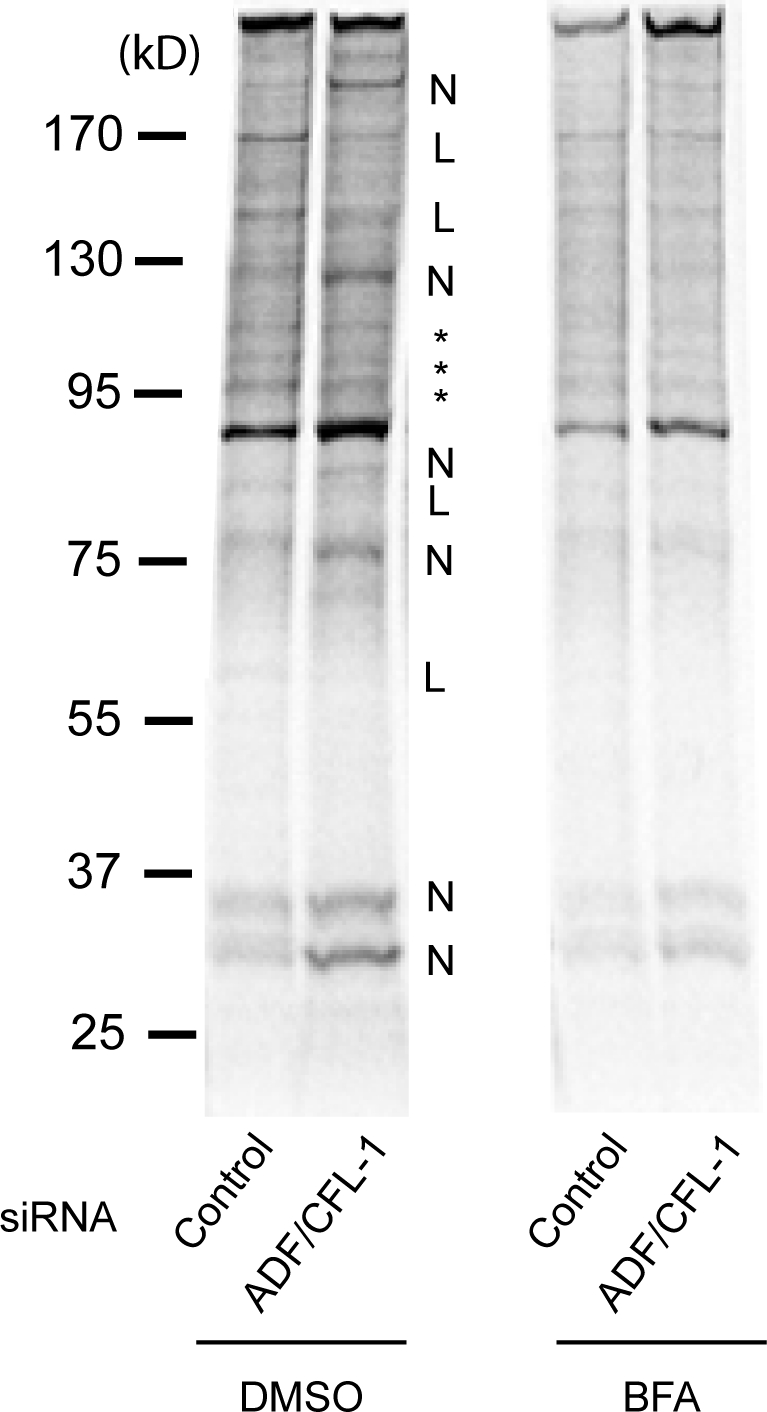
A subset of plasma membrane proteins are missorted upon ADF and cofilin1 knockdown. ADF and cofilin1 (CFL-1) knockdown affects the polypeptide composition of plasma membrane. HeLa cells were transfected with control or ADF + cofilin1 siRNA. The cells were pulsed with 35S-labeled methionine for 15 min and chased with unlabeled L-methionine for 2 h with or without BFA. Subsequently, intact cells were surface biotinylated with NHS-biotin and lysed. Equal amounts of lysates (based on [35S]methionine content) from control and ADF/cofilin1 knockdown cells were incubated with avidin-conjugated beads. The plasma membrane population of polypeptides in ADF + cofilin1 knockdown cells was different compared with control cells. N, new proteins; L, less or missing; *, no obvious change.
TGN is swollen upon ADF and cofilin1 knockdown
To gain further insight into the effect of ADF and cofilin1 knockdown on the ultrastructure of the Golgi membranes, HeLa cells (control and ADF and cofilin1 knockdown) were visualized by electron microscopy (Fig. 9 A). The Golgi membranes retain their morphology of stacks of cisternae upon knockdown of ADF and cofilin1. Interestingly, there was an accumulation of large, swollen membranes juxtaposed to the Golgi stacks (Fig. 9 A). A section of the Golgi membrane in ADF and cofilin1 knockdown cells is enlarged to reveal the features of swollen Golgi cisternae (Fig. 9 A). To identify these swollen membranes, HeLa cells treated with control or ADF and cofilin1 siRNA were cotransfected with a TGN-specific marker, TGN38 tagged with HRP (TGN38-HRP). The localization of TGN38-HRP for immunoelectron microscopy was determined as described previously (Polishchuk et al., 2000). 30 cells were processed for this experiment, and a typical section is shown (Fig. 9 B). HRP-conjugated TGN38 was localized in swollen cisternae, which provides strong evidence that the swollen membranes in cells depleted of ADF and cofilin1 are the TGN.
Figure 9.

ADF and cofilin1 knockdown causes swelling of the TGN. (A) HeLa cells were transfected with control or ADF + cofilin1 (CFL-1) siRNA. The cells were fixed and processed for electron microscopy. (B) HeLa treated with control or ADF + cofilin1 siRNA were transfected with HRP-TGN38, and the organization of Golgi membranes and the localization of TGN38 were monitored by electron microscopy as described previously (Bossard et al., 2007). Bars, 180 nm.
Discussion
Actin is required for a large number of cellular processes such as cytokinesis, cell shape, and migration. Actin has also been reported in the events leading to membrane fission in endocytosis and exocytosis (Egea et al., 2006; Sun et al., 2006; Massarwa et al., 2009). Our findings reveal a surprising role of actin in cargo sorting at the TGN. Knockdown of the actin-severing proteins ADF and cofilin1 arrested HRP in Golgi membranes of HeLa cells. A SILAC-based analysis of the secreted proteins revealed a more precise role of ADF and cofilin1 at the Golgi. Knockdown or inactivation of cofilin had the following effects on the secretory cargo and Golgi membranes. (a) Cab45 and the lysosomal hydrolase cathepsin D were secreted. Cab45 is a soluble lumenal Ca2+-binding protein in the Golgi (Scherer et al., 1996). Cab45 is normally not secreted, implying that in the knockdown cells, it must be exported from the TGN because of a defect in sorting. ADF and cofilin1 knockdown did not affect the steady-state distribution of mannose 6-phosphate receptor (unpublished data) but caused export of cathepsin D in an immature form. These data suggest a defect in sorting that resulted in secretion of cathepsin D instead of its transport to the lysosomes. (b) Export of secretory proteins such as TIMP1 from the TGN was accelerated upon ADF and cofilin1 knockdown. (c) Secretogranin-2 and collagens were not secreted. Secretogranin-2 is somehow sorted into secretory storage granules at the TGN and exported from cells. However, collagens do not enter the secretory storage granule for secretion. In other words, proteins normally destined for secretion by different routes were retained in the TGN upon ADF and cofilin1 knockdown. (d) The TGN was swollen. The TGN is a highly fenestrated compartment, but the significance of this feature in sorting and cargo export is not known. Treatment with carboxylic ionophore monensin causes efflux of protons in exchange for sodium ions, and this leads to the osmotic swelling of acidic compartments such as the TGN (Dinter and Berger, 1998). Unlike monensin treatment, which neutralizes the acidity of TGN, ADF and cofilin1 knockdown did not affect the intraluminal pH of TGN (unpublished data). Therefore, the defective export of proteins documented in this study was unlikely caused by pH changes in the TGN. (e) The composition of integral membrane proteins at the cell surface was perturbed upon ADF and cofilin1 knockdown. The identity of these proteins is not known, and the changes observed could be the result of a combination of defects in exocytosis and perhaps endocytosis. However, trafficking of Shiga toxin B from the cell surface to the endosomes, Golgi, and the ER was not affected by ADF and cofilin1 knockdown, and the organization of endosomal and lysosomal membranes was normal in these cells (unpublished data). Collectively, these findings strongly indicate that ADF and cofilin1 are required for sorting of cargo in the TGN. In this regard, it is noteworthy that carboxypeptidase Y, a vacuolar protein, is secreted in yeast harboring a temperature-sensitive mutant of cofilin (Okreglak and Drubin, 2007). We suggest that this could be the result of missorting at the Golgi, as we have found to be the case in mammalian cells.
HA-tagged LIMK1 has been localized to the Golgi and was reported to play a role in Golgi organization and transport to the cell surface in neurons (Rosso et al., 2004). More recently, it was reported that overexpression of activated cofilin, LIMK1-kd but not LIMK2-kd, or knockdown of LIMK1 delayed the transport of p75 from Golgi to the apical surface in polarized MDCK cells (Salvarezza et al., 2009). Transport of a basolateral plasma membrane protein and GPI-YFP was unaffected under these conditions. Under their experimental conditions, Salvarezza et al. (2009) suggest a defect in cutting of apically targeted transport carriers at the TGN. We did not observe any obvious defect in the transport of p75 to the cell surface in HeLa cells upon ADF and cofilin knockdown. Therefore, the delay in p75 export from the TGN reported by Salvarezza et al. (2009) could also be the result of defective sorting of the overexpressed p75 (by nuclear injection of a plasmid) in MDCK cells under conditions that promoted actin depolymerization.
An unexpected role for actin at the TGN
COPII vesicles to the ER–Golgi intermediate compartment/Golgi transport almost all secretory cargo that exits the ER. The ER has only one kind of export site of which numerous copies are spread throughout the ER. However, the TGN has numerous export routes. We hypothesize that the TGN is compartmentalized into different domains for cargo export and that those involved in export to the cell surface are organized by dynamic actin. We suggest that actin is bound to the molecular machinery sorters, which influences the capture of secretory cargo for export. Two layers of components are therefore required for cargo sorting at the TGN: the actin domain, which organizes the sorters, and the latter that binds directly to cargo. The sorters could be channels or pumps that regulate Ca2+ concentration or the lipid composition within the actin-based sorting domain. We suggest that these hyper-actin patches generated as a result of ADF/cofilin1 knockdown are pulled by myosin in all directions. This pulling causes membrane swelling, and the membrane tension created modifies the physicochemical properties of the sorters, thus changing their sorting properties.
Although we await the identification of the molecular sorters, there is some evidence already in support of this model. Treatment of post-ADF and cofilin1 knockdown cells with LatA restored HRP secretion from cells. Although LatA rescued defects in HRP secretion, it is important to note that treatment of control cells with this reagent caused missorting of soluble secretory cargo (unpublished data). This means that an actin patch has to be assembled and disassembled in a regulated manner to organize molecular sorters. Because actin is a key in this scheme of protein sorting, regulators of actin would influence this reaction. This might explain the requirement of a large number of actin-binding proteins for protein export from the Golgi (Kroschewski et al., 1999; Stamnes, 2002; Egea et al., 2006; Hayes and Pfeffer, 2008). A dynamic, actin-based process is reported to segregate GPI-anchored proteins at the plasma membrane (Goswami et al., 2008), and this kind of mechanism for protein concentration and sorting might be more widely applicable than presently appreciated.
Materials and methods
Antibodies, plasmids, and cell culture
Monoclonal antibodies against cofilin1, HA clone (15CA5), HRP, and TIMP1 were obtained from Abcam, polyclonal antibodies against cofilin2 and cofilin phospho-Ser3 were provided by S. Aznar (Center for Genomic Regulation, Barcelona, Spain), polyclonal anti-ADF and monoclonal anti–β-actin were obtained from Sigma-Aldrich, sheep anti–human TGN46 were obtained from AbD Serotec, monoclonal anti–cathepsin D antibody was obtained from Cell Signaling Technology, and anti-Cab45 monoclonal antibody was obtained from Transduction Laboratories. The monoclonal antibody 8G5F11 against the extracellular domain of VSV-G protein was provided by D. Lyles (Wake Forest University School of Medicine, Winston-Salem, NC). Secondary antibodies for immunofluorescence microscopy, Western blotting, and allophycocyanin-labeled secondary mouse antibody were obtained from Invitrogen. ss-HRP–Flag and GFP–VSV-G plasmids were described previously (Bossard et al., 2007), Discosoma sp. Red–ss-RFP was provided by H.-P. Hauri (Biozentrum University of Basel, Basel, Switzerland) and described previously (Ben-Tekaya et al., 2005), and HA–LIMK1-wt or HA–LIMK-kd were described previously (Sumi et al., 1999). GFP-p75 neurotrophin receptor was provided by G. Egea (University of Barcelona, Barcelona, Spain; Lázaro-Diéguez et al., 2007).
Cell culture and transfection
Drosophila S2 cells stably expressing ss-HRP (Bard et al., 2006) were grown in Schneider's medium (Invitrogen) supplemented with 10% FCS at 20°C without CO2. HeLa cells stably expressing GFP-tagged M2 (HeLa-GFP-M2) and GFP-ts045–VSV-G were grown in DME (Invitrogen) containing 10% FCS at 37°C at 7% CO2 incubator. The cells were transfected with Lipofectamine 2000 (Invitrogen), FuGENE 6 (Roche), or the Nucleofector solution (Cell Line kit V; Lonza) following the manufacturers' recommendation.
Generation of dsRNAs
To generate twinstar-specific dsRNAs, the cDNA fragments corresponding to the positions 176–344 (dstsr-1) and 31–523 (dstsr-2) were amplified by PCR with sequence-specific primers containing T7 promoter at both ends using the pMT-DEST48-twinstar as a template. To generate the dsRNA against syntaxin 5, the cDNA fragment corresponding to the position 676–1,182 was obtained by PCR using the pMT-DEST48– syntaxin 5 as a template. Full-length dLIMK and dSSH were PCR amplified from a Drosophila cDNA library. Different cDNA fragments with T7 promoter were generated as described for the dstsr: dLIMK-R1, position 2,757–3,097; dLIMK-R2, position 4,725–4,957; dSSH-R1, position 2,305–2,564; and dsSSH-R2, position 2,275–2,592. dsRNAs were generated by in vitro transcription of the T7 containing cDNA fragments using the MEGAscriptT7 kit (Applied Biosystems) following the manufacturer's instructions.
dsRNA transfection and HRP secretion assay in S2 cells
To knockdown twinstar, syntaxin 5, dLIMK, and dSSH, S2 cells stably expressing ss-HRP on a copper-inducible promoter were seeded with 3 µg dsRNA in triplicates in medium without serum. Serum was added after 4 h incubation. After 5 d, HRP secretion was induced by replacing culture medium with medium containing 0.5 mM CuSO4. 4 h later, 50 µl of medium was mixed with ECL reagent (Thermo Fisher Scientific), and luminescence was measured with a multi-label counter (WALLAC1420; PerkinElmer).
RT-PCR
RNAs from control and dsRNA-treated S2 cells were prepared using the RNAeasy Mini kit (QIAGEN) following the manufacturer's instructions. cDNAs were synthesized with the Cloned AMV First-Strand cDNA Synthesis kit (Invitrogen). Finally, twinstar was amplified with specific primers recognizing a 226-bp fragment of twinstar.
siRNA transfection in HeLa cells
The day before transfection, HeLa cells were plated to ensure 50% confluence on the day of transfection. Functionally validated Stealth RNA directed against ADF (oligo1, 5′-GCTTTGTATGATGCAAGCTTTGAAA-3′; oligo2, 5′-AGAAGATCTCAATCGGGCTTGTATT-3′) and cofilin1 (oligo1, 5′-GGATCAAGCATGAATTGCAAGCAAA-3′; oligo2, 5′-CCTACGCCACCTTTGTCAAGATGCT-3′) and Stealth RNA negative control were obtained from Invitrogen. ADF and cofilin1 siRNAs (150 nM each) or control siRNA were transfected using HiPerfect (QIAGEN).
HRP transport assay
30 h after transfection with siRNA, HeLa cells were transfected with ss-HRP–Flag using Lipofectamine 2000. 50 µl of the medium was harvested 48 h after the initial siRNA transfection. HRP activity was measured by the chemiluminescence assay as described for S2 cells. For normalization, cells were lysed with RIPA buffer, and internal HRP activity was measured. Lat A or Jasp was added to a final concentration of 500 nM for 4 h at 37°C, and HRP secretion was monitored.
Immunofluorescence microscopy of fixed samples
All fixed samples were analyzed with a confocal microscope (SPE; Leica) using the 63× Plan Apo NA 1.3 objective. For detection, the following laser lines were applied: DAPI, 405 nm; Discosoma sp. Red and Alexa Fluor 594, 532 nm; and Alexa Fluor 488 and GFP, 488 nm. Pictures were aquired using the Leica software and converted to TIFF files in ImageJ (version 1.37; National Institutes of Health).
VSV-G transport assay
Cells stably expressing GFP-ts045–VSV-G were transfected with siRNA. After 12 h, the cells were shifted to 40°C for an additional 34 h. To accumulate VSV-G in the Golgi, the cells were incubated for 2 h in the presence of 100 µm CHX at 20°C. Cells were shifted to 32°C, and at various time points, as indicated in Fig. 4, were fixed and incubated with the antibody 8G5F11 that recognizes the extracellular domain of VSV-G for 1 h and further processed for immunofluorescence microscopy.
FACS analysis of VSV-G surface exposure
To monitor the arrival of VSV-G at the cell surface quantitatively, the cells were processed as described for the VSV-G transport assay and subsequently harvested after 90 min at 32°C with a cell dissociation buffer (Invitrogen). After blocking with PBS containing 1.5% serum and 0.1% sodium azide, the labeling of surface VSV-G–GFP was performed for a 30-min incubation at 4°C with the anti–VSV-G 8G5F11 antibody. After washing with the blocking buffer, the cells were incubated with the allophycocyanin-labeled anti–mouse IgG antibody (Invitrogen) for 30 min at 4°C. After washing, the cells were analyzed with a flow cytometer (FACSCalibur; BD).
Imaging of HRP, RFP, p75, and TIMP1
30 h after transfection of HeLa (for HRP and p75) or HeLa GFP-M2 (for RFP) cells with siRNA, cells were transfected with a GFP-p75, ss-RFP, or ss-HRP–Flag. To yield a high transfection efficacy, ss-HRP and ss-RFP were transfected using Nucleofector solution (Lonza). 100 µg/ml CHX was added before a 2-h incubation at 20°C. Cells were fixed after different time points, as indicated in Figs. 3, 4, and S2, after the shift to 32°C, incubated with primary antibodies (anti-HRP and -TGN46), and further processed for immunofluorescence microscopy.
Live cell imaging and iFRAP analysis
iFRAP experiments were performed with a confocal laser-scanning microscope (SP5; Leica) using a 63× Plan Apo NA 1.4 objective equipped with an environmental control system set to 37°C and 5% CO2 atmosphere. All experiments were performed at 32°C and 5% CO2. For visualization of GFP, a 488-nm laser line (emission range detection of 500–600 nm) was used with the confocal pinhole set at 4.94 Airy units to minimize changes in fluorescence caused by protein GFP displacement from the plane of focus. The whole cytoplasm, except the Golgi of GFP fusion protein–transfected cells, was photobleached using 50–80 scans with the 488-nm laser line at full power. After bleaching, images were monitored at 5-s intervals for 15 min. The excitation intensity was attenuated to ∼5% of the half-laser power to avoid significant photobleaching. Longer recording times were also performed to monitor the arrival of cargo at the plasma membrane and the subsequent absence of fluorescence in the Golgi. Decay curves were background subtracted, fitted, and calculated as described previously (Lázaro-Diéguez et al., 2007).
Metabolic labeling
Control and siRNA-treated cells were cultured in DME without L-methionine and L-cysteine for 60 min and pulsed with 100 µCi [35S]methionine (PerkinElmer) for 15 min. The cells were washed five times with PBS and chased with DME containing 10 mM unlabeled methionine for 2 h. For BFA treatment, 10 µg/ml BFA was added to the medium for the last 30 min of incubation and kept for the whole experiment. Cells were lysed with RIPA buffer (100 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, and 1% deoxycholic acid, pH 7.4), 20 µl of cell extracts was mixed with scintillation cocktail, and the radioactivity was determined for normalization. The collected medium was precipitated with TCA and analyzed by SDS-PAGE/autoradiography.
Surface biotinylation of membrane-associated proteins
To analyze cell surface proteins, cells were pulse chased as described for metabolic labeling. After 2 h chase, cells were washed twice with ice-cold PBS and subsequently labeled with 250 µg/ml sulfo-NHS-SS-biotin (Thermo Fisher Scientific) for 30 min on ice. The biotin-labeled cells were incubated with 150 mM glycine solution for 20 min to quench unlabeled free biotin followed by an ice-cold PBS wash. Cells were lysed in RIPA buffer including complete protease inhibitor mixture (Roche), and the lysates were affinity purified using immobilized NeutrAvidin beads (Thermo Fisher Scientific) overnight at 4°C. Finally, the beads were washed with RIPA buffer followed by the addition of SDS-PAGE sample loading buffer and analyzed by SDS-PAGE/autoradiography.
SILAC cell culture and sample preparation
Ready to use SILAC DME containing unlabeled arginine and lysine (R0K0; light) and media containing heavy (13C6) arginine and (13C14N) lysine (R6K8; heavy) were purchased from Dundee Cell Products. Dialyzed FBS was obtained from Invitrogen. HeLa cells were grown in light and heavy medium for five passages until full incorporation of heavy amino acids according to previously described protocols (Ong and Mann, 2006; Harsha et al., 2008). The incorporation of heavy amino acids was analyzed by liquid chromatography/MS/MS.
Control (heavy-labeled cells) or siRNA (unlabeled cells)-transfected cells (∼5 × 107 cells/condition) were washed five times with serum-free medium. The cells were grown in serum-free medium, and after 2 h, the medium was collected. Cells were counted and lysed in RIPA buffer. Media from control and siRNA-treated cells were filtered using a 0.45-µm filter (Millipore) centrifuged at 5,000 g for 15 min, and the resulting supernatant was spun at 100,000 g for 2 h. Samples were concentrated using a 3,000-D molecular mass cutoff spin column. The concentrated medium from control (heavy) and ADF and cofilin1 (light) siRNA–treated cells was mixed according to cell counts and SILAC-based analysis of the cells after collection. The mixed proteins were hereafter separated by 1D gel electrophoresis, and the bands were excised and trypsinized (Promega) according to established protocols (Shevchenko et al., 2006). Extracted peptides were analyzed by reverse-phase HPLC (1200 nanoflow pump; Agilent Technologies) coupled to a mass spectrometer (Orbitrap XL; Thermo Fisher Scientific). MS/MS data were extracted using MaxQuant (version 1.0.12.31; Cox and Mann, 2008). MS/MS data were searched against forward and reversed International Protein Index Human database using Mascot (version 2.2; Matrix Science Inc.). 10 ppm and 0.5 D were used as MS and MS/MS mass accuracies, respectively. N-terminal acetylation and oxidation of methionine residues were allowed as variable modifications. A false discovery rate of 0.01 was used as the identification threshold. Matched peptides were assigned to MS spectra using MaxQuant, and fold differences between heavy and light peptides were calculated without normalization.
Transmission electron microscopy
For transmission electron microscopy, HeLa cells were transfected with control or ADF and cofilin1 siRNA. 48 h after the transfection, cells were fixed with 1% glutaraldehyde in Hepes buffer embedded in epon 812 and processed for electron microscopy. Thin sections were analyzed by an electron microscope (Philips Tecnai-12; FEI). Images were obtained with a charge-coupled device digital camera (UltraView; Olympus). Morphometric analysis of Golgi stacks was performed in 30 cells for each experimental condition using image analysis software (Olympus).
Immunoelectron microscopy
HeLa cells were transfected with control or ADF and cofilin1 siRNA. After 36 h, cells were transfected with TGN38-HRP. 12 h later, cells were fixed with 1% glutaraldehyde in 0.2 M Hepes for 20 min. After washing with PBS and 0.1 M Tris-HCl, pH 7.4, buffers, cells were incubated in 0.5 mg/ml DAB/H2O2 solution for 15 min. After washing with 0.1 M Tris and PBS, cells were incubated with 1% glutaraldehyde in 0.2 M Hepes for 5 min. Finally, cells were placed in 1% BSA solution, scraped, and pelleted. Finally, cells were processed and imaged by electron microscopy.
Statistical analysis
Statistical significance was tested in an unpaired Student's t test using Prism software (GraphPad Software, Inc.). Compared datasets were termed as statistically significant when P < 0.01.
Online supplemental material
Fig. S1 shows that expression of LIMK-wt or phosphorylated cofilin1 inhibited HRP secretion. Fig. S2 shows that RFP is arrested in the TGN upon ADF/cofilin1 knockdown. Fig. S3 reveals that overexpression of LIMK-wt or cofilin S3E did not affect transport of VSV-G protein to the cell surface. Fig. S4 shows that Cab45 is a luminal Golgi protein. Fig. S5 shows that ADF/cofilin1 knockdown did not affect protein synthesis of Cab45. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200908040/DC1.
Acknowledgments
We thank members of the Malhotra laboratory, Scott Emr, Misha Kozlov, and Gustavo Egea for insightful discussions. We thank Carolina de la Torre for help with proteomics and Tobias Maier for suggesting the use of SILAC technology to analyze the secreted proteins, Timo Zimmermann and Raquel García for help with laser-scanning microscopy, Josse van Galen for providing normal rat kidney Golgi membranes, and Felix Campelo for support with the preparation of the manuscript. We also thank Dr. Marina Bacac (University of Lausanne, Lausanne, Switzerland) for the generation of the stable VSV-G HeLa cell line.
The work is supported by a consolider grant from Plan de Nacional of Spanish ministry to V. Malhotra. J. von Blume is supported by a European Molecular Biology Organization fellowship, J.M. Duran is supported by a Juan De la Cierva fellowship, and V. Malhotra is an Institució Catalana de Recerca i Estudis Avançats professor at the Center for Genomic Regulation.
Footnotes
Abbreviations used in this paper:
- ADF
- actin-depolymerizing factor
- BFA
- brefeldin A
- CHX
- cycloheximide
- dLIMK
- Drosophila LIMK
- dsRNA
- double-stranded RNA
- dSSH
- Drosophila SSH
- GPI
- glycosylphosphatidylinositol
- iFRAP
- inverse FRAP
- Jasp
- Jasplakinolide
- LatA
- latrunculin A
- LIMK
- LIM kinase
- LIMK-kd
- LIMK–kinase dead
- LIMK-wt
- LIMK–wild type
- MS
- mass spectrometry
- SILAC
- stable isotope labeling by amino acids in cell culture
- ss-HRP
- signal sequence HRP
- ss-RFP
- signal sequence RFP
- SSH
- slingshot
- VSV-G
- vesicular stomatitis virus G
References
- Ang A.L., Fölsch H., Koivisto U.M., Pypaert M., Mellman I. 2003. The Rab8 GTPase selectively regulates AP-1B–dependent basolateral transport in polarized Madin-Darby canine kidney cells. J. Cell Biol. 163:339–350 10.1083/jcb.200307046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang A.L., Taguchi T., Francis S., Fölsch H., Murrells L.J., Pypaert M., Warren G., Mellman I. 2004. Recycling endosomes can serve as intermediates during transport from the Golgi to the plasma membrane of MDCK cells. J. Cell Biol. 167:531–543 10.1083/jcb.200408165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S., Barbayannis F.A., Hanser H., Schneider C., Stanyon C.A., Bernard O., Caroni P. 1998. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 393:805–809 10.1038/31729 [DOI] [PubMed] [Google Scholar]
- Bamburg J.R. 1999. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell Dev. Biol. 15:185–230 10.1146/annurev.cellbio.15.1.185 [DOI] [PubMed] [Google Scholar]
- Bard F., Casano L., Mallabiabarrena A., Wallace E., Saito K., Kitayama H., Guizzunti G., Hu Y., Wendler F., Dasgupta R., et al. 2006. Functional genomics reveals genes involved in protein secretion and Golgi organization. Nature. 439:604–607 10.1038/nature04377 [DOI] [PubMed] [Google Scholar]
- Ben-Tekaya H., Miura K., Pepperkok R., Hauri H.P. 2005. Live imaging of bidirectional traffic from the ERGIC. J. Cell Sci. 118:357–367 10.1242/jcs.01615 [DOI] [PubMed] [Google Scholar]
- Bossard C., Bresson D., Polishchuk R.S., Malhotra V. 2007. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J. Cell Biol. 179:1123–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C., Beltzner C.C., Pollard T.D. 2009. Cofilin dissociates Arp2/3 complex and branches from actin filaments. Curr. Biol. 19:537–545 10.1016/j.cub.2009.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26:1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- Dinter A., Berger E.G. 1998. Golgi-disturbing agents. Histochem. Cell Biol. 109:571–590 10.1007/s004180050256 [DOI] [PubMed] [Google Scholar]
- Egea G., Lázaro-Diéguez F., Vilella M. 2006. Actin dynamics at the Golgi complex in mammalian cells. Curr. Opin. Cell Biol. 18:168–178 10.1016/j.ceb.2006.02.007 [DOI] [PubMed] [Google Scholar]
- Fölsch H., Ohno H., Bonifacino J.S., Mellman I. 1999. A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell. 99:189–198 10.1016/S0092-8674(00)81650-5 [DOI] [PubMed] [Google Scholar]
- Fölsch H., Mattila P.E., Weisz O.A. 2009. Taking the scenic route: biosynthetic traffic to the plasma membrane in polarized epithelial cells. Traffic. 10:972–981 10.1111/j.1600-0854.2009.00927.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P., Griffith J., Geuze H.J., Kornfeld S. 2003. Mammalian GGAs act together to sort mannose 6-phosphate receptors. J. Cell Biol. 163:755–766 10.1083/jcb.200308038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieselmann V., Hasilik A., von Figura K. 1985. Processing of human cathepsin D in lysosomes in vitro. J. Biol. Chem. 260:3215–3220 [PubMed] [Google Scholar]
- Glick B.S., Malhotra V. 1998. The curious status of the Golgi apparatus. Cell. 95:883–889 10.1016/S0092-8674(00)81713-4 [DOI] [PubMed] [Google Scholar]
- Glick B.S., Nakano A. 2009. Membrane traffic within the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 25:113–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami D., Gowrishankar K., Bilgrami S., Ghosh S., Raghupathy R., Chadda R., Vishwakarma R., Rao M., Mayor S. 2008. Nanoclusters of GPI-anchored proteins are formed by cortical actin-driven activity. Cell. 135:1085–1097 10.1016/j.cell.2008.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsha H.C., Molina H., Pandey A. 2008. Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat. Protoc. 3:505–516 10.1038/nprot.2008.2 [DOI] [PubMed] [Google Scholar]
- Hayes G.L., Pfeffer S.R. 2008. WHAMMing into the Golgi. Dev. Cell. 15:171–172 10.1016/j.devcel.2008.07.011 [DOI] [PubMed] [Google Scholar]
- Hotulainen P., Paunola E., Vartiainen M.K., Lappalainen P. 2005. Actin-depolymerizing factor and cofilin-1 play overlapping roles in promoting rapid F-actin depolymerization in mammalian nonmuscle cells. Mol. Biol. Cell. 16:649–664 10.1091/mbc.E04-07-0555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos R., Pozsonyi K., Nevalainen E., Lappalainen P., Nyitrai M., Hild G. 2009. The effects of ADF/cofilin and profilin on the conformation of the ATP-binding cleft of monomeric actin. Biophys. J. 96:2335–2343 10.1016/j.bpj.2008.12.3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld S., Mellman I. 1989. The biogenesis of lysosomes. Annu. Rev. Cell Biol. 5:483–525 10.1146/annurev.cb.05.110189.002411 [DOI] [PubMed] [Google Scholar]
- Kroschewski R., Hall A., Mellman I. 1999. Cdc42 controls secretory and endocytic transport to the basolateral plasma membrane of MDCK cells. Nat. Cell Biol. 1:8–13 10.1038/8977 [DOI] [PubMed] [Google Scholar]
- Kueh H.Y., Charras G.T., Mitchison T.J., Brieher W.M. 2008. Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 182:341–353 10.1083/jcb.200801027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázaro-Diéguez F., Colonna C., Cortegano M., Calvo M., Martínez S.E., Egea G. 2007. Variable actin dynamics requirement for the exit of different cargo from the trans-Golgi network. FEBS Lett. 581:3875–3881 10.1016/j.febslet.2007.07.015 [DOI] [PubMed] [Google Scholar]
- Lee M.C., Miller E.A., Goldberg J., Orci L., Schekman R. 2004. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20:87–123 10.1146/annurev.cellbio.20.010403.105307 [DOI] [PubMed] [Google Scholar]
- Massarwa R., Schejter E.D., Shilo B.Z. 2009. Apical secretion in epithelial tubes of the Drosophila embryo is directed by the Formin-family protein Diaphanous. Dev. Cell. 16:877–888 10.1016/j.devcel.2009.04.010 [DOI] [PubMed] [Google Scholar]
- Matsuura-Tokita K., Takeuchi M., Ichihara A., Mikuriya K., Nakano A. 2006. Live imaging of yeast Golgi cisternal maturation. Nature. 441:1007–1010 10.1038/nature04737 [DOI] [PubMed] [Google Scholar]
- Mellman I., Warren G. 2000. The road taken: past and future foundations of membrane traffic. Cell. 100:99–112 10.1016/S0092-8674(00)81687-6 [DOI] [PubMed] [Google Scholar]
- Nishimura N., Plutner H., Hahn K., Balch W.E. 2002. The delta subunit of AP-3 is required for efficient transport of VSV-G from the trans-Golgi network to the cell surface. Proc. Natl. Acad. Sci. USA. 99:6755–6760 10.1073/pnas.092150699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa R., Nagata-Ohashi K., Takeichi M., Mizuno K., Uemura T. 2002. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 108:233–246 10.1016/S0092-8674(01)00638-9 [DOI] [PubMed] [Google Scholar]
- Ohashi K., Hosoya T., Takahashi K., Hing H., Mizuno K. 2000. A Drosophila homolog of LIM-kinase phosphorylates cofilin and induces actin cytoskeletal reorganization. Biochem. Biophys. Res. Commun. 276:1178–1185 10.1006/bbrc.2000.3599 [DOI] [PubMed] [Google Scholar]
- Okreglak V., Drubin D.G. 2007. Cofilin recruitment and function during actin-mediated endocytosis dictated by actin nucleotide state. J. Cell Biol. 178:1251–1264 10.1083/jcb.200703092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.E., Mann M. 2006. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 1:2650–2660 10.1038/nprot.2006.427 [DOI] [PubMed] [Google Scholar]
- Patterson G.H., Hirschberg K., Polishchuk R.S., Gerlich D., Phair R.D., Lippincott-Schwartz J. 2008. Transport through the Golgi apparatus by rapid partitioning within a two-phase membrane system. Cell. 133:1055–1067 10.1016/j.cell.2008.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polishchuk R.S., Polishchuk E.V., Marra P., Alberti S., Buccione R., Luini A., Mironov A.A. 2000. Correlative light-electron microscopy reveals the tubular-saccular ultrastructure of carriers operating between Golgi apparatus and plasma membrane. J. Cell Biol. 148:45–58 10.1083/jcb.148.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso S., Bollati F., Bisbal M., Peretti D., Sumi T., Nakamura T., Quiroga S., Ferreira A., Cáceres A. 2004. LIMK1 regulates Golgi dynamics, traffic of Golgi-derived vesicles, and process extension in primary cultured neurons. Mol. Biol. Cell. 15:3433–3449 10.1091/mbc.E03-05-0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K., Chen M., Bard F., Chen S., Zhou H., Woodley D., Polischuk R., Schekman R., Malhotra V. 2009. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 136:891–902 10.1016/j.cell.2008.12.025 [DOI] [PubMed] [Google Scholar]
- Salvarezza S.B., Deborde S., Schreiner R., Campagne F., Kessels M.M., Qualmann B., Caceres A., Kreitzer G., Rodriguez-Boulan E. 2009. LIM kinase 1 and cofilin regulate actin filament population required for dynamin-dependent apical carrier fission from the trans-Golgi network. Mol. Biol. Cell. 20:438–451 10.1091/mbc.E08-08-0891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer P.E., Lederkremer G.Z., Williams S., Fogliano M., Baldini G., Lodish H.F. 1996. Cab45, a novel (Ca2+)-binding protein localized to the Golgi lumen. J. Cell Biol. 133:257–268 10.1083/jcb.133.2.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1:2856–2860 10.1038/nprot.2006.468 [DOI] [PubMed] [Google Scholar]
- Stamnes M. 2002. Regulating the actin cytoskeleton during vesicular transport. Curr. Opin. Cell Biol. 14:428–433 10.1016/S0955-0674(02)00349-6 [DOI] [PubMed] [Google Scholar]
- Sumi T., Matsumoto K., Takai Y., Nakamura T. 1999. Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho- and Cdc42-activated LIM-kinase 2. J. Cell Biol. 147:1519–1532 10.1083/jcb.147.7.1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Martin A.C., Drubin D.G. 2006. Endocytic internalization in budding yeast requires coordinated actin nucleation and myosin motor activity. Dev. Cell. 11:33–46 10.1016/j.devcel.2006.05.008 [DOI] [PubMed] [Google Scholar]
- Wang C.W., Hamamoto S., Orci L., Schekman R. 2006. Exomer: a coat complex for transport of select membrane proteins from the trans-Golgi network to the plasma membrane in yeast. J. Cell Biol. 174:973–983 10.1083/jcb.200605106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeaman C., Ayala M.I., Wright J.R., Bard F., Bossard C., Ang A., Maeda Y., Seufferlein T., Mellman I., Nelson W.J., Malhotra V. 2004. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat. Cell Biol. 6:106–112 10.1038/ncb1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi N., Maurer A., Nieke S., Kalbacher H. 2008. Cathepsin D: a cellular roadmap. Biochem. Biophys. Res. Commun. 376:5–9 10.1016/j.bbrc.2008.08.099 [DOI] [PubMed] [Google Scholar]