

Abstract
To identify functional contacts between HIV-1 integrase (IN) and its viral DNA substrate, we devised a new experimental strategy combining the following two methodologies. First, disulfide-mediated cross-linking was used to site-specifically link select core and C-terminal domain amino acids to respective positions in viral DNA. Next, surface topologies of free IN and IN-DNA complexes were compared using Lys- and Arg-selective small chemical modifiers and mass spectrometric analysis. This approach enabled us to dissect specific contacts made by different monomers within the multimeric complex. The footprinting studies for the first time revealed the importance of a specific N-terminal domain residue, Lys-14, in viral DNA binding. In addition, a DNA-induced conformational change involving the connection between the core and C-terminal domains was observed. Site-directed mutagenesis experiments confirmed the importance of the identified contacts for recombinant IN activities and virus infection. These new findings provided major constraints, enabling us to identify the viral DNA binding channel in the active full-length IN multimer. The experimental approach described here has general application to mapping interactions within functional nucleoprotein complexes.
HIV-1 integrase (IN)4 is commonly viewed as an important therapeutic target for the following reasons: its catalytic activities are required for viral replication, there is no closely related cellular equivalent of IN, and specific IN inhibitors are likely to be effective against viral strains resistant to currently available therapies targeting reverse transcriptase (RT), protease, and virus-cell fusion. Detailed structural information on functional IN-DNA complexes could aid drug design efforts. For example, the promising diketo acid class of inhibitors preferentially bind to the assembled IN-viral DNA complex rather than the free protein (1-4).
The two chemical reactions catalyzed by HIV-1 IN, 3′ processing and DNA strand transfer, have been characterized in detail (reviewed in Ref. 5). First, IN removes two nucleotides from each 3′-end of the viral DNA synthesized by reverse transcriptase. In the following step, concerted transesterification reactions covalently join the viral DNA ends into the host genome (6). In vivo, the enzyme acts in the context of a large nucleoprotein complex with a number of viral and host proteins contributing to the integration process (7-21).
HIV-1 IN is composed of three distinct structural and functional domains: the N-terminal domain (NTD) (residues 1–50) that contains an HHCC zinc binding motif, the catalytic core domain (CCD) (residues 51–212) containing the DDE motif essential for coordinating catalytic divalent metals, and the C-terminal domain (CTD) (residues 213–288) that is thought to provide a platform for DNA binding. Crystallographic or NMR structural data are available for each of the individual domains (22-26). In addition, two-domain CCD/ CTD (27) and NTD/CCD (28) crystal structures have been determined. However, efforts to obtain detailed structures for the full-length protein or IN-DNA complexes have been impeded by relatively poor protein solubility and/or full-length enzyme flexibility.
To better understand IN-DNA interactions, a number of biochemical approaches have been employed. Site directed mutagenesis experiments indicated that different monomers within the IN multimer provide complementary rather than symmetrical contacts to DNA (29-31). Photo and chemical cross-linking studies revealed several potential DNA binding residues (32-37). These biochemical experiments together with crystallographic determination of the two-domain structures prompted molecular modeling research. However, the IN-DNA models obtained by different groups vary significantly, indicating that the available experimental data comprises an insufficient number of constraints for formulating a common outcome (28, 37-43).
Here we employed a new experimental strategy combining two established methodologies of site specific protein-DNA cross-linking through disulfide bond formation and mass spectrometric (MS) protein footprinting. This approach enabled us to identify monomer selective contacts with substrate DNA. Importantly, our studies uncovered an essential role for the NTD in binding to the viral DNA substrate. We also found that upon DNA binding, IN undergoes a conformational change involving the α-helical connection between the core and C-terminal domains. The functional importance of the identified residues were confirmed by site-directed mutagenesis. Our data provide new and important details on how HIV-1 IN interacts with its viral DNA substrate.
EXPERIMENTAL PROCEDURES
Preparation of Recombinant HIV-1 IN, DNA Oligonucleotides, and Cross-linked Products
Full-length IN and mutant proteins were expressed in E. coli and purified as described previously (35). The catalytic activities of recombinant proteins were monitored according to the reported procedure (35).
Phosphoramidites (O6-phenyl-dI, O4-triazolyl-dU and 2-F-dI) were purchased from Glen Research (Sterling, VA) and synthetically incorporated in the viral DNA sequences using an Applied Biosystems 392 DNA synthesizer. Post-synthetic attachment of the carbon tether (cystamine) to the modified bases, and subsequent activation and purification of oligonucleotides were performed as published previously (35).
For cross-linking reactions, 10 μm IN was incubated with equimolar DNA duplex in 50 mm HEPES, pH 7.0, 100 mm NaCl, 5 mm MgCl2, and 10% glycerol at 37 °C for 20 min. The reactions were quenched by 20 mm methyl methanethiosulfonate and subjected to surface topology analysis described below.
MS Footprinting
Free IN and cross-linked IN-DNA complexes were treated at 37 °C with NHS-biotin or p-Hydroxyphenylglyoxal (HPG) for 30 and 60 min, respectively. The reagent concentrations are indicated in the text and figure legends. The reactions were quenched by excess Lys and Arg using free amino acid forms. Cross-linked products were separated by denaturing SDS-PAGE using nonreducing gel loading and separation buffers. Bands were visualized by Microwave Blue stain (Protiga, Gaithersburg, MD), excised, and destained according to the described procedure (44-49). In-gel proteolysis was performed using 0.5 μg of trypsin to generate small peptide peaks amenable to MS and MS/MS analysis. Tryptic peptides were monitored by a Kratos MALDI-ToF instrument equipped with a curved field reflectron feature (Kratos Analytical Instruments, Manchester, UK), enabling generation of postsource decay (PSD) amino acid sequence data. MALDI-ToF experiments were performed using α-cyano-4-hydroxy-cinnamic acid as a matrix. For accurate quantitation of the modified peptide peaks, the intensities of unmodified IN tryptic peptides were used as internal controls.
CD Spectroscopy
CD spectra were recorded on an AVIV 202 circular dichroism spectrometer. A 0.01-cm pathlength cell and 1-nm bandwidth were used to make measurements in the near (215–320 nm) ultraviolet region. The protein concentration was 10 μm in 20 mm PIPES, pH 6.8, 750 mm NaCl, 2 mm β-mercaptoethanol, 0.1 mm EDTA, and 1 mm CHAPS.
Virus Infectivity
The SpeI-EcoRI fragment from pNL4-3 containing the IN coding sequence subcloned into pBlueScript (Invitrogen) was subjected to QuikChange mutagenesis (Stratagene, La Jolla, CA), and the mutated fragments were returned to pNL4–3. AgeI-PflMI fragments were subsequently exchanged for the IN-coding fragment in the NLX.Luc.R-strain that carries the luciferase reporter gene (50). Normalized levels of viral infectivities using SupT1 target cells were calculated as previously described (50).
RESULTS
We chose to employ MS protein footprinting (44-49) to study IN-viral DNA interactions for the following reasons. The methodology proved to be effective for identifying contact amino acids to cognate nucleic acids for a number of nucleoprotein complexes not amenable to conventional structural biology analyses such as x-ray crystallography or NMR. Of note, the MS footprinting consistently revealed functionally essential interactions (44-49). Briefly, the approach is based on comparing surface topologies of free protein versus pre-assembled nucleoprotein complexes using amino acid specific reagents such as NHS-biotin and HPG that modify exposed lysine and arginine residues, respectively. The basic residues are targeted due to their likely role in electrostatic contacts with nucleic acids. Reagent concentrations are carefully optimized to ensure mild modification conditions where the integrity of the functional complex is preserved. Subsequent MS analyses reveal the surface residues readily labeled in free protein yet shielded from modification in the complex due to bound nucleic acid.
Unlike previously examined nucleoprotein complexes, IN-DNA interactions presented us with a new challenge as homologous protein monomers are predicted to provide complementary rather than symmetrical contacts to cognate DNA. Indeed, earlier complementation studies (29, 30) suggested that the viral DNA is coordinated by the CCD of one monomer and the CTD of another monomer. Because of this, it was essential to separate individual monomers based on their differential interactions with DNA to accurately assign contacts within full-length multimeric IN. For this, we employed a new experimental strategy combining two technologies established in our laboratories: site-specific protein-DNA cross-linking through disulfide bridging (reviewed in Ref. 51) and MS footprinting (44-49) (Fig. 1). Our goal was to obtain two different cross-linked complexes: one with the CCD tethered to the DNA end, and the other with the CTD linked to a subterminal position in the DNA substrate. The two complexes were then exposed to selective amino acid modifying reagents. Subsequent SDS-PAGE enabled us to separate the cross-linked complexes from free protein. As depicted in Fig. 1, monomer 1 cross-linked to DNA through CCD residue E152C was separated from monomer 2 (Fig. 1, middle). In a parallel experiment, the monomer 2-DNA complex cross-linked through a selective CTD residue was separated from monomer 1 (Fig. 1, bottom). Cross-linked products were then processed and analyzed separately using MS. We predicted that protection patterns within monomer 1 and monomer 2 would differ, and the identified amino acids in the respective monomers would reveal a DNA binding channel in the context of full-length multimeric IN.
FIGURE 1. Experimental strategy.
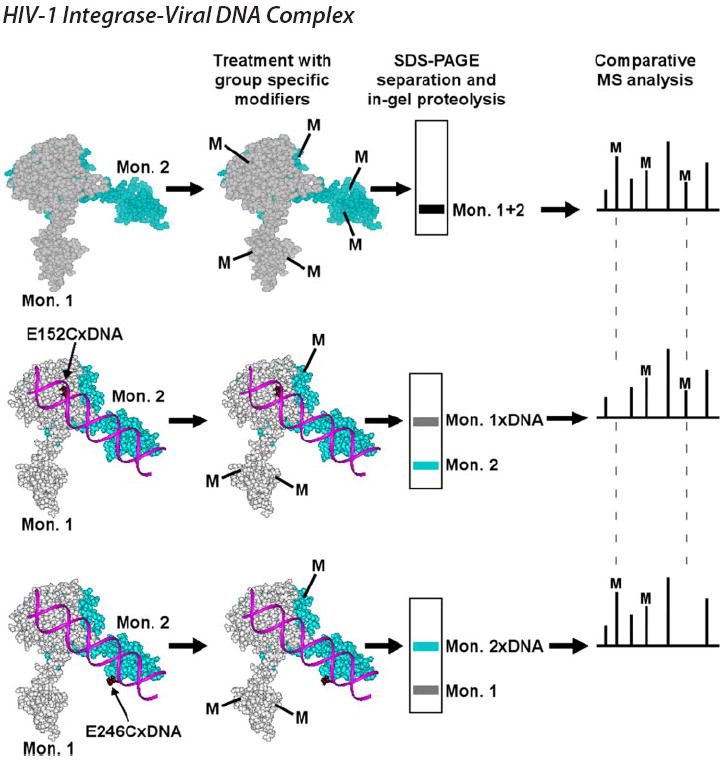
Free IN (space-filling model of the two domain CCD/CTD structure is used for illustration), and the two cross-linked IN-DNA complexes are examined in parallel experiments. The two IN mutants each containing a reactive Cys residue in either the CCD (E152C) or CTD (E246C) are cross-linked to DNA substrates that contain modified nucleotides placed at respective positions. Free IN and the two complexes are subjected to treatment by small chemical modifiers (M). Surface residues in free IN and complexes are modified, but amino acids interacting with DNA are shielded from modification. Complexes and free IN monomers are separated by SDS-PAGE. Monomer-DNA complex bands are excised and subjected to in-gel proteolysis. Subsequent comparative MS analyses reveal modification patterns in free protein and individual monomers.
To form disulfide-bridged IN-DNA complexes, the alkanethiol tether was placed at selected sites in the viral DNA sequence. DNA oligonucleotides were prepared through synthetic incorporation of commercially available analogs of A, C, and G. Crystallographic and NMR studies have shown that placements of cross-linkable base analogs in double stranded DNA do not alter Watson-Crick base pairing or global nucleic acid structures (reviewed in Ref. 51). Furthermore, viral sequences containing the modified bases were effective IN substrates (Fig. 2A).
FIGURE 2.
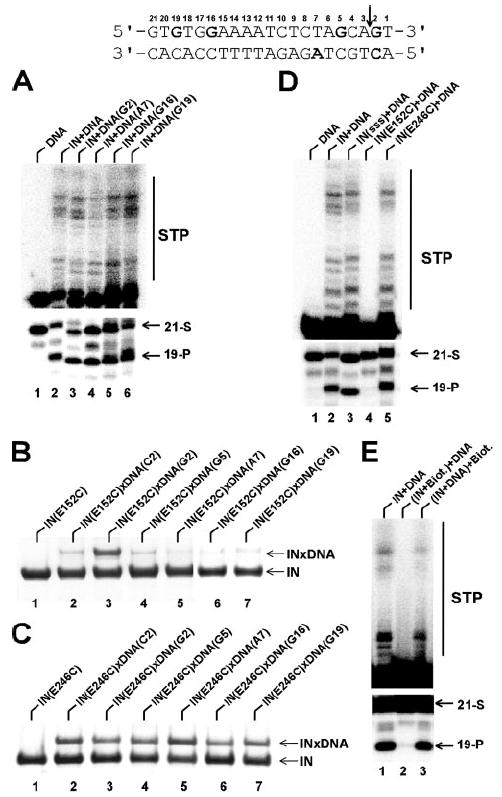
The upper panel shows the 21-mer U5 viral DNA end substrate used in our studies. The modified nucleotides are in bold and the IN cleavage site is indicated by a vertical arrow. A, wild type IN 3′-processing and strand transfer activities using native and thiol cross-linkable oligonucleotides. Lane 1, no IN control; lane 2, unmodified DNA sequence+IN; lane 3, DNA(G2)+IN; lane 4, DNA(A7)+IN; lane 5, DNA(G16)+IN; lane 6, DNA(G19)+IN. B, cross-linking efficacy of IN(E152C) to the following modified DNAs: 1) no DNA control, 2) DNA(C2), 3), DNA(G2), 4) DNA(G5), 5) DNA(A7), 6) DNA(G16), and 7) DNA(G19). C, cross-linking efficacy of IN(E246C) to the following modified DNAs: 1) no DNA control, 2) DNA(C2), 3), DNA(G2), 4) DNA(G5), 5) DNA(A7), 6) DNA(G16), and 7) DNA(G19). D, 3′-processing and strand transfer activities of the following proteins: lane 1, no IN control; lane 2, wild type IN; lane 3, the background triple (C56S, C65S, and C280S) mutant IN; lane 4, the IN(E152C) mutant; lane 5, the IN(E246C) mutant. Of note, while E152C(lane 4)andE246C(lane 5)mutationswhereintroducedinthebackground SSS sequence (lane 3), these proteins for simplicity are referred in the figure and text as IN(E152C) and IN(E246C). E, effects of the modifying reagent on IN function. Lane 1, reactions without any modifications; lane 2, IN was pretreated with NHS-biotin (IN/NHS-biotin ratio of 1/50) prior to DNA substrate addition. Lane 3, NHS-biotin added after IN-DNA complex formation. 21-mer substrate (21-S), 19-mer cleavage (19-P) and strand transfer (STP) products are indicated.
IN was prepared for cross-linking using a previously reported two-step procedure (35, 39). First, three surface Cys residues (Cys-56, Cys-65, and Cys-280) were replaced with Ser to rid nonspecific cross-linking between the wild type protein and modified DNA. Of the remaining three cysteines, Cys-40 and Cys-43 coordinate the structural Zn2+ ion, and Cys-130 is hindered from the protein surface (27, 28). Indeed, the triple mutant (C56S, C65S, and C280S) exhibited minimal reactivity with cross-linkable DNA (35). In the second step, the reactive Cys residue was introduced at a surface position predictive of specific nucleoprotein complex formation. Based on available mechanistic and structural studies, we chose E152C to tether IN to DNA modified at base G2 (numbering as in Fig. 2, upper panel). Glu-152 is the catalytic Glu of the DDE motif, and G2 is located at the scissile phosphodiester bond. Furthermore, the reactive SH group in the G analog points to the minor groove (51), stabilizing the IN catalytic site immediately adjacent to the scissile bond. To confirm specificity, binding of E152C protein to DNA modified at other positions was examined. The results reveal a striking preference for formation of the IN(E152C)xDNA(G2) complex (Fig. 2B). The reduced reactivity for position 2 in the lower strand could be explained by the fact that the SH group of the C-analog points to the major groove, away from the cleavage site. The observation that the other sites yield only residual cross-linking (Fig. 2B) indicates the specific nature of the IN(E152C)xDNA(G2) interaction.
Previous studies by Bushman and co-workers (39) identified IN(E246C) as a plausible CTD contact to DNA. Using TNB-thiotethered substrates, the authors observed robust but nonspecific cross-linking of IN(E246C) to various nucleotide positions. Further experiments with less reactive nucleotide analogs uncovered the relative preference of IN(E246C) for position A7 albeit significantly reduced formation of the nucleoprotein complex. To generate sufficient amounts of cross-linked products for our MS footprinting we used TNB-activated DNAs. In addition to IN(E246C) cross-linking to A7 (Fig. 2C, lane 5) we probed interactions with additional nucleotides including G16 and G19 located at the distal region of the DNA substrate (Fig. 2C, lanes 6 and 7). The data indicated comparable reactivities of IN(246C) with various cross-linkable nucleotides (Fig. 2C). While these nonspecific interactions were expected, the available crystal structure of the CCD-CTD fragment (27) allowed us to delineate functional from non-productive IN-DNA interactions. For example, based on the spatial separation between the CCD and CTD, we could predict that crosslinking IN(E246C) to nucleotides adjacent to the scissile bond (Fig. 2C, lanes 2 and 3) would position the CCD toward the opposite terminus of the DNA substrate. In contrast, in the functional complex the CCD would have to directly interact with the 3′-processing site, apposing the CTD to the distal regions of the DNA substrate. Therefore, for the footprinting studies we chose to examine IN(E246C) cross-links to positions A7, G16, and G19.
Of note, the triple (C56S, C65S, and C280S) background mutant and E246C proteins were catalytically active, indicating these mutations did not significantly alter IN folding or its interactions with viral DNA (Fig. 2D). As expected, replacement of active site residue E152 compromised IN activity, due to the inability of the substituent amino acid to coordinate the essential magnesium ion for catalysis. Therefore, the integrity of E152C IN was assessed as follows. First, E152C efficiently crosslinked with DNA(G2) (Fig. 2B, lane 3), indicating that its global structure is intact. Chemical modification and subsequent MS analyses importantly revealed very similar surface topologies for the wild type and E152C INs. Furthermore, the two proteins yielded overlapping CD spectra. This collection of data supports the notion that substituting Cys for Glu-152 does not overtly alter IN structure.
The modification reactions were carried out under mild conditions such that the integrity of the nucleoprotein complex was preserved. For this, effects of increasing concentration of NHS-biotin and HPG modifiers on wild type IN activities were examined. Representative data in Fig. 2E show that treatment of free IN with NHS-biotin using the 1:50 ratio of protein/reagent inactivated IN function, probably due to modification of essential lysine residues in the vicinity of the enzyme active site (32). To ensure that this was the case as compared with gross disruption of IN structure, we subjected the pre-assembled IN-DNA complex to the same modification. The result was a remarkable retention of original IN activity, indicating that under these conditions, the integrity of the functional complex is preserved (Fig. 2E, lane 3). The optimal IN/HPG ratio was analogously defined as 1:1000. Importantly, centrifugation assays (15,800 × g for 1 h) indicated that the crosslinked complexes were fully soluble (data not shown).
The two nucleoprotein complexes IN(E152C)xDNA(G2) and IN(E246C)xDNA(A7) were subjected to MS footprinting analysis according to the scheme depicted in Fig. 1. Representative segments of the mass spectra are depicted in Fig. 3. C1, C2, and C3 peaks are unmodified IN peptides that provide internal controls for accurate quantitation of modified peptides. The peptide peak 265–269 containing biotinylated Lys-266 is another control, as this lysine is equally exposed in the free protein and IN-DNA complexes (Fig. 3A). Fig. 3B reveals monomer selective protection of Lys-160. This residue is readily modified in free IN. However, upon formation of the IN(E152C)xDNA(G2) complex, Lys-160 is shielded from modification. In contrast, the residue remains surface-exposed in the IN(E246C)xDNA(A7) complex. A converse picture is observed for CTD residue Lys-264 (Fig. 3C). The intensity of the modified peak persists in the IN(E152C)xDNA(G2) complex, but was significantly diminished in the IN(E246C)xDNA(A7) complex. Taken together, these results alongside the summary in Table 1 demonstrate that separate monomers differentially contact substrate DNA. DNA cross-linked via E152C specifically protected Lys-14, Lys-159, Lys-160, and Arg-199, whereas residues Lys-219, Lys-244, Arg-263, Lys-264, and Arg-269 were footprinted only via the E246C link. The protection patterns of E246C with positions A7, G16, and G19 were very similar.
FIGURE 3. Representative segments of MALDI-ToF data.
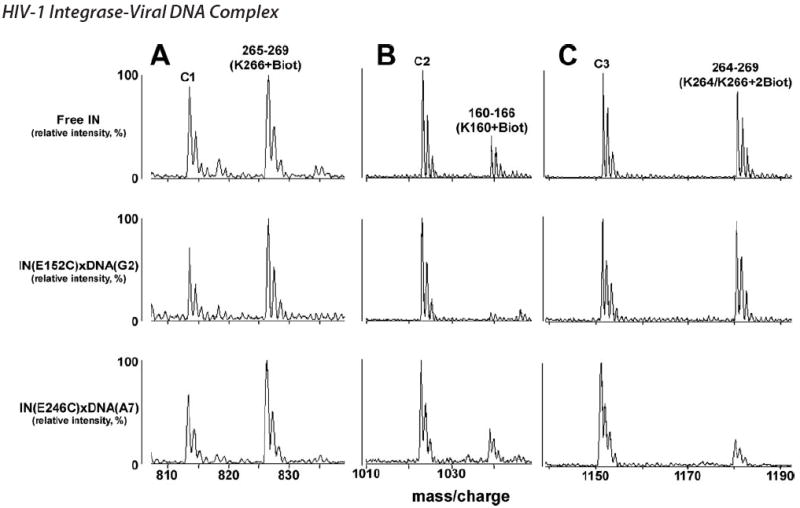
Free IN, IN(E152C)xDNA(G2), and IN(E246C)xDNA(A7) complexes were treated with NHS-biotin (A–C). IN peptides as well as the affected residues are indicated. Peaks C1, C2, and C3 are unmodified tryptic peptide peaks of IN, which serve as internal controls.
TABLE 1. Susceptibility of IN basic residues to modification.
Amino acids that become shielded from modifiers in the nucleoprotein complex are highlighted.
| Residue | Free IN | IN(E152C)xDNA(G2) | IN(E246C)xDNA(A7)a |
|---|---|---|---|
| K14 | +b | -c | + |
| K111 | + | + | + |
| K159/K160 | + | - | + |
| K160 | + | - | + |
| R166 | + | + | + |
| K188 | + | + | + |
| R199 | + | - | +++d |
| K211 | + | + | + |
| K219 | + | + | - |
| R224 | + | + | + |
| R228 | + | + | + |
| K244 | + | + | - |
| R262 | + | + | + |
| R262/R263 | + | + | - |
| K264/K266 | + | + | - |
| K266 | + | + | + |
| R269 | + | + | - |
| K273 | + | + | + |
IN(E246C) complexes with DNA(A7), DNA(G16), and DNA(G19) yielded very similar protection patterns.
Surface-exposed residues readily modified.
Amino acids inaccessible to modification.
Increased reactivity (see Fig. 4).
Interestingly, a unique modification pattern was observed for Arg-199 (Fig. 4). This residue was only marginally modified in the context of free IN. The reactivity was further reduced in the IN(E152C)xDNA(G2) complex. Of note, we observed a remarkable increase in the extent of Arg-199 modification within the IN(E246C)xDNA(A7) complex (Fig. 4C), indicating that subunit-specific DNA binding significantly exposes this amino acid to the protein surface. Arg-199 was also more reactive in the IN(E246C)xDNA(G16) and IN(E246C)xDNA(G19) complexes as compared with the free protein. However, a pattern could be noticed wherein the extent of Arg-199 modification decreased as the cross-linking points were further distanced from the cleavable DNA end (compare Fig. 4, C–E).
FIGURE 4. Representative segments of MALDI-ToF data.
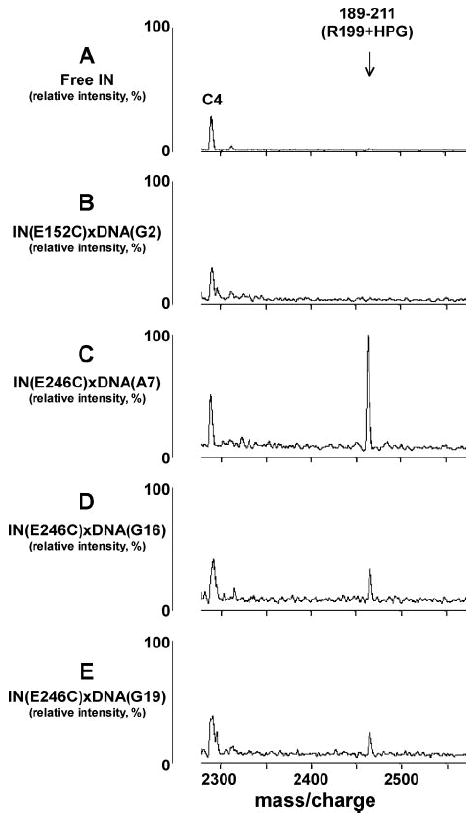
Free IN (A), IN(E152C)xDNA(G2) (B), IN(E246C)xDNA(A7) (C), IN(E246C)xDNA(G16) (D), and IN(E246C)xDNA(G19) (E) complexes were treated with HPG in parallel reactions. The amino acid 189 –211 peptide containing HPG-modified Arg-199 is indicated. Peak C4 is an unmodified tryptic peptide of IN, which served as an internal control.
To address the specificity of the IN-DNA interactions, E152C was cross-linked with nonspecific double-stranded and single-stranded DNA. The nonspecific double strand did not yield sufficient cross-linked species for quantitative MS analysis. These results together with the data depicted in Fig. 2B indicate that in nonspecific IN-DNA complexes, E152C is not positioned close enough to establish disulfide cross-links with the DNA. The single-stranded oligonucleotide resulted in higher yields of cross-linked product than its double-stranded counterpart, but constituted only ~25% of the specific IN(E152C)xDNA(G2) complex (data not shown). MS comparison of IN(E152C)-ssDNA and IN(E152C)xDNA(G2) products indicated sharp differences in protein-DNA contacts within the two complexes. The residues protected in the latter complex remained surface exposed in the former species. Representative data showing differential accessibilities of Lys-14 in the two complexes are depicted in Fig. 5. These results suggest that detectable cross-linking between E152C and ssDNA was due probably to the highly flexible nature of the oligonucleotide, rather than its specific binding to IN.
FIGURE 5. Representative MALDI-TOF spectra showing specific protection of Lys-14.
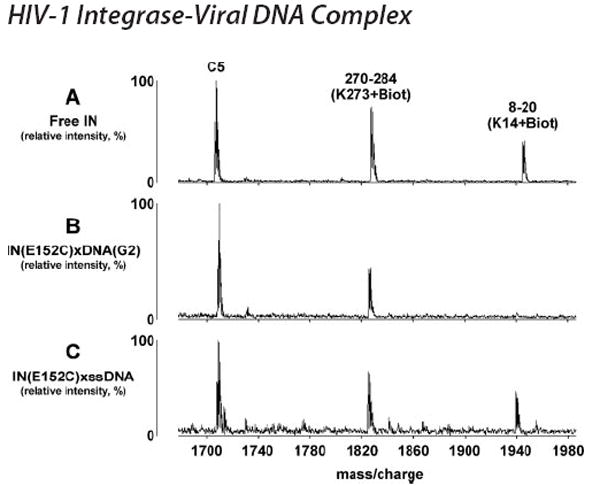
In parallel experiments IN(E152C)xDNA(G2) and IN(E152C)-ssDNA complexes were subjected to NHS-biotin treatment. The presented spectra indicate that Lys-273 remained surface assessable in both complexes, while Lys-14 was specifically protected in IN(E152C)xDNA(G2) but not IN(E152C)-ssDNA. C5 is an unmodified tryptic peptide of IN, which served as an internal control.
We next used site-directed mutagenesis to confirm the functional importance of the identified amino acids. In fact, several interacting residues revealed from our footprinting results have previously been examined by mutagenesis due to their high degree of conservation among various HIV strains, and were shown to be essential for recombinant IN function and HIV-1 replication (32, 50, 52). Therefore, our efforts focused on analyzing two novel contacts: Lys-14, the first NTD residue to be implicated in viral DNA binding, and Arg-199, involved in the protein conformational change in one subunit while interacting with DNA in the other monomer (Table 1). The substitution of Ala or Glu was engineered at each position within the wild type IN background. MS surface mapping and CD spectroscopy revealed proper folding of the recombinant mutant proteins. The results in Fig. 6 show that the K14A/E and R199A/E mutants exhibited residual levels of specific activity (3′-processing and DNA strand transfer) and generated nonspecific (20-mer) cleavage products. Interestingly, the same phenomenon was observed with mutations of certain IN residues (for example Gln-148) implicated in specific recognition of the viral DNA substrate (35). An alternative scenario that our preparations could be contaminated with Escherichia coli nucleases is less tenable as the D116N active site mutant purified according to the identical procedure did not generate the 20-mer product (Fig. 6, lane 7). Also noteworthy is that the Ala mutants displayed significantly reduced specific activity, while the Glu mutations yielded more pronounced affects (Fig. 6, compare lane 4 to lane 3 and lane 6 to lane 5). Neutralizing the positive charge that is needed to stabilize DNA at the specific site by Ala substitutions could reduce binding affinity, while the placement of Glu may result in more efficient displacement of the nucleic acid from its specific site due to electrostatic repulsions.
FIGURE 6. Effects of amino acid substitutions on recombinant IN activities.
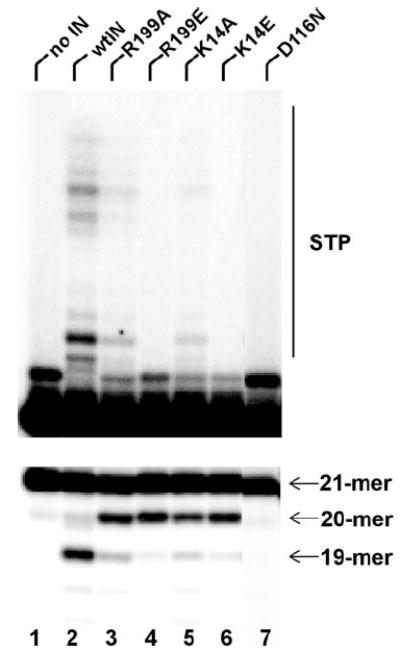
The upper image depicts strand transfer activities. Positions of 21-mer substrate and reaction products (STP) are indicated. Lower image displays 3′-processing activities. The positions of 21-mer substrate, and specific (19-mer) and nonspecific (20-mer) products are shown.
Each of the four mutations also significantly impacted the infectivity of the virus. The R199A mutant retained ~0.24% of wild-type HIV-1NL4–3 function in a single-round infection, whereas the other viruses failed to yield detectable infectivities (Table 2).
TABLE 2. Infectivities of IN mutant viruses.
Percent wild-type infectivity (2.7 × 105 relative light units/μg total cell protein using 4 × 107 RT-cpm of HIV-1NLX. Luc. R- and 5 × 106 SupT1 cells) of three independent infections with standard error. V165A and D64N/D116N were included as prototypical class II and class I IN mutant viruses, respectively (50).
| Mutant | Infectivity |
|---|---|
| V165A | 0.01 ± 0.01 |
| K14A | < 0.01 |
| K14E | < 0.01 |
| R199A | 0.24 ± 0.13 |
| R199E | < 0.01 |
| D64N/D116N | 0.05 ± 0.02 |
Our experimental results were used to dissect the viral DNA binding channel in the functional IN nucleoprotein complex. For this, a full-length IN model was created by superimposing the two available crystal structures: NTD/CCD and CCD/CTD (27, 28). Then the residues identified by our footprinting analysis were located on the IN monomers (Fig. 7). The assignment of the amino acids to the respective IN subunits was critical to visualize a plausible DNA binding channel. The DNA substrate was readily positioned along the path of the basic residues (Fig. 7) with all three protein domains providing direct contacts to nucleic acid.
FIGURE 7. Dissecting a viral DNA binding channel in full-length IN.
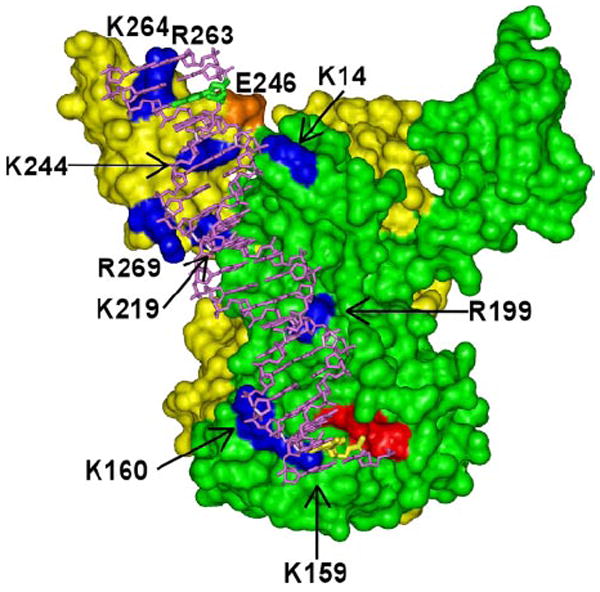
The molecular model of full-length HIV-1 IN was generated by overlaying the CCDs within PDB structures 1EX4 (27) and 1K6Y (28) using Insight II software package (Accelrys). Subunit-specific protected residues within their respective IN monomers (yellow and green) are highlighted in blue. One viral DNA end is readily positioned over the channel revealed by the basic contacts. The three acidic active site (DDE) residues including Glu-152 that coordinate catalytic Mg2+ions are in red, the DNA substrate is in magenta, the G2 nucleotide is in yellow, the G19 nucleotide is in green, and the CTD residue Glu-246 is in orange.
DISCUSSION
We have investigated interactions between full-length HIV-1 IN and its viral DNA substrate using a novel experimental strategy combining two established methodologies: site-specific protein-DNA cross-linking through disulfide bridging, and MS protein footprinting. The cross-linking technology has been successfully used in combination with x-ray crystallography to obtain atomic structures of a number of nucleoprotein complexes including the covalently trapped complex of RT with a DNA primer-template and a deoxynucleoside triphosphate (51, 53). Analogous efforts for the HIV-1 IN-DNA complex have so far been hampered by poor protein solubility and/or full-length enzyme flexibility. Combining site-specific cross-linking and MS footprinting offered a powerful alternative and led us to the following important findings. Asymmetric contacts between IN and viral DNA were identified, which provided major constraints to dissect the viral DNA binding channel in the functional nucleoprotein complex. In addition, our experiments uncovered a subunit-specific conformational change induced upon DNA binding.
Our work was based on and expanded previous optimization of IN-DNA cross-linking through disulfide bridging (39). We complemented the cross-linking approach with MS surface topology analysis of basic amino acid residues. Our results reveal the importance of these technologies for identification of asymmetric contacts between IN and viral DNA. While the observed protections near the cross-linking points were expected, the method also revealed subunit selective contacts distant from the disulfide linkages. For example, Lys-14 within the NTD was protected when CCD residue E152C was cross-linked to DNA(G2). In addition, increased reactivity rather than protection of Arg-199 was observed in the IN(E246C) complexes with DNA substrates. Site-specific cross-linking was also essential to delineate specific from nonspecific IN-DNA interactions. IN-DNA binding could yield a mixture of active and non-productive complexes. However, functional site-specific modification of IN and DNA enabled us to covalently link and separate specific complexes from non-productive IN-DNA interactions, the latter of which dissociated under denaturing SDS-PAGE (Fig. 2B). Although our method does not distinguish whether the observed amino acid protections are due to direct protein-nucleic acid or protein-protein contacts, the protections nonetheless strictly depend on the presence of the specific HIV-1 DNA substrate in the reaction mixture. The surface basic residues identified in this study are moreover likely to engage in electrostatic interactions with the DNA substrate. Indeed, all the interacting amino acids align in a plausible DNA binding channel (Fig. 7).
Our results for the first time implicate the NTD in direct contact with viral DNA. Previous studies revealed the importance of the NTD for IN catalytic activities via testing mixtures of different protein domains in functional trans-complementation assays (29-31). This approach, however, fell short of demonstrating direct NTD-viral substrate interactions. We in contrast observed specific protection of NTD residue Lys-14 in the IN(E152C)xDNA(G2) complex, but importantly not with the nonspecific IN(E152C)-ssDNA complex (Fig. 5) nor IN(E246C) complexes (Table 1). Other studies reported that the N-terminal 11 residues of HIV-1 IN cross-linked to the target DNA component rather than viral sequences in a dumbbell disintegration substrate (36, 37). Given that the authors examined a limited number of cross-linkable sites in the viral DNA, the contribution by Lys-14 could easily have been missed. In contrast, the Lys and Arg modifying reagents employed here provide a significantly more detailed picture of individual amino acid residues. The alternative scenario that Lys-14 became protected at a point in the reaction pathway that lay significantly downstream of IN-viral DNA complex formation for example during assembly of the DNA strand transfer complex could be ruled out for the following reasons. While our in vitro assays could yield strand transfer reaction products after 3′-processing of the blunt-ended 21-mer substrate, our crosslinking and modification reactions were performed for relatively short periods of time under which no 3′-processing or strand transfer products were detected (data not shown). In addition, the IN(E152C)xDNA(G2) complex, in which Lys-14 was protected from modification, was catalytically inactive. Therefore, our findings suggest that Lys-14 plays an important role in interacting with viral DNA. This conclusion is further supported by the site-directed mutagenesis studies, which showed that substituting Lys-14 with Ala or Glu compromised both 3′-processing and strand transfer activities in vitro and abrogated HIV-1 infectivity in cell culture. Interestingly, the recombinant mutant proteins generated -1 cleavage products (Fig. 6), a phenotype reminiscent of the pattern observed with mutations of certain IN residues involved in selective interactions with viral DNA (35). Fig. 7 indicates that Lys-14 is positioned in the viral DNA binding channel, where it could indeed contribute to cognate DNA recognition.
Another key finding of this study is a DNA-induced protein conformational change involving the extended alpha helix that connects the CCD to the CTD. In a previous study, Asante-Appiah and Skalka identified a metal induced reorganization of HIV-1 IN structure, which affected the recognition of the CCD and CTD, but not the NTD, by domain selective antibodies (54). Bushman and co-workers (39) observed differential cross-linking of CTD residues with blunt ended and processed DNA substrates, suggesting protein structural changes upon cleavage of the viral DNA terminus. Roth and co-workers (55) found that a 19-amino acid sequence insertion at E212, located on the helix connecting the CCD and CTD, was tolerated by IN as this large structural change did not significantly impact enzyme function. Our observation of the increased reactivity of Arg-199 in the IN-viral DNA complex (Fig. 4) provides important clues regarding the nature of protein structural changes. Arg-199 is located on the ridged helix (amino acids 196–221) bridging the CCD to the CTD (see supplemental Fig. S1). The helix is flanked by a flexible loop (amino acids 187–195) and the highly basic CTD on its N and C termini, respectively. Such structural organization enables the flexibility of the loop to be translated to and exploited by the CTD upon formation of the functional nucleoprotein complex.
Our interpretation of the stepwise decrease in Arg-199 reactivity within the IN(E246C)xDNA(G16) and IN(E246C)xDNA(G19) complexes as compared with IN(E246C)xDNA(A7) is the following. Linking the CTD on A7 with the CCD concomitantly committed to the scissile bond at the 3′-end of the cleaved strand would require a significant conformational change in the protein. Accordingly, the CTD accommodated distal G16 and G19 sites with less overall structural rearrangements (Fig. 4). This observation reflects an extraordinary degree of flexibility within this region of HIV-1 IN. The residual reactivity of R199 in the IN(E246C)xDNA(G16) and IN(E246C)xDNA(G19) complexes could reflect relatively minor adjustments of the CTD to bound DNA. We would argue that the CTD interacts with the energetically more favorable distal regions of the DNA substrate. In Fig. 7, G19 is located nearby protected Lys-244 and crosslinkable Glu-246 residues. However, the binding site of the CTD can apparently vary. Unlike the CCD that needs to precisely position over the cleavable scissile bond, the CTD is highly flexible and thus engages in nonspecific contacts with DNA in vitro (Fig. 2C). In this respect it is noteworthy that insertion of 19 amino acids at the CCD-CTD connection, which could have very well altered the positioning of the CTD on viral DNA, did not significantly affect IN activity (55). We propose that this reflects an inherent flexibility of the CCD-CTD connector loop, which could be essential for the productive assembly and stability of the nucleoprotein complex.
Why does viral DNA bind to only one surface of the IN multimer while the opposite side of the protein remains unaligned? There are at least the following two factors to consider. The crystal structure of the CCD-CTD fragment indicates that the extended α-helix (amino acids 196–221) is a canonical cylinder in one monomer and is bent by ~45 degrees at Gln-209 in the second monomer, thus rendering the IN dimer asymmetric ((27), also see supplemental Fig. S1). Our findings indicate that upon DNA binding only one of the two monomers undergoes a conformational change. Thus, the DNA bound and surface exposed sites within the IN multimer differ significantly. We suggest that the conformational change together with the kink observed in the crystal structure have biological relevance and are important for the assembly of the functional nucleoprotein complex.
IN dimers have been shown to bind each viral DNA end and catalyze the 3′-processing reactions (56, 57). Following GT dinucleotide removal IN remains stably associated with cognate DNA (56) and the two viral DNA-bound dimers presumably form a tetramer to carry out the integration of both viral DNA ends, as occurs during infection (58). Fig. 7 depicts the IN dimer bound to one viral DNA end. Using this model we were able to assemble the synaptic complex (supplemental Fig. S2), where two NTDs, two CCDs, and four CTDs within the IN tetramer complement each other to coordinate the two viral DNA substrates. The prediction that either of two CTDs within the tetramer can bind a single viral DNA end is fully consistent with the promiscuous behavior of this domain in functional trans-complementation assays (29, 30). The model depicted in supplemental Fig. S2 is however one possible scenario, and further experimental data elucidating protein-protein interfaces are needed to validate higher order interactions at play during the concerted integration of both viral DNA ends. Conditions for effective full-site integration by recombinant IN and two-dimensional gel electrophoretic isolation of synaptic complexes have been reported (58, 59). These techniques can be combined with MS surface topology analyses to better understand the protein-protein interactions essential for the two-ended integration reaction. It will also be intriguing to apply our footprinting approach to study effects of physiologically relevant cell factors like LEDGF/p75 on IN-DNA interactions. Our identification of a restricted, single viral end-specific DNA binding channel by the combined use of subunit-specific protein-DNA cross-linking and MS footprinting is an important step toward these long-term goals. In addition, the new experimental strategy reported here can be employed by other research groups to analyze a large variety of nucleoprotein complexes.
The new findings reported here also have implications for exploiting the IN-viral DNA structure as a therapeutic target. For example, the most promising diketo class of inhibitors are known to specifically interact with the pre-assembled IN-viral DNA complex and impair the DNA strand transfer activity (1, 2, 60). More recently, IN-DNA interfacial quinolonyl diketo acid derivatives, which potently inhibit both 3′-processing and strand transfer, were reported (61). The novel IN-viral DNA contacts uncovered herein could help to elucidate the mechanisms of these known compounds and facilitate rational evolution of new potent inhibitors.
Acknowledgments
We thank Robert Craigie for critical reading of the manuscript. We thank Allison Johnson and Yves Pommier for sharing their experiences with IN-DNA cross-linking experiments.
Footnotes
This work was supported in part by NIAID, National Institutes of Health Grants AI062520 (to M. K.) and AI039394 (to A. E.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
The abbreviations used are: IN, integrase; NTD, N-terminal domain; CCD, catalytic core domain; CTD, C-terminal domain; RT, reverse transcriptase; MS, mass spectrometry; HPG, p-hydroxyphenylglyoxal; PIPES, 1,4-pipera-zinediethanesulfonic acid; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M705241200/DC1
References
- 1.Espeseth AS, Felock P, Wolfe A, Witmer M, Grobler J, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ. Proc Natl Acad Sci U S A. 2000;97:11244–11249. doi: 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grobler JA, Stillmock K, Hu B, Witmer M, Felock P, Espeseth AS, Wolfe A, Egbertson M, Bourgeois M, Melamed J, Wai JS, Young S, Vacca J, Hazuda DJ. Proc Natl Acad Sci U S A. 2002;99:6661–6666. doi: 10.1073/pnas.092056199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Science. 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 4.Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang L, Fisher TE, Embrey M, Guare JP, Jr, Egbertson MS, Vacca JP, Huff JR, Felock PJ, Witmer MV, Stillmock KA, Danovich R, Grobler J, Miller MD, Espeseth AS, Jin L, Chen IW, Lin JH, Kassahun K, Ellis JD, Wong BK, Xu W, Pearson PG, Schleif WA, Cortese R, Emini E, Summa V, Holloway MK, Young SD. Proc Natl Acad Sci U S A. 2004;101:11233–11238. doi: 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown PO. In: Retroviruses. Coffin JM, Hughes SH, Varmus HE, editors. Cold Spring Harbor Laboratory; Plain-view, NY: 1997. pp. 161–204. [PubMed] [Google Scholar]
- 6.Engelman A, Mizuuchi K, Craigie R. Cell. 1991;67:1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- 7.Llano M, Vanegas M, Fregoso O, Saenz D, Chung S, Peretz M, Poeschla EM. J Virol. 2004;78:9524–9537. doi: 10.1128/JVI.78.17.9524-9537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llano M, Saenz DT, Meehan A, Wongthida P, Peretz M, Walker WH, Teo W, Poeschla EM. Science. 2006;314:461–464. doi: 10.1126/science.1132319. [DOI] [PubMed] [Google Scholar]
- 9.Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. PLoS Pathog. 2006;2:e60. doi: 10.1371/journal.ppat.0020060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farnet CM, Haseltine WA. Proc Natl Acad Sci U S A. 1990;87:4164–4168. doi: 10.1073/pnas.87.11.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukrinsky MI, Sharova N, McDonald TL, Pushkarskaya T, Tarpley WG, Stevenson M. Proc Natl Acad Sci U S A. 1993;90:6125–6129. doi: 10.1073/pnas.90.13.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buckman JS, Bosche WJ, Gorelick RJ. J Virol. 2003;77:1469–1480. doi: 10.1128/JVI.77.2.1469-1480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carteau S, Batson SC, Poljak L, Mouscadet JF, de Rocquigny H, Darlix JL, Roques BP, Kas E, Auclair C. J Virol. 1997;71:6225–6229. doi: 10.1128/jvi.71.8.6225-6229.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carteau S, Gorelick RJ, Bushman FD. J Virol. 1999;73:6670–6679. doi: 10.1128/jvi.73.8.6670-6679.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Engelman A. Proc Natl Acad Sci U S A. 1998;95:15270–15274. doi: 10.1073/pnas.95.26.15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MS, Craigie R. Proc Natl Acad Sci U S A. 1994;91:9823–9827. doi: 10.1073/pnas.91.21.9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee MS, Craigie R. Proc Natl Acad Sci U S A. 1998;95:1528–1533. doi: 10.1073/pnas.95.4.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cherepanov P, Maertens G, Proost P, Devreese B, Van Beeumen J, Engelborghs Y, De Clercq E, Debyser Z. J Biol Chem. 2003;278:372–381. doi: 10.1074/jbc.M209278200. [DOI] [PubMed] [Google Scholar]
- 19.Cherepanov P, Sun ZY, Rahman S, Maertens G, Wagner G, Engelman A. Nat Struct Mol Biol. 2005;12:526–532. doi: 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- 20.Shun MC, Raghavendra NK, Vandegraaff N, Daigle JE, Hughes S, Kellam P, Cherepanov P, Engelman A. Genes Dev. 2007;21:1767–1778. doi: 10.1101/gad.1565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engelman A. Future HIV Ther. 2007;1:415–426. [Google Scholar]
- 22.Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM. Nat Struct Biol. 1997;4:567–577. doi: 10.1038/nsb0797-567. [DOI] [PubMed] [Google Scholar]
- 23.Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. Science. 1994;266:1981–1986. doi: 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- 24.Goldgur Y, Dyda F, Hickman AB, Jenkins TM, Craigie R, Davies DR. Proc Natl Acad Sci U S A. 1998;95:9150–9154. doi: 10.1073/pnas.95.16.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eijkelenboom AP, Lutzke RA, Boelens R, Plasterk RH, Kaptein R, Hard K. Nat Struct Biol. 1995;2:807–810. doi: 10.1038/nsb0995-807. [DOI] [PubMed] [Google Scholar]
- 26.Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, Gronenborn AM. Biochemistry. 1995;34:9826–9833. doi: 10.1021/bi00031a002. [DOI] [PubMed] [Google Scholar]
- 27.Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. Proc Natl Acad Sci U S A. 2000;97:8233–8238. doi: 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang JY, Ling H, Yang W, Craigie R. EMBO J. 2001;20:7333–7343. doi: 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Gent DC, Vink C, Groeneger AA, Plasterk RH. EMBO J. 1993;12:3261–3267. doi: 10.1002/j.1460-2075.1993.tb05995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engelman A, Bushman FD, Craigie R. EMBO J. 1993;12:3269–3275. doi: 10.1002/j.1460-2075.1993.tb05996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van den Ent FM, Vos A, Plasterk RH. J Virol. 1999;73:3176–3183. doi: 10.1128/jvi.73.4.3176-3183.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins TM, Esposito D, Engelman A, Craigie R. EMBO J. 1997;16:6849–6859. doi: 10.1093/emboj/16.22.6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esposito D, Craigie R. EMBO J. 1998;17:5832–5843. doi: 10.1093/emboj/17.19.5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazumder A, Neamati N, Pilon AA, Sunder S, Pommier Y. J Biol Chem. 1996;271:27330–27338. doi: 10.1074/jbc.271.44.27330. [DOI] [PubMed] [Google Scholar]
- 35.Johnson AA, Santos W, Pais GC, Marchand C, Amin R, Burke TR, Jr, Verdine G, Pommier Y. J Biol Chem. 2006;281:461–467. doi: 10.1074/jbc.M511348200. [DOI] [PubMed] [Google Scholar]
- 36.Heuer TS, Brown PO. Biochemistry. 1997;36:10655–10665. doi: 10.1021/bi970782h. [DOI] [PubMed] [Google Scholar]
- 37.Heuer TS, Brown PO. Biochemistry. 1998;37:6667–6678. doi: 10.1021/bi972949c. [DOI] [PubMed] [Google Scholar]
- 38.De Luca L, Vistoli G, Pedretti A, Barreca ML, Chimirri A. Biochem Biophys Res Commun. 2005;336:1010–1016. doi: 10.1016/j.bbrc.2005.08.211. [DOI] [PubMed] [Google Scholar]
- 39.Gao K, Butler SL, Bushman F. EMBO J. 2001;20:3565–3576. doi: 10.1093/emboj/20.13.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karki RG, Tang Y, Burke TR, Jr, Nicklaus MC. J Comput Aided Mol Des. 2004;18:739–760. doi: 10.1007/s10822-005-0365-5. [DOI] [PubMed] [Google Scholar]
- 41.Chen A, Weber IT, Harrison RW, Leis J. J Biol Chem. 2006;281:4173–4182. doi: 10.1074/jbc.M510628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podtelezhnikov AA, Gao K, Bushman FD, McCammon JA. Biopolymers. 2003;68:110–120. doi: 10.1002/bip.10217. [DOI] [PubMed] [Google Scholar]
- 43.Wielens J, Crosby IT, Chalmers DK. J Comput Aided Mol Des. 2005;19:301–317. doi: 10.1007/s10822-005-5256-2. [DOI] [PubMed] [Google Scholar]
- 44.Kvaratskhelia M, Miller JT, Budihas SR, Pannell LK, Le Grice SF. Proc Natl Acad Sci U S A. 2002;99:15988–15993. doi: 10.1073/pnas.252550199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Kvaratskhelia M, Hess S, Qu Y, Zou Y. J Biol Chem. 2005;280:32775–32783. doi: 10.1074/jbc.M505705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shell SM, Hess S, Kvaratskhelia M, Zou Y. Biochemistry. 2005;44:971–978. doi: 10.1021/bi048208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shkriabai N, Datta SA, Zhao Z, Hess S, Rein A, Kvaratskhelia M. Biochemistry. 2006;45:4077–4083. doi: 10.1021/bi052308e. [DOI] [PubMed] [Google Scholar]
- 48.Datta SA, Zhao Z, Clark PK, Tarasov S, Alexandratos JN, Campbell SJ, Kvaratskhelia M, Lebowitz J, Rein A. J Mol Biol. 2007;365:799–811. doi: 10.1016/j.jmb.2006.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchand C, Krajewski K, Lee HF, Antony S, Johnson AA, Amin R, Roller P, Kvaratskhelia M, Pommier Y. Nucleic Acids Res. 2006;34:5157–5165. doi: 10.1093/nar/gkl667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu R, Limon A, Devroe E, Silver PA, Cherepanov P, Engelman A. J Virol. 2004;78:12735–12746. doi: 10.1128/JVI.78.23.12735-12746.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verdine GL, Norman DP. Annu Rev Biochem. 2003;72:337–366. doi: 10.1146/annurev.biochem.72.121801.161447. [DOI] [PubMed] [Google Scholar]
- 52.Lu R, Limon A, Ghory HZ, Engelman A. J Virol. 2005;79:2493–2505. doi: 10.1128/JVI.79.4.2493-2505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang H, Chopra R, Verdine GL, Harrison SC. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 54.Asante-Appiah E, Skalka AM. J Biol Chem. 1997;272:16196–16205. doi: 10.1074/jbc.272.26.16196. [DOI] [PubMed] [Google Scholar]
- 55.Puglia J, Wang T, Smith-Snyder C, Cote M, Scher M, Pelletier JN, John S, Jonsson CB, Roth MJ. J Virol. 2006;80:9497–9510. doi: 10.1128/JVI.00856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guiot E, Carayon K, Delelis O, Simon F, Tauc P, Zubin E, Gottikh M, Mouscadet JF, Brochon JC, Deprez E. J Biol Chem. 2006;281:22707–22719. doi: 10.1074/jbc.M602198200. [DOI] [PubMed] [Google Scholar]
- 57.Hayouka Z, Rosenbluh J, Levin A, Loya S, Lebendiker M, Veprintsev D, Kotler M, Hizi A, Loyter A, Friedler A. Proc Natl Acad Sci U S A. 2007;104:8316–8321. doi: 10.1073/pnas.0700781104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li M, Mizuuchi M, Burke TR, Jr, Craigie R. EMBO J. 2006;25:1295–1304. doi: 10.1038/sj.emboj.7601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinha S, Pursley MH, Grandgenett DP. J Virol. 2002;76:3105–3113. doi: 10.1128/JVI.76.7.3105-3113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pommier Y, Johnson AA, Marchand C. Nat Rev Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 61.Di Santo R, Costi R, Roux A, Artico M, Lavecchia A, Marinelli L, Novellino E, Palmisano L, Andreotti M, Amici R, Galluzzo CM, Nencioni L, Palamara AT, Pommier Y, Marchand C. J Med Chem. 2006;49:1939–1945. doi: 10.1021/jm0511583. [DOI] [PMC free article] [PubMed] [Google Scholar]