

Accumulation of autophagosomes because of impaired autophagy during valosin-containing protein (VCP)–linked dementia is explained by the absence or reduced activity of VCP.
Abstract
Mutations in valosin-containing protein (VCP) cause inclusion body myopathy (IBM), Paget's disease of the bone, and frontotemporal dementia (IBMPFD). Patient muscle has degenerating fibers, rimmed vacuoles (RVs), and sarcoplasmic inclusions containing ubiquitin and TDP-43 (TARDNA-binding protein 43). In this study, we find that IBMPFD muscle also accumulates autophagosome-associated proteins, Map1-LC3 (LC3), and p62/sequestosome, which localize to RVs. To test whether VCP participates in autophagy, we silenced VCP or expressed adenosine triphosphatase–inactive VCP. Under basal conditions, loss of VCP activity results in autophagosome accumulation. After autophagic induction, these autophagosomes fail to mature into autolysosomes and degrade LC3. Similarly, IBMPFD mutant VCP expression in cells and animals leads to the accumulation of nondegradative autophagosomes that coalesce at RVs and fail to degrade aggregated proteins. Interestingly, TDP-43 accumulates in the cytosol upon autophagic inhibition, similar to that seen after IBMPFD mutant expression. These data implicate VCP in autophagy and suggest that impaired autophagy explains the pathology seen in IBMPFD muscle, including TDP-43 accumulation.
Introduction
Valosin-containing protein (VCP) is a ubiquitously expressed protein belonging to the AAA+ (ATPases associated with various activities) protein family (Halawani and Latterich, 2006). VCP is implicated in multiple cellular processes, including cell cycle regulation, nuclear envelope formation, Golgi biogenesis, and the ubiquitin proteasome system (UPS; Halawani and Latterich, 2006). Of these functions, degradation of misfolded substrates synthesized through the secretory pathway is the best characterized (Ye et al., 2001). VCP in a complex with Ufd1 and Npl4 participates in the retrotranslocation of ER-associated degradation (ERAD) substrates (Ye et al., 2001). Loss of VCP activity leads to the accumulation of ubiquitinated proteins and impaired ERAD (Dalal et al., 2004; Wójcik et al., 2004). VCP also facilitates protein aggregate trafficking to an inclusion body. The absence of VCP disrupts aggresome formation and the degradation of expanded polyglutamine-containing proteins (Kobayashi et al., 2007).
Consistent with VCP's involvement in these processes, dominantly inherited mutations cause inclusion body myopathy (IBM), Paget's disease of the bone (PDB), and frontotemporal dementia (FTD [IBMPFD]), which is a rare multisystem degenerative disorder with three variably penetrant phenotypic features (Watts et al., 2004). 90% of patients develop disabling weakness by the fourth to fifth decade. At this same age, 51% of patients develop PDB. And 32% of patients develop FTD in the fifth to sixth decade. Other features in some families include cataracts, neuropathy, and cardiomyopathy, suggesting that IBMPFD pathogenesis may relate to other common age-associated diseases (Weihl et al., 2009).
Several shared pathological features exist in the disparate tissues affected in IBMPFD. Muscle and brain contain ubiquitin- and TDP-43 (TARDNA-binding protein 43)–positive inclusions (Schröder et al., 2005; Forman et al., 2006; Hübbers et al., 2007; Neumann et al., 2007; Weihl et al., 2008). In brain tissue, ubiquitinated and TDP-43 inclusions are intranuclear, classifying IBMPFD as a frontotemporal lobar degeneration with ubiquitinated inclusions (FTLD-U) subtype (Schröder et al., 2005; Forman et al., 2006). In muscle, ubiquitinated inclusions are myonuclear and sarcoplasmic (Guyant-Maréchal et al., 2006; Hübbers et al., 2007), and TDP-43 is diminished from the nuclei of inclusion-bearing myofibers (Weihl et al., 2008; Salajegheh et al., 2009). Why TDP-43 accumulates in the cytosol of IBMPFD patients is unknown, but it is consistent with the behavior of TDP-43 in other FTLD-Us and sporadic amyotrophic lateral sclerosis (Neumann et al., 2006).
Another striking pathology in IBMPFD muscle is sarcoplasmic vacuoles (Watts et al., 2004). These vacuoles have characteristics of rimmed vacuoles (RVs) found in other muscle degenerative disorders such as sporadic inclusion body myositis (sIBM) and hereditary IBM. The term RV is actually a misnomer because RVs are not lined by a limiting membrane and do not contain any specific cellular organelle contents. They are more appropriately described as an accumulation of membranous and proteinaceous debris within a myofiber. During tissue processing, this membranous accumulation is extracted, leaving an RV. Ultrastructural analysis demonstrates that RVs contain discontinuous membranous whorls admixed with tubulofilamentous and electron-dense material (Fernandez et al., 2005). The origin of RVs in sIBM and hereditary IBM is unclear but has been assumed to be autophagic.
How disease mutations in VCP affect its function is not fully understood. All described mutations reside within the N-terminal and D1 domains, which are regions proposed to be involved in substrate and cofactor association (Watts et al., 2004; Weihl et al., 2009). One initial study shows that IBMPFD mutants still bind to Ufd1 and Npl4, which are essential for UPS function (Hübbers et al., 2007). Biochemical characterization of several disease mutations finds that they form stable hexamers and have increased ATPase activity (Weihl et al., 2006; Halawani et al., 2009). The increased ATPase activity may reflect structural changes upon ATP binding (Halawani et al., 2009). Skeletal muscle–restricted expression of IBMPFD mutant VCP-R155H (VCP-RH) in transgenic mice recapitulates many of the features seen in IBMPFD muscle, including weakness, myopathic features, ubiquitinated inclusions, and vacuolation (Weihl et al., 2007). Expression of IBMPFD mutations in cell culture has shown mixed results. Some studies find an increase in ubiquitinated proteins (Weihl et al., 2006; Ju et al., 2008), whereas others identify minimal or no change in its levels (Hübbers et al., 2007; Gitcho et al., 2009). How well cell culture can effectively model IBMPFD may relate to cell type and protein expression level. ERAD and UPS substrates may also accumulate in IBMPFD mutant cells. The levels of the ERAD substrate ΔF508 cystic fibrosis transmembrane regulator (CFTR; Weihl et al., 2006) and UPS substrate Unc-45B (Janiesch et al., 2007) are increased in IBMPFD mutant cells, similar to that with an ATPase-inactive VCP or after proteasome inhibition (Dalal et al., 2004). Finally, the clearance of an aggregated polyglutamine is slowed in IBMPFD mutant cells (Ju et al., 2008). This was caused by impaired protein aggregate trafficking to an aggresome (Ju et al., 2008).
None of these previous findings explain all of the pathology seen in IBMPFD patient muscle. Whether VCP is involved in autophagy is not known. In this study, we evaluate autophagosome formation, maturation, and flux in cells and animals devoid of functional VCP or expressing IBMPFD mutant VCP. In addition, we assess the accumulation of cytosolic TDP-43 in IBMPFD mutant VCP–expressing cells and animals as well as under conditions of impaired autophagy.
Results
It is speculated that RVs are of autophagic origin. To see whether autophagic protein markers are elevated in IBMPFD muscle, we immunoblotted quadriceps muscle lysates from 9–12-mo-old control or VCP wild-type (WT)– or two different VCP-RH–expressing transgenic mouse lines (RH9 or RH12) using antibodies to p62 or LC3. There was an increase in p62 (Fig. 1 A) and specifically the autophagosome-associated cleavage product of LC3, LC3II, by approximately twofold in the RH9 and RH12 muscle (Fig. 1 B). The increase in p62 and LC3II levels is similar to that seen after the acute treatment of control mice with 50 mg/kg intraperitoneal hydroxychloroquine (chloroquine) per day for 48 h or chronic treatment for 4 wk (Fig. S1). Chloroquine is an inhibitor of autophagosome–lysosome fusion, and treatment leads to a decrease in the degradation of autophagic substrates such as LC3II and p62. An immunoblot of muscle lysates from nine different IBMPFD patients muscle biopsies showed an approximately fivefold increase in p62 protein levels when compared with normal patient muscle (Fig. S2). As previously described, sIBM muscle had an increase in p62 protein levels by approximately threefold (Nogalska et al., 2009). Further characterization of IBMPFD mutant–expressing transgenic mice found that RH9 and RH12 muscle accumulated subsarcolemmal, diffuse, and punctate sarcoplasmic p62 and LC3 (Fig. 1, C and D). This pattern of p62 accumulation is similar to that seen after acute treatment of control mice with 50 mg/kg intraperitoneal chloroquine per day for 48 h (Ju et al., 2009) or chronic treatment for 4 wk (Fig. S5 D). In some instances, p62 and LC3 localized to the center of a myofiber at a region consistent with an RV (Fig. 1 D). No similar pattern was seen in control or VCP-WT mouse muscle (Fig. 1 C). IBMPFD patient muscle had identical findings (Fig. S3). These data demonstrate that the autophagosome markers p62 and LC3 are increased in IBMPFD muscle and localize to RVs.
Figure 1.

LC3 and p62 accumulate in IBMPFD muscle tissue. (A and B) Immunoblot of 9-mo-old quadriceps muscle lysates from control (cont) or VCP-WT, -RH9, or -RH12 mutant transgenic lines with p62 (A) or LC3 (B) antibodies. Note the increase in p62 and LC3II isoforms in mutant animals. Densitometric quantification is from more than six 9-mo-old animals/group. LC3 and p62 levels are normalized to loading control. Error bars represent the standard error from six independent experiments. **, P < 0.001. AU, arbitrary unit. (C) p62 immunostaining of tibialis anterior (TA) or quadriceps (Quad) muscle from 9-mo-old control, VCP-WT, or one of two VCP-RH transgenic lines (RH9 or RH12). Note accumulations of p62 in the middle of myofibers or along subsarcolemmal regions. (D) Histochemistry and immunohistochemistry of quadriceps muscle from VCP-RH–expressing transgenic mice. Hematoxylin and eosin (H&E) staining of a 15-mo-old animal with an RV, p62 immunostaining of an RV from a 12-mo-old animal, LC3 immunostaining of an RV, and subsarcolemmal accumulations of LC3 from a 12-mo-old animal. The bracket highlights one LC3-positive myofiber. A single muscle fiber is outlined in white. (C and D) Arrows denote vacuoles or accumulations of p62 or LC3. Bars, 30 µm.
Because mutations in VCP lead to IBMPFD, we evaluated the effect of a loss of VCP on autophagy. After 4 d of VCP-targeted siRNA oligonucleotide treatment (knockdown [KD]), VCP protein levels decreased by ∼60–70% in U20S cells (Fig. 2 A). Immunoblots for p62 and LC3 using these same lysates demonstrated increased LC3II and p62 protein levels compared with scrambled control siRNA treatment (Scr; Fig. 2 A). Transfection of similarly treated cells with a GFP-LC3 expression construct showed increased GFP-LC3 puncta under basal conditions in VCP-KD cells compared with control Scr cells (Fig. 2 B). We further explored the effect of VCP on autophagy by using previously characterized tetracycline-inducible U20S cells that express myc-tagged VCP-WT, ATPase inactive VCP-E578Q (VCP-EQ) or one of two IBMPFD mutants, VCP-RH or VCP-A232E (VCP-AE), at approximately two to three times the endogenous VCP (Ju et al., 2008). Immunoblots for LC3 or p62 from cell lysates of 16 h–induced U20S cells identified an increase in p62 and the LC3II fragment of LC3 in IBMPFD mutant (RH and AE)– and VCP-EQ–expressing cells (Fig. 2 C). Lentiviral transfection of the parental U20S cell line with VCP-WT–, VCP-EQ–, or IBMPFD mutant–expressing constructs gave identical results, suggesting that our finding was not caused by the clonal nature of the U20S stable cells (unpublished data). Just as with VCP-KD, there was an increase in basal GFP-LC3 puncta in VCP-EQ or IBMPFD mutant cells when compared with control or VCP-WT cells (Fig. 2, D and E). The increase in GFP-LC3 puncta was similar to that seen when VCP-WT or control cells were treated with bafilomycin A (Baf), an inhibitor of autophagosome–lysosome fusion and lysosomal vacuolar-type H+-ATPase, for 4 h (Fig. 2, D and E). Interestingly, VCP-EQ– and IBMPFD mutant–expressing cells did not significantly increase the number of GFP-LC3–positive puncta upon treatment with Baf for 4 h (Fig. 2 E). These data suggest that loss of VCP activity or IBMPFD mutant VCP expression does not disrupt autophagosome formation and results in their accumulation.
Figure 2.

LC3 and p62 accumulate in VCP-KD and IBMPFD mutant–expressing cells. (A) Lysates from scrambled siRNA or VCP-targeted siRNA–treated U20S cells immunoblotted with antibodies to VCP, LC3, p62, or α-tubulin. Densitometric analysis is graphically represented from four independent experiments. LC3 and p62 levels are normalized to loading control. (B) Fluorescent microscopy images of siRNA scrambled control (Scr) or VCP-targeted siRNA KD cells expressing GFP-LC3. Basal numbers of GFP-LC3 puncta/cell were counted. (C) Lysates from control or myc-tagged VCP-WT, ATPase-inactive VCP-EQ, or one of two IBMPFD mutants, VCP-RH or -AE, expressed for 16 h from stably transfected tetracycline-inducible U20S cells and immunoblotted with antibodies to myc, LC3, p62, or α-tubulin. Densitometric analysis is graphically represented from four independent experiments. LC3 and p62 levels are normalized to loading control. Note the increase in LC3II and p62 in VCP siRNA KD and mutant-expressing cells. (D) Fluorescent microscopy images of tetracycline-inducible U20S cells expressing GFP-LC3 treated with and without Baf for 4 h (DMSO control [left] or Baf+ [right]). Cells are control U20S cells and VCP-WT, -RH, -AE, and -EQ. (E) The number of GFP-LC3 puncta/cell was counted for control U20S and VCP-WT, -EQ, -RH, and -AE with and without Baf for 4 h (DMSO or Baf). Note the increase in basal GFP-LC3 puncta in mutant-expressing cells. (A–C and E) Error bars represent the standard error from four independent experiments. *, P < 0.01; and **, P < 0.001. Bars, 25 µm.
Normally, an autophagosome acquires acidic properties after fusion with organelles such as lysosomes. Once fused, they become autolysosomes. One way to distinguish an autophagosome from an autolysosome is to use a tandemly tagged monomeric RFP (mRFP)–GFP-LC3 reporter (tfLC3; Kimura et al., 2007). This reporter fluoresces green and red in the neutral environment of the autophagosome, distinguishing it from the acidic environment of an autolysosome which quenches the green fluorescence, leaving only the red fluorescence detectable. We transfected control, Scr control, VCP-KD, or cells expressing VCP-WT, -EQ, -RH, or -AE with the tfLC3 reporter. As a positive control, cells were treated with Baf to inhibit autolysosome formation. To induce autophagosome maturation, cells were incubated with rapamycin for 2 h, a time point determined to be optimal for the visualization of autolysosomes. Rapamycin is capable of inducing autophagy in U20S cells, as measured by GFP-LC3–positive puncta and the conversion of LC3I to LC3II (unpublished data). Control, Scr control, and VCP-WT–expressing cells had fewer green/red autophagosomes and more red autolysosomes than cells cotreated with Baf for 2 h (Fig. 3, A and B). The correlation coefficient of red dots to green dots was calculated for control cells as ∼0.5, whereas this measure was ∼0.8 in Baf-treated cells (Fig. 3 C). Similar to the Baf-treated cells, VCP-KD and -EQ and both IBMPFD mutants had an increase in red/green-positive structures and a colocalization coefficient of >0.75 (Fig. 3, A and C). To confirm that autolysosome formation was impaired, we immunoblotted lysates from cells expressing VCP-WT or mutant VCP with and without Baf for 4 h. In normal cells, Baf increases the levels of LC3II protein, whereas in cells with a defect in autophagosome–lysosome fusion, it will not increase LC3II levels (Rubinsztein et al., 2009). The LC3II species in control and VCP-WT–expressing cells increased with Baf, whereas both IBMPFD mutant–expressing cells did not increase LC3II protein levels further (Fig. 3 D). These data are consistent with the results in Fig. 2 (D and E), in which Baf treatment failed to increase the amount of GFP-LC3–positive puncta further than that already seen under basal conditions. These data suggest that loss of VCP function or IBMPFD mutant VCP expression results in the accumulation of nondegradative autophagosomes.
Figure 3.

Functional VCP is required for autophagosome maturation. (A) Epifluorescent images for GFP and mRFP in U20S or VCP-WT–, VCP-RH–, VCP-AE–, or VCP-EQ–expressing cells transfected with mRFP-GFP-LC3 (tfLC3) and treated with rapamycin for 2 h to induce autolysosome formation. (B) siRNA control (Scr) or VCP-KD– or Baf-treated control U20S cells transfected with tfLC3 and treated with rapamycin for 2 h to induce autolysosome formation. (A and B) Open arrows denote autophagosomes (both GFP and mRFP fluorescence), whereas closed arrows highlight autolysosomes (mRFP only fluorescence). (C) The graph represents Pearson's coefficient of GFP and mRFP colocalization from 10 independent fields of cells in two different experiments. Error bars represent the standard error from 20 fields in two independent experiments. *, P < 0.001. (D) Lysates from U20S or tetracycline-inducible VCP-WT, -RH, or -AE cells treated with vehicle or Baf for 4 h and immunoblotted for LC3 and α-tubulin. Note that Baf treatment does not increase the LC3II levels in IBMPFD mutant (RH and AE)–expressing cells. Bars, 15 µm.
We evaluated degradation of an autophagic substrate in VCP-inactive or IBMPFD mutant–expressing cells via an LC3 immunoblot. The degradation of the autophagosome-associated LC3II fragment is a specific marker of autophagic flux in cells (for review see Klionsky et al., 2008). Either Scr control or VCP-KD cells had autophagy induced via nutrient deprivation and were harvested for immunoblotting at 0, 2, or 4 h. An immunoblot of Scr control cell lysates for LC3 demonstrates a decrease in LC3II protein over the 4 h (Fig. 4 A). We did not see a transient increase in LC3II levels indicative of a conversion of LC3I to LC3II upon autophagic induction because the earliest time point evaluated was 2 h. Previous studies show that this conversion in cell culture occurs before 2 h and would therefore not be seen under our assay conditions (for review see Klionsky et al., 2008). In contrast, VCP-KD–treated cells failed to degrade LC3II (Fig. 4 A). A similar experiment was performed using control, VCP-WT and -EQ, and IBMPFD mutant cells. For these experiments, endogenous LC3II was detected after immunoblot of cell lysates after nutrient deprivation for up to 6 h. Although control or VCP-WT cells had a decrease in LC3II, VCP-EQ and IBMPFD mutant VCP cells failed to degrade LC3II as rapidly (Fig. 4 B). This is shown graphically after densitometric analysis from three independent experiments (Fig. 4 C).
Figure 4.
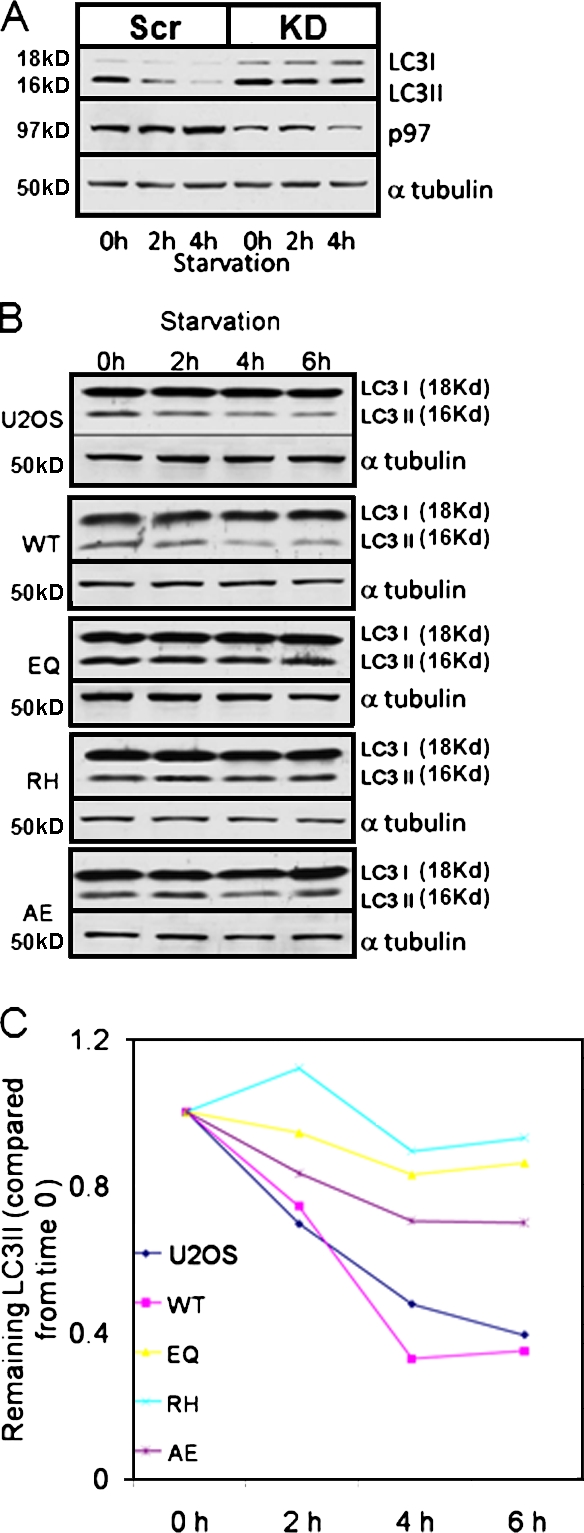
Functional VCP is required for autophagic protein degradation. (A) Lysates from siRNA-treated U20S cells (scramble control [Scr] or VCP siRNA KD) after autophagic induction via nutrient deprivation for 0, 2, or 4 h and immunoblotting for LC3 and α-tubulin. LC3II degrades over time in control but not in KD cells. One of two experiments is shown. (B) Lysates from U20S or tetracycline-inducible VCP-WT, -EQ, -RH, or -AE cells after autophagic induction via nutrient deprivation for 0, 2, 4, or 6 h and immunoblotting for LC3 and α-tubulin. LC3II degrades in control and VCP-WT but degrades less efficiently in mutant (EQ, RH, or AE)-expressing cells. (C) Graphical representation of densitometric evaluation of LC3II and α-tubulin at each time point from three independent experiments.
Next, we evaluated the colocalization of GFP-LC3 puncta with endosomal/lysosomal markers, LysoTracker red (LTR), or endogenous Lamp1 after a 2-h treatment with rapamycin to see whether autophagosome–lysosome fusion is defective. Control or VCP-WT cells had GFP-LC3 puncta that colocalized or were adjacent to LTR (Fig. 5 A)- or Lamp1 (Fig. 5 C)-positive vesicles, with a colocalization coefficient of ∼0.5 (Fig. 5, B and D). In VCP-EQ, IBMPFD mutant VCP, or control cells cotreated with Baf, there was a decrease in the colocalization of GFP-LC3 puncta and LTR (Fig. 5 A) or Lamp1 (Fig. 5 C), with a colocalization coefficient of ∼0.3–0.4 (Fig. 5, B and D). Similar results were obtained using coexpression of an mRFP-LAMP1 fusion protein (unpublished data). To demonstrate that GFP-LC3–positive structures were consistent with accumulating autophagosomes, we used electron microscopy. Vacuolated structures consistent with autophagosomes and lysosomes were identified in VCP-RH– and VCP-AE–expressing cells (Fig. 5 E). These data suggest that IBMPFD mutant VCP may lead to dysfunctional fusion of autophagosomes with lysosomes.
Figure 5.

Autophagosomes have decreased localization with lysosomal markers in IBMPFD mutant–expressing cells. (A) Epifluorescent images for GFP-LC3 (LC3) and LTR of VCP-WT–, VCP-RH–, VCP-AE–, or VCP-EQ–expressing cells transfected with GFP-LC3 and treated with rapamycin for 2 h to induce autolysosome formation. Open arrows highlight autolysosomes (GFP and LTR colocalized), and closed arrows show autophagosomes (GFP only). (B) Pearson's coefficient of GFP and LTR colocalization from 10 independent fields of cells in two different experiments. (C) Epifluorescent images for GFP-LC3 and endogenous Lamp1 immunohistochemistry of U20S, VCP-WT–, VCP-RH–, VCP-AE–, or VCP-EQ–expressing cells or U20S cells cotreated with Baf transfected with GFP-LC3 and treated with rapamycin for 2 h to induce autolysosome formation. Arrows highlight autolysosomes (GFP and Lamp1 colocalized). (A and C) The boxed regions in the merge field are enlarged in the adjacent panels. (D) Pearson's coefficient of GFP and Lamp1 colocalization from 10 independent fields of cells in two different experiments. (B and D) Error bars represent the standard error from 20 fields in two independent experiments. *, P < 0.001. (E) Electron microscopy of tetracycline-inducible control or VCP-WT–, VCP-RH–, or VCP-AE–expressing U20S cells induced for 16 h and then treated with rapamycin for 2 h. Note the accumulation of autophagic structures in the mutant-expressing cell lines. Arrows denote autophagic structures. Bars: (A and C) 15 µm; (E) 1 µm.
We reasoned that RVs may be similar accumulations of nondegradative autophagosomes seen in cell culture. Immunohistochemistry of quadriceps muscle with p62 and Lamp1 revealed deposition in 9-mo-old VCP-RH mouse muscle within the central region of myofibers and around RV-like structures in some fibers that was not apparent in VCP-WT or control animals (Fig. 6 A). This colocalization did not completely overlap, with some p62 puncta being distinct from Lamp1-positive puncta. A similar pattern was seen using IBMPFD patient muscle in which a p62-positive RV contained Lamp2-positive vesicles (Fig. 6 A). Consistent with the accumulation of nondegradative autophagosomes, RVs in IBMPFD- and VCP-RH–expressing mice failed to label with hydrolytic enzyme stains such as acid phosphatase and nonspecific esterase (Fig. S3, C and D). Ultrastructural analysis identified structures containing accumulations of membranous vacuoles in VCP-RH muscle (Fig. 6, B and C).
Figure 6.

Autophagic and lysosomal markers localize to RVs in IBMPFD tissue. (A) Immunofluorescence images of quadriceps muscle using p62 and Lamp1 or -2 antibodies. Quadriceps section from a 9-mo-old VCP-WT mouse, age-matched VCP-RH transgenic, 12-mo-old VCP-RH transgenic, and IBMPFD patient muscle biopsy are shown. Open arrows highlight p62 and Lamp1/2 colocalization, and closed arrows show regions of only p62 immunofluorescence. Single myofibers containing RVs are outlined in the merge panels. (B and C) Transmission electron microscopy of tibialis anterior muscle from one of two independent lines (B shows RH12, and C shows RH9 lines) of 12-mo-old VCP-RH–expressing transgenic mice. Note the large vacuolated structures that are perinuclear or subsarcolemmal. Bars: (A) 30 µm; (B and C) 500 nm.
To validate a functional consequence of impaired autophagy, we used a quantitative luciferase-based assay that measures selective protein aggregate degradation upon autophagic stimulation (Ju et al., 2009). This assay utilizes two luciferase reporters fused to a nonaggregating polyglutamine repeat (polyQ19-luciferase) or an aggregation-prone expanded polyglutamine repeat (polyQ80-luciferase) electroporated into the left and right tibialis anterior of 6-mo-old control or VCP-WT or -RH (RH9 and RH12) transgenic mice. Using in vivo bioluminescent imaging, baseline measurements of luciferase activity were taken 2 d after electroporation (Fig. 7 A). Animals were then starved for 24 h, and repeat luciferase activity measurements were recorded (Fig. 7 A). The change in the ratio of polyQ80-luciferase/polyQ19-luciferase was calculated for three animals/treatment condition. Control and VCP-WT animals had a mean decrease in polyQ80-luciferase/polyQ19-luciferase of ∼25% and 18%, respectively (Fig. 7 B). In contrast, RH12 and RH9 transgenic lines had an increase in the ratio of polyQ80-luciferase/polyQ19-luciferase. These data suggest that VCP-RH animals have a defect in the autophagic clearance of protein aggregates.
Figure 7.

Impaired autophagic substrate degradation in IBMPFD mouse muscle. (A) Representative images using in vivo bioluminescence of control or IBMPFD mutant RH–expressing mice (RH12) 2 d after electroporation (baseline) or after 24 h of nutrient deprivation (starvation) of polyQ80 (Q80)- or polyQ19-luciferase (Q19) in the right and left tibialis anterior, respectively. The ratio of polyQ80-luciferase/polyQ19-luciferase is indicated below each image set. Values are in ×104 photons per second per square centimeter per steradian. (B) Box and whisker plot of the change (Δ) in the ratio of polyQ80-luciferase/polyQ19-luciferase activity in the left and right tibialis anterior muscle of control, VCP-WT, or one of two VCP-RH (RH12 or RH9) transgenic mouse lines after 24 h of starvation. The graph is representative of three animals per group. The p-value for RH12 was 0.05 and 0.06 when compared with control animals or VCP-WT transgenic. The p-value for RH9 was 0.03 when compared with either control or VCP-WT transgenic. The p-value was 0.01 and 0.02 when combined RH12 and RH9 animals were compared with control or VCP-WT groups, respectively. There was no statistical difference between control or VCP-WT groups.
One feature of IBMPFD is the loss of nuclear TDP-43 and accumulation cytoplasmic TDP-43 inclusions (Neumann et al., 2007; Weihl et al., 2008). To see whether TDP-43 accumulated in the cytoplasm of IBMPFD mutant–expressing cells, we transfected a human TDP-43 with an N-terminal mCherry tag (mCherry–TDP-43) into our tetracycline-inducible U20S cells. After 48 h of mCherry–TDP-43 expression, we quantitated the percentage of mCherry-positive cells having nuclear only or cytosolic fluorescence (Fig. 8 A). Because TDP-43 leaves the nucleus during cell division and cell death (unpublished data), we costained with DAPI to visualize mitotic figures and pyknotic nuclei. This allowed us to only count cells with intact nuclei. Similar to a previously reported study, IBMPFD mutant– and VCP-EQ–expressing cells had an increase in cytosolic TDP-43 (Fig. 8, A and B; Gitcho et al., 2009). Similarly, treatment of control U20S cells with agents known to impair autophagosome maturation such as chloroquine and Baf for 4 h resulted in the cytosolic accumulation of transfected TDP-43 (Fig. 8, A and B). Lysates from similarly transfected cells were separated into nuclear and cytosolic fractions. These fractions were immunoblotted for mCherry–TDP-43 along with lamin A/C and actin to confirm similar protein loading and the integrity of the fractions. mCherry–TDP-43 was enriched in IBMPFD mutant– and VCP-EQ–expressing cell cytoplasm when compared with VCP-WT–expressing cells (Fig. 8 C). There was no clear difference in the amount of nuclear TDP-43 in these same cells. These data were similar to that seen when cell fractionation was performed on lysates from U20S cells treated with chloroquine or Baf for 4 h (Fig. 8 C) and suggests that a block in autophagosome maturation can increase the amount of cytosolic TDP-43.
Figure 8.

IBMPFD mutant expression or autophagic inhibition redistributes TDP-43 to the cytosol in cells. (A) Quantitation of mCherry–TDP-43 distribution (nuclear only or cytoplasmic) in control U20S, VCP-WT–, VCP-RH–, VCP-AE–, or VCP-EQ–expressing cells, or control U20S cells treated with 10 µM Baf or 50 µM chloroquine (Chlq) for 4 h from 10 different fields from two independent experiments. Error bars represent the standard error from 20 fields in two independent experiments. *, P < 0.02 when compared with VCP-WT–expressing cells. (B) Epifluorescent images for mCherry–TDP-43 (red) and DAPI (blue) in control U20S, VCP-WT–, VCP-RH–, or VCP-AE–expressing cells, or control U20S cells treated with Baf or chloroquine for 4 h. Arrows denote cytosolic TDP-43 and perinuclear TDP-43 inclusions. (C, top) Immunoblot for TDP-43, lamin A/C, and actin from nuclear or cytosolic lysate fractions of mCherry–TDP-43–transfected VCP-WT–, VCP-RH–, VCP-AE–, or VCP-EQ–expressing cells. Note the increase in cytosolic TDP-43 from IBMPFD mutant– and EQ-expressing cells. (bottom) Immunoblot for TDP-43, lamin A/C, and actin from nuclear or cytosolic lysate fractions of untreated U20S cells transfected with mCherry–TDP-43 or similarly transfected cells treated with 30 µg/ml (cq1) or 120 µg/ml (cq2) chloroquine diphosphate or 200 ng/ml Baf. Data are representative of three independent experiments. Bar, 15 µm.
Next, we evaluated TDP-43 immunolocalization in control and VCP-WT– and IBMPFD mutant–expressing animals. Notably, fixation of skeletal muscle with acetone abolished all nuclear TDP-43, whereas fixation with PFA and then acetone resulted in TDP-43 immunofluorescence in ∼80–90% of nuclei from mouse skeletal muscle (Fig. S4). TDP-43 was predominantly within nuclei from 15-mo-old control and VCP-WT–expressing animal muscle, similar to that previously reported for human skeletal muscle (Fig. 9 A; Weihl et al., 2008; Salajegheh et al., 2009). In contrast, skeletal muscle from similarly aged RH9 and RH12 transgenic mice had a reduction of TDP-43 immunofluorescence within nuclei and an increase in perinuclear sarcoplasmic TDP-43 (Fig. 9 A). In some fibers, TDP-43 immunostaining was concentrated in small punctuate inclusions throughout the sarcoplasm or in central regions of the myofiber consistent with an RV (Fig. 9 A). This pattern is identical to that seen in IBMPFD patient skeletal muscle (Fig. S3, M and N) and never seen in control, VCP-WT–expressing mice, or normal patients. Rodents chronically administered chloroquine develop an RV myopathy with LC3- and acid phosphatase–positive vacuoles presumably caused by the impaired lysosomal degradation of autophagic components (Ikezoe et al., 2009). We evaluated the localization of TDP-43 in 3-mo-old mice treated with 50 mg/kg chloroquine per day for 1 mo compared with age-matched control mice treated with daily injections of buffered saline. Consistent with a previous study (Ikezoe et al., 2009), chloroquine treatment caused vacuolation, myofiber degeneration, and the accumulation of p62 and LC3II (Figs. S1 and S4). TDP-43 immunostaining in chloroquine-treated animals was similar to that seen in IBMPFD mutant–expressing animals and patients. Specifically, myofibers accumulated TDP-43 in the sarcoplasm adjacent to nuclei from chloroquine-treated animals (Fig. 9 B). These data suggest that a block in autophagic protein degradation recapitulates TDP-43 pathology in IBMPFD.
Figure 9.

IBMPFD mutant expression or autophagic inhibition redistributes TDP-43 to the cytosol in mouse skeletal muscle. (A) TDP-43 immunostaining of quadriceps skeletal muscle from 15-mo-old control (Cont) or VCP-WT, -RH9, or -RH12 transgenic mice. A single myofiber is outlined in white. (B) TDP-43 immunostaining of quadriceps skeletal muscle from 3-mo-old mice treated with saline or 50 mg/kg/d intraperitoneal chloroquine (Chlq) for 4 wk. (A and B) Insets show one myonuclei from each field. Bars, 30 µm.
Discussion
We demonstrate that loss of VCP activity impairs autophagy. Specifically, LC3-positive vacuoles fail to mature into autolysosomes, resulting in the accumulation of nondegradative autophagosomes. Moreover, measures of autophagic flux are impaired in cells lacking a functional VCP. Expression of the IBMPFD mutant VCP-RH or -AE leads to similar results with dysfunctional autophagosomes accumulating at RVs in IBMPFD patient and transgenic mouse muscle. TDP-43 accumulation in the cytosol occurs in IBMPFD mutant–expressing cells and animals as well as with chemical inhibition of autophagosome maturation in vitro and in vivo. Our data suggest that IBMPFD is a disorder of autophagosome maturation resulting in dysfunctional autophagy.
A defect in autophagosome maturation explains many of the pathological features seen in IBMPFD muscle. For example, ubiquitinated aggregates and p62 inclusions are present in autophagy-deficient tissue (Hara et al., 2006; Komatsu et al., 2007). RVs, which are speculated to be of autophagic origin, are indeed accumulations of p62- and LC3-positive autophagosomes in IBMPFD. Autophagic inhibition via pharmacologic treatment with chloroquine, which is an inhibitor of lysosomal proteases, in mice or patients results in a myopathy with vacuoles and protein accumulation (Suzuki et al., 2002). Similarly, toxic administration of the microtubule-disrupting drugs colchicine and vinblastine results in a myopathy with RVs (Kuncl et al., 2003). The mechanism of action with these drugs is proposed to be a defect in autophagosome/lysosome trafficking.
The role of autophagy in skeletal muscle is unclear. Skeletal muscle–specific knockout of the autophagy essential protein ATG5 in mice leads to type II muscle fiber atrophy with an accumulation of ubiquitin and p62 within these fibers (Raben et al., 2008). In contrast, skeletal muscle atrophy induced by constitutively active FOXO3 (forkhead box O3) was reduced when muscle was treated with silencing RNAs directed to LC3 (Mammucari et al., 2007). Impairment in autophagosomal–lysosomal degradation is the proposed mechanism in other vacuolar myopathies such as Danon's disease (Nishino et al., 2000) and X-linked myopathy with excessive autophagy (Ramachandran et al., 2009). These myopathies, similar to IBMPFD, accumulate undegraded protein in association with membranous structures. Presumably, mice deficient in skeletal muscle autophagy genes such as ATG5 do not make autophagosomes, whereas in the case of IBMPFD, autophagosomes are formed yet fail to mature and be degraded. Perhaps the cytoskeletal derangements and membranous accumulations that occur in vacuolar myopathies are deleterious to skeletal muscle function and explain the pathological disconnect between autophagy-deficient and -defective muscle.
TDP-43 redistribution from the nucleus to the cytosol and subsequent aggregation is a feature seen in IBMPFD patient muscle (Weihl et al., 2008). Sarcoplasmic TDP-43 colocalizes with ubiquitin, suggesting that its degradation is affected (Weihl et al., 2008). This feature is seen in other muscle disorders with RVs, including sIBM and hIBM2 (Weihl et al., 2008; Küsters et al., 2009; Salajegheh et al., 2009). The cause of RVs in these disorders is not known but has been presumed to be secondary to autophagy. We demonstrate that TDP-43 accumulates in the cytoplasm of IBMPFD mutant–expressing cells and transgenic mouse muscle. This pathological finding is recapitulated under conditions of impaired autophagosome–lysosome fusion after chloroquine and Baf treatment. We suggest that IBMPFD mutants disrupt autophagy and that this subsequently leads to the accumulation of cytosolic TDP-43. One recent study found that VCP and TDP-43 could interact in cell culture and patient tissue (Gitcho et al., 2009). They proposed that TDP-43 may be a substrate for VCP. It has been speculated that TDP-43 or its fragments may be degraded via autophagy and accumulate when autophagy is dysfunctional (Filimonenko et al., 2007; Caccamo et al., 2009; Kadokura et al., 2009; Kim et al., 2009). How TDP-43 redistribution affects skeletal muscle and whether TDP-43 or its fragments are bona fide autophagy substrates remain to be determined.
This study focuses principally on the skeletal muscle pathogenesis of IBMPFD. Muscle weakness is the presenting feature in >50% of IBMPFD patients, and 90% of IBMPFD patients develop IBM with significantly less phenotypically expressing PDB and FTD (∼51% and ∼32%, respectively; Weihl et al., 2009). Moreover, the pathologies in IBMPFD patient muscle and neurons are distinctively different. For example, IBMPFD brain tissue has predominantly nuclear ubiquitinated inclusions with rare cytoplasmic inclusions (Schröder et al., 2005; Forman et al., 2006), whereas IBMPFD skeletal muscle tissue has ubiquitin-positive nuclei and abundant sarcoplasmic ubiquitinated inclusions (Hübbers et al., 2007; Gidaro et al., 2008; Weihl et al., 2009). Similarly, IBMPFD brain tissue has nuclear TDP-43 inclusions as well as a reduction of TDP-43 from the nuclei of affected cortical neurons (Neumann et al., 2007), whereas IBMPFD skeletal muscle has the exclusive accumulation of sarcoplasmic TDP-43 and absence from the myonuclei of inclusion-bearing myofibers (Weihl et al., 2008; Küsters et al., 2009; Salajegheh et al., 2009). The reason for these pathological differences is unknown but may relate to how muscle and brain tissue handle autophagic insults.
A disruption in autophagy is consistent with data from other diseases that share phenotypic features with IBMPFD (i.e., PDB and FTLD-U). Mutations in the autophagosome protein p62 cause PDB (Laurin et al., 2002). p62 binds ubiquitinated proteins and LC3 (Pankiv et al., 2007). Under conditions of impaired autophagy, p62 mediates the aggregation of ubiquitinated proteins (Komatsu et al., 2007) and sequesters them from the UPS (Korolchuk et al., 2009). Interestingly, VCP improves UPS function in the setting of p62 overexpression, identifying a potential point of intersection between the UPS and autophagy (Korolchuk et al., 2009). Mutations in CHMP2B (charged multivesicular body protein 2B) cause FTLD-U (Skibinski et al., 2005). CHMP2B is a member of the ESCRT (endosomal-sorting complex required for transport) pathway, which facilitates the endosomal/lysosomal degradation of plasma membrane proteins (Skibinski et al., 2005). ESCRT family members, including CHMP2B, participate in autophagosome–lysosome fusion, and loss of ESCRTI and ESCRTIII family members leads to TDP-43 accumulation (Filimonenko et al., 2007).
This study does not preclude the possibility that the UPS or the degradation of muscle-specific UPS substrates is impaired in IBMPFD. We previously reported that steady-state levels of ΔF508CFTR are increased in IBMPFD mutant VCP myoblasts (Weihl et al., 2006). We suggested that this was caused by a defect in UPS-mediated degradation. However, our current data open the possibility that ΔF508CFTR accumulates because of IBMPFD mutant's effect on autophagy. Whether this is via direct degradation of the ERAD substrate by autophagy or a secondary impairment in the UPS as seen with chronic autophagic inhibition is not known (Korolchuk et al., 2009). Regardless, it is conceivable that previous studies evaluating the degradation of UPS substrates and accumulation of ubiquitinated proteins reflect a primary defect in autophagy mediated by IBMPFD mutants rather than dysfunctional UPS (Weihl et al., 2006; Janiesch et al., 2007; Ju et al., 2008).
The mechanism by which VCP regulates autophagy remains to be elucidated. Our data suggest that loss of VCP does not affect the induction of autophagy but instead impairs autophagosome maturation to acidic autolysosomes. This can be caused by defects in autophagosome/lysosome trafficking or autophagosome–lysosome fusion. We have previously shown that IBMPFD mutations in VCP affect the trafficking of protein aggregates to an inclusion body (Ju et al., 2008). This leads to impaired autophagic degradation of aggregated polyglutamine-containing protein. Whether IBMPFD mutants could also affect the trafficking of autophagosomes to inclusion bodies is not known but would be consistent with our current data. VCP performs its functions via interactions with ubiquitinated intermediates or by association with ubiquitin-like domains (Schuberth and Buchberger, 2008). These cofactors contain a homologous 76–80-aa sequence with structural similarities to ubiquitin. Several autophagosome-associated proteins (LC3, GABARAP, and GATE-16) also have domains with homology to ubiquitin (Sou et al., 2006). Whether VCP forms a complex with these proteins to mediate autophagosome–lysosome fusion is speculative. However, another AAA+ protein, NSF, associates with GATE-16 (Sagiv et al., 2000). NSF mediates the assembly of GATE-16 and GOS-28, a Golgi v-SNARE required for homotypic membrane fusion (Muller et al., 2002). Our study identifies an essential role for VCP in autophagy and implicates dysfunctional autophagy in the pathogenesis of IBMPFD, its phenotypic variants (IBM, PDB, and FTLD-U), and perhaps other TDP-43 proteinopathies.
Materials and methods
Plasmids and cell culture
PolyQ19- and polyQ80-luciferase constructs were previously described (Ju et al., 2009). LC3-GFP plasmid and tfLC3 tandemly tagged with GFP and mRFP construct were obtained from the laboratory of H. Virgin (Washington University, St. Louis, MO). mCherry–TDP-43 was constructed as follows: a cDNA encoding human full-length TDP-43 with an N-terminal Flag tag was generated by PCR using the full-length human TDP-43 cDNA (Thermo Fisher Scientific) as a template. The Flag sequence was placed immediately after the start methionine, with the final sequence MDYKDDDKSEYIR. This PCR product was cloned into the XhoI and BamHI sites in frame with the mCherry red fluorescent protein in the vector pCherry-C1, which was generated by replacing EGFP with mCherry on the pEGFP-C1 backbone. U20S TRex stable cell lines (VCP-WT, -EQ, -RH, and -AE point mutations) were established and cultured as described previously (Ju et al., 2008). The cells were maintained in DME supplemented with penicillin/streptomycin, 10% fetal bovine serum, 50 µg/ml hygromycin B, and 65 µg/ml Zeocin at 37°C with 5% CO2. The stable cell lines were seeded in appropriate plates and grown to ∼80% confluence on the day of transfection. Transfections were performed with Lipofectamine 2000 (Invitrogen) or, in the case of mCherry–TDP-43, with Fugene 6 (Roche) according to manufacturers' protocol. For amino acid starvation, cells were extensively washed with HBSS and further incubated in HBSS for the indicated time periods. Rapamycin was used at a final concentration of 10 µg/ml for the indicated times. Baf was used at a concentration of 200 ng/ml, and chloroquine diphosphate was used at a concentration of 120 µg/ml.
Transfection of siRNA oligonucleotides
KD of VCP was achieved by treatment of U20S cells with siRNA duplexes, corresponding to the VCP RNA sequence (QIAGEN) or nontargeting siRNA duplexes (QIAGEN). Cells were transfected according to manufacturer's instructions (HiPerFect transfection reagent; QIAGEN). In brief, cells were seeded onto 6-well plates 1 d before transfection to reach 70% confluence in a DME containing 10% FBS without antibiotics. Targeting siRNA or negative control siRNA were mixed with HiPerFect transfection reagent in OptiMEM (Invitrogen) for 15 min at room temperature and then added to the culture medium with a final concentration of 50 nM of either oligonucleotide. Cells were incubated at 37°C for 48 h. Cells were retransfected with either siRNA duplex in an analogous manner. After 48 h of second transfection, protein expression was determined by Western blotting using an antibody for VCP, which revealed an ∼60–70% reduction of VCP.
Fluorescence and electron microscopy
U20S cells grown on glass coverslips were transiently transfected with plasmid or siRNA constructs. For stably expressing cells, 1 µg/µl tetracycline was added the day after transfection. 24–48 h after transfection, cells were washed twice with PBS, fixed with 3% PFA in PBS for 15 min, permeabilized with 0.1% Triton X-100 in PBS for 10 min, washed, blocked with 3% goat serum for 1 h, and stained for fluorescence microscopy. For skeletal muscle immunohistochemistry, isolated skeletal muscle was mounted using tragacanth gum and quick frozen in liquid nitrogen–cooled 2-methylbutane. Frozen biopsy samples were sectioned into 8-µM thick sections and fixed in ice-cold acetone or 4% PFA and then acetone in the case of TDP-43 immunostaining (Fig. S5). Specimens were examined using a fluorescent microscope (80i upright; Nikon) and charge-coupled device camera (EZ monochrome; Roper Industries) with deconvolution software analysis (NIS Elements; Nikon). Nonfluorescent images were taken with a 5-megapixel color charge-coupled device (Nikon). Image processing and analysis were performed with NIS Elements 4.0 software and Photoshop CS3 (Adobe). All images were performed on fixed cells or tissue at room temperature using Prolong Gold mounting solution (Invitrogen). Objectives used for immunofluorescence were Apochromat 20× and 40×. Conjugated secondary antibody fluorophores used were Alexa Fluor 488 and 543 (Invitrogen). For colocalization analysis, 10 random fields of cells were selected, each containing at least two transfected cells. The Pearson's colocalization coefficient was determined for each cell using NIS Elements 4.0 software. Results are from two independent experiments. LC3 puncta were calculated from four independent experiments in which five to eight low power fields were counted. All images were taken at the same gain and exposure intensity. Images were postprocessed using ImageJ software (National Institutes of Health) and the TOPHAT filter (http://u759.curie.u-psud.fr/softwaresu759.html) before the counting of dot intensity.
For mCherry–TDP-43 quantitation, U20S cells were seeded at 50,000 cells per 12-well coverslips in antibiotic-free media and transfected on the next day. After transfection, cells were induced or treated with the indicated drugs. After treatment, the coverslips were washed with PBS three times, fixed in 4% PBS/PFA for 20 min, washed again, and mounted in fluorescence mounting medium that contained DAPI to visualize nuclei. Deconvoluted images from 10 random fields, each one containing at least 10 transfected cells, were acquired with a microscope (Eclipse 80i; Nikon) using an Apochromat 20× objective. Acquisition was performed at the same gain and exposure setting. Cells were visualized with the NIS Elements Advanced Research 3.0 software (Nikon) and categorized as nuclear only or cytoplasmic containing based on the presence of Cherry fluorescence outside the boundaries of the nucleus. Only cells carrying healthy nuclei were considered for counting purposes, based on a homogeneous DAPI staining. Dividing cells were excluded from the current analysis.
For thin-section electron microscopy, transiently transfected cells were grown to 70% confluence in a 100-mm dish for 36 h. For sample preparation, trypsinized cells were collected, washed in PBS, fixed in 2.5% glutaraldehyde in Na cacodylate, embedded, sectioned, and stained with uranyl acetate according to standard procedures.
Western blotting
Muscle tissues and U20S culture cells were homogenized in ice-cold radioimmunoprecipitation assay lysis buffer with protease inhibitor cocktail (Sigma-Aldrich), and lysates were centrifuged at 14,000 g for 10 min. Soluble proteins were quantitated using a BCA protein assay kit (Thermo Fisher Scientific). Aliquots of homogenate were further solubilized in Laemmli sample buffer, and 20–50 µg of protein was subjected to SDS-PAGE (10–12% resolving gel). Proteins were transferred to nitrocellulose membranes (Trans-Blot; 0.2-µm nitrocellulose; Bio-Rad Laboratories). Membranes were blocked in a solution of Tris-buffered saline containing 5% nonfat dry milk. The following antibodies were used: anti-p97 (1:2,000), anti-Lamp1, and anti–lamin A/C (1:2,000; BD); anti-LC3 (1:2,000); anti-p62 (1:4,000), anti-GFP, and antiactin (Sigma-Aldrich); anti-myc and antitubulin (Cell Signaling Technology); and anti–TDP-43 (1:2,000; N terminus; Proteintech). After incubation with the appropriate secondary antibody (horseradish peroxidase–conjugated IgG), bands were visualized by ECL. Immunoblots were scanned with a film scanner (Perfection 1359; Epson), and images were collected in Photoshop CS3. Densitometry was measured with ImageJ.
Nuclear cytoplasmic fractionation
Cells were plated in 10-cm dishes. 8 h after transfection, tetracycline induction was performed, and cells were grown for an additional 36 h. For autophagy inhibition experiments, transfected U20S cells were grown for 24 h and then treated with the indicated concentrations of chloroquine, Baf, or vehicle alone for 6 h before. After treatment, cells were washed twice, scraped in cold PBS, and collected by centrifugation at 700 g for 10 min. The cell pellet was resuspended in 400 µl of buffer A (10 mM, Hepes-KOH, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, and 0.5 mM DTT) supplemented with 0.1 mM PMSF and a protease inhibitor cocktail (Sigma-Aldrich), and the cells were allowed to swell on ice for 30 min and then vortexed for 10 s and passed seven times through a 29-gauge needle. Samples were centrifuged at 700 g for 10 min to pellet nuclei, and the postnuclear supernatant was collected in a new tube. To prevent carryover, the nuclear pellet was washed once in buffer A, and the postnuclear supernatant was additionally centrifuged at top speed for 10 s to remove all light debris and nuclei. To obtain the soluble cytosolic fraction, the postnuclear supernatant was centrifuged at 100,000 g for 1 h, and the soluble cytosolic fraction was concentrated by acetone precipitation. The cytosolic fraction was then resuspended in buffer B (20 mM Hepes-KOH, pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 0.5 mM DTT) supplemented with 1 mM PMSF and a protease inhibitor cocktail. To obtain the nuclear fraction, nuclear pellets were resuspended in 50–70 µl of buffer B supplemented with 1 mM PMSF and a protease inhibitor cocktail and incubated on ice for 20 min for high salt extraction. The nuclear fraction was collected after top speed centrifugation in a new tube. Protein concentration on each fraction was determined by the Bradford assay (Bio-Rad Laboratories), and 2 µg of nuclear and 8 µg of cytosolic fractions were subjected to immunoblotting.
Luciferase assays
Autophagic protein degradation in vivo was measured using in vivo electroporation and bioluminescent imaging of mice, as described previously (Ju et al., 2009). In brief, mice were electroporated with polyQ19- and polyQ80-luciferase constructs into the left and right tibialis anterior skeletal muscle, respectively. After 2 d of recovery, mice were anesthetized (isoflurane inhalation), injected with 150 µg/g d-luciferin, and imaged with a charge-coupled device camera (IVIS imaging system; Caliper) for pretreatment. Regions of interest were defined manually over the tibialis anterior muscles for determining total photon flux (photons per second). The ratio of polyQ80-luciferase/polyQ19-luciferase activity was determined for each animal. Imaging was performed after 24 h of starvation, and the ratio of polyQ80-luciferase/polyQ19-luciferase activity was again determined for each animal. The Δ of the ratio was plotted for comparative purposes.
Statistical analysis
Data were evaluated by the paired Student's t test and/or analysis of variance followed by Fisher LSD post hoc comparisons at P < 0.05.
Online supplemental material
Fig. S1 shows p62 and LC3 immunoblot analysis of skeletal muscle lysates from chloroquine-treated animals. Fig. S2 shows p62 immunoblots from IBMPFD, sIBM, and control patient muscle. Fig. S3 shows histochemical and immunohistochemical analysis of IBMPFD patient skeletal muscle biopsies. Fig. S4 demonstrates TDP-43 immunofluorescence in normal mouse muscle. Fig. S5 shows skeletal muscle histochemistry and p62 immunohistochemistry of chloroquine-treated mice. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200908115/DC1.
Acknowledgments
We thank Dr. Paul Taylor for his helpful comments. We also thank the Alafi Imaging Laboratory at Washington University for confocal microscopy support. Dr. Herbert Virgin supplied the GFP-LC3 and the tfLC3 expression vectors. Dr. Alan Pestronk assisted with human muscle tissue biopsy processing.
Funding for this project was from National Institutes of Health (NIH) grants R01AG031867 (to C.C. Weihl), 5K08AG026271 (to C.C. Weihl), and P50 CA94056 (to D. Piwnica-Worms). We also received support from NIH Neuroscience Blueprint Core grant P30 NS057105 (to Washington University) and Washington University Alzheimer's Disease Research Center grant P50AG05681.
Footnotes
Abbreviations used in this paper:
- Baf
- bafilomycin A
- CFTR
- cystic fibrosis transmembrane regulator
- ERAD
- ER-associated degradation
- FTD
- frontotemporal dementia
- FTLD-U
- frontotemporal lobar degeneration with ubiquitinated inclusions
- IBM
- inclusion body myopathy
- IBMPFD
- IBM, PDB, and FTD
- KD
- knockdown
- LTR
- LysoTracker red
- PDB
- Paget's disease of the bone
- RV
- rimmed vacuole
- sIBM
- sporadic inclusion body myositis
- UPS
- ubiquitin proteasome system
- VCP
- valosin-containing protein
- WT
- wild type
References
- Caccamo A., Majumder S., Deng J.J., Bai Y., Thornton F.B., Oddo S. 2009. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. J. Biol. Chem. 284:27416–27424 10.1074/jbc.M109.031278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal S., Rosser M.F., Cyr D.M., Hanson P.I. 2004. Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol. Biol. Cell. 15:637–648 10.1091/mbc.E03-02-0097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez C., Figarella-Branger D., Meyronet D., Cassote E., Tong S., Pellissier J.F. 2005. Electron microscopy in neuromuscular disorders. Ultrastruct. Pathol. 29:437–450 10.1080/01913120500323175 [DOI] [PubMed] [Google Scholar]
- Filimonenko M., Stuffers S., Raiborg C., Yamamoto A., Malerød L., Fisher E.M., Isaacs A., Brech A., Stenmark H., Simonsen A. 2007. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 179:485–500 10.1083/jcb.200702115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman M.S., Mackenzie I.R., Cairns N.J., Swanson E., Boyer P.J., Drachman D.A., Jhaveri B.S., Karlawish J.H., Pestronk A., Smith T.W., et al. 2006. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J. Neuropathol. Exp. Neurol. 65:571–581 10.1097/00005072-200606000-00005 [DOI] [PubMed] [Google Scholar]
- Gidaro T., Modoni A., Sabatelli M., Tasca G., Broccolini A., Mirabella M. 2008. An Italian family with inclusion-body myopathy and frontotemporal dementia due to mutation in the VCP gene. Muscle Nerve. 37:111–114 10.1002/mus.20890 [DOI] [PubMed] [Google Scholar]
- Gitcho M.A., Strider J., Carter D., Taylor-Reinwald L., Forman M.S., Goate A.M., Cairns N.J. 2009. VCP mutations causing frontotemporal lobar degeneration disrupt localization of TDP-43 and induce cell death. J. Biol. Chem. 284:12384–12398 10.1074/jbc.M900992200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyant-Maréchal L., Laquerrière A., Duyckaerts C., Dumanchin C., Bou J., Dugny F., Le Ber I., Frébourg T., Hannequin D., Campion D. 2006. Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology. 67:644–651 10.1212/01.wnl.0000225184.14578.d3 [DOI] [PubMed] [Google Scholar]
- Halawani D., Latterich M. 2006. p97: The cell's molecular purgatory? Mol. Cell. 22:713–717 10.1016/j.molcel.2006.06.003 [DOI] [PubMed] [Google Scholar]
- Halawani D., LeBlanc A.C., Rouiller I., Michnick S.W., Servant M.J., Latterich M. 2009. Hereditary inclusion body myopathy-linked p97/VCP mutations in the NH2 domain and the D1 ring modulate p97/VCP ATPase activity and D2 ring conformation. Mol. Cell. Biol. 29:4484–4494 10.1128/MCB.00252-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 441:885–889 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- Hübbers C.U., Clemen C.S., Kesper K., Böddrich A., Hofmann A., Kämäräinen O., Tolksdorf K., Stumpf M., Reichelt J., Roth U., et al. 2007. Pathological consequences of VCP mutations on human striated muscle. Brain. 130:381–393 10.1093/brain/awl238 [DOI] [PubMed] [Google Scholar]
- Ikezoe K., Furuya H., Arahata H., Nakagawa M., Tateishi T., Fujii N., Kira J. 2009. Amyloid-beta accumulation caused by chloroquine injections precedes ER stress and autophagosome formation in rat skeletal muscle. Acta Neuropathol. 117:575–582 10.1007/s00401-009-0488-1 [DOI] [PubMed] [Google Scholar]
- Janiesch P.C., Kim J., Mouysset J., Barikbin R., Lochmüller H., Cassata G., Krause S., Hoppe T. 2007. The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat. Cell Biol. 9:379–390 10.1038/ncb1554 [DOI] [PubMed] [Google Scholar]
- Ju J.S., Miller S.E., Hanson P.I., Weihl C.C. 2008. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J. Biol. Chem. 283:30289–30299 10.1074/jbc.M805517200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J.S., Miller S.E., Jackson E., Cadwell K., Piwnica-Worms D., Weihl C.C. 2009. Quantitation of selective autophagic protein aggregate degradation in vitro and in vivo using luciferase reporters. Autophagy. 5:511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura A., Yamazaki T., Kakuda S., Makioka K., Lemere C.A., Fujita Y., Takatama M., Okamoto K. 2009. Phosphorylation-dependent TDP-43 antibody detects intraneuronal dot-like structures showing morphological characters of granulovacuolar degeneration. Neurosci. Lett. 463:87–92 10.1016/j.neulet.2009.06.024 [DOI] [PubMed] [Google Scholar]
- Kim S.H., Shi Y., Hanson K.A., Williams L.M., Sakasai R., Bowler M.J., Tibbetts R.S. 2009. Potentiation of amyotrophic lateral sclerosis (ALS)-associated TDP-43 aggregation by the proteasome-targeting factor, ubiquilin 1. J. Biol. Chem. 284:8083–8092 10.1074/jbc.M808064200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S., Noda T., Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 3:452–460 [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Abeliovich H., Agostinis P., Agrawal D.K., Aliev G., Askew D.S., Baba M., Baehrecke E.H., Bahr B.A., Ballabio A., et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 4:151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Manno A., Kakizuka A. 2007. Involvement of valosin-containing protein (VCP)/p97 in the formation and clearance of abnormal protein aggregates. Genes Cells. 12:889–901 10.1111/j.1365-2443.2007.01099.x [DOI] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Koike M., Sou Y.S., Ueno T., Hara T., Mizushima N., Iwata J., Ezaki J., Murata S., et al. 2007. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 131:1149–1163 10.1016/j.cell.2007.10.035 [DOI] [PubMed] [Google Scholar]
- Korolchuk V.I., Mansilla A., Menzies F.M., Rubinsztein D.C. 2009. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell. 33:517–527 10.1016/j.molcel.2009.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuncl R.W., Bilak M.M., Craig S.W., Adams R. 2003. Exocytotic “constipation” is a mechanism of tubulin/lysosomal interaction in colchicine myopathy. Exp. Cell Res. 285:196–207 10.1016/S0014-4827(03)00034-X [DOI] [PubMed] [Google Scholar]
- Küsters B., van Hoeve B.J., Schelhaas H.J., Ter Laak H., van Engelen B.G., Lammens M. 2009. TDP-43 accumulation is common in myopathies with rimmed vacuoles. Acta Neuropathol. 117:209–211 10.1007/s00401-008-0471-2 [DOI] [PubMed] [Google Scholar]
- Laurin N., Brown J.P., Morissette J., Raymond V. 2002. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70:1582–1588 10.1086/340731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C., Milan G., Romanello V., Masiero E., Rudolf R., Del Piccolo P., Burden S.J., Di Lisi R., Sandri C., Zhao J., et al. 2007. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6:458–471 10.1016/j.cmet.2007.11.001 [DOI] [PubMed] [Google Scholar]
- Muller J.M., Shorter J., Newman R., Deinhardt K., Sagiv Y., Elazar Z., Warren G., Shima D.T. 2002. Sequential SNARE disassembly and GATE-16-GOS-28 complex assembly mediated by distinct NSF activities drives Golgi membrane fusion. J. Cell Biol. 157:1161–1173 10.1083/jcb.200202082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 314:130–133 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Neumann M., Mackenzie I.R., Cairns N.J., Boyer P.J., Markesbery W.R., Smith C.D., Taylor J.P., Kretzschmar H.A., Kimonis V.E., Forman M.S. 2007. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J. Neuropathol. Exp. Neurol. 66:152–157 10.1097/nen.0b013e31803020b9 [DOI] [PubMed] [Google Scholar]
- Nishino I., Fu J., Tanji K., Yamada T., Shimojo S., Koori T., Mora M., Riggs J.E., Oh S.J., Koga Y., et al. 2000. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 406:906–910 10.1038/35022604 [DOI] [PubMed] [Google Scholar]
- Nogalska A., Terracciano C., D'Agostino C., King Engel W., Askanas V. 2009. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 118:407–413 10.1007/s00401-009-0564-6 [DOI] [PubMed] [Google Scholar]
- Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–24145 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- Raben N., Hill V., Shea L., Takikita S., Baum R., Mizushima N., Ralston E., Plotz P. 2008. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 17:3897–3908 10.1093/hmg/ddn292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran N., Munteanu I., Wang P., Aubourg P., Rilstone J.J., Israelian N., Naranian T., Paroutis P., Guo R., Ren Z.P., et al. 2009. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell. 137:235–246 10.1016/j.cell.2009.01.054 [DOI] [PubMed] [Google Scholar]
- Rubinsztein D.C., Cuervo A.M., Ravikumar B., Sarkar S., Korolchuk V., Kaushik S., Klionsky D.J. 2009. In search of an “autophagomometer”. Autophagy. 5:585–589 [DOI] [PubMed] [Google Scholar]
- Sagiv Y., Legesse-Miller A., Porat A., Elazar Z. 2000. GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J. 19:1494–1504 10.1093/emboj/19.7.1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salajegheh M., Pinkus J.L., Taylor J.P., Amato A.A., Nazareno R., Baloh R.H., Greenberg S.A. 2009. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 40:19–31 10.1002/mus.21386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder R., Watts G.D., Mehta S.G., Evert B.O., Broich P., Fliessbach K., Pauls K., Hans V.H., Kimonis V., Thal D.R. 2005. Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann. Neurol. 57:457–461 10.1002/ana.20407 [DOI] [PubMed] [Google Scholar]
- Schuberth C., Buchberger A. 2008. UBX domain proteins: major regulators of the AAA ATPase Cdc48/p97. Cell. Mol. Life Sci. 65:2360–2371 10.1007/s00018-008-8072-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibinski G., Parkinson N.J., Brown J.M., Chakrabarti L., Lloyd S.L., Hummerich H., Nielsen J.E., Hodges J.R., Spillantini M.G., Thusgaard T., et al. 2005. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37:806–808 10.1038/ng1609 [DOI] [PubMed] [Google Scholar]
- Sou Y.S., Tanida I., Komatsu M., Ueno T., Kominami E. 2006. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J. Biol. Chem. 281:3017–3024 10.1074/jbc.M505888200 [DOI] [PubMed] [Google Scholar]
- Suzuki T., Nakagawa M., Yoshikawa A., Sasagawa N., Yoshimori T., Ohsumi Y., Nishino I., Ishiura S., Nonaka I. 2002. The first molecular evidence that autophagy relates rimmed vacuole formation in chloroquine myopathy. J. Biochem. 131:647–651 [DOI] [PubMed] [Google Scholar]
- Watts G.D., Wymer J., Kovach M.J., Mehta S.G., Mumm S., Darvish D., Pestronk A., Whyte M.P., Kimonis V.E. 2004. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36:377–381 10.1038/ng1332 [DOI] [PubMed] [Google Scholar]
- Weihl C.C., Dalal S., Pestronk A., Hanson P.I. 2006. Inclusion body myopathy-associated mutations in p97/VCP impair endoplasmic reticulum-associated degradation. Hum. Mol. Genet. 15:189–199 10.1093/hmg/ddi426 [DOI] [PubMed] [Google Scholar]
- Weihl C.C., Miller S.E., Hanson P.I., Pestronk A. 2007. Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum. Mol. Genet. 16:919–928 10.1093/hmg/ddm037 [DOI] [PubMed] [Google Scholar]
- Weihl C.C., Temiz P., Miller S.E., Watts G., Smith C., Forman M., Hanson P.I., Kimonis V., Pestronk A. 2008. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry. 79:1186–1189 10.1136/jnnp.2007.131334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihl C.C., Pestronk A., Kimonis V.E. 2009. Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul. Disord. 19:308–315 10.1016/j.nmd.2009.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wójcik C., Yano M., DeMartino G.N. 2004. RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J. Cell Sci. 117:281–292 10.1242/jcs.00841 [DOI] [PubMed] [Google Scholar]
- Ye Y., Meyer H.H., Rapoport T.A. 2001. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 414:652–656 10.1038/414652a [DOI] [PubMed] [Google Scholar]