

Abstract
The cannabinoid 1 (CB1) and cannabinoid 2 (CB2) receptor agonist, Δ9-tetrahydrocannabinol (THC), has been shown to be a broad range inhibitor of cancer in culture and in vivo, and is currently being used in a clinical trial for the treatment of glioblastoma. It has been suggested that other plant-derived cannabinoids, which do not interact efficiently with CB1 and CB2 receptors, can modulate the actions of Δ9-THC. However, there are conflicting reports as to what extent other cannabinoids can modulate Δ9-THC activity, and most importantly, it is not clear whether other cannabinoid compounds can either potentiate or inhibit the actions of Δ9-THC. We therefore tested cannabidiol (CBD), the second most abundant plant derived cannabiniod, in combination with Δ9-THC. In U251 and SF126 glioblastoma cell lines, Δ9-THC and CBD acted synergistically to inhibit cell proliferation. The treatment of glioblastoma cells with both compounds led to significant modulations of the cell cycle and induction of reactive oxygen species (ROS) and apoptosis as well as specific modulations of extracellular signal-regulated kinase (ERK) and caspase activities. These specific changes were not observed with either compound individually, indicating that the signal transduction pathways affected by the combination treatment were unique. Our results suggest that the addition of CBD to Δ9-THC may improve the overall effectiveness of Δ9-THC in the treatment of glioblastoma in cancer patients.
Keywords: cannabinoid, synergism, proliferation, invasion, apoptosis
Introduction
Δ9-THC and other cannabinoids can act as direct anticancer agents in multiple types of cancer in culture and in vivo (1). Specifically, activation by Δ9-THC of the two cloned cannabinoid receptors, CB1 and CB2, can lead to the inhibition of cell proliferation, invasion and induction of apoptosis in cancer cell lines resulting in the reduction of tumor burden in vivo (2–4). The promising preclinical therapeutic potential of Δ9-THC, as an inhibitor of glioblastoma, has prompted a human clinical trial (5).
The CB1 and CB2 receptors are members of the G-protein coupled receptor (GPCR) superfamily, and can interact with five structurally distinct classes of compounds. These include the plant-derived classical cannabinoids, such as Δ9-THC; the non-classical bicyclic cannabinoids, such as CP55,940; the endogenous cannabinoids, such as anandamide (AEA); the aminoalkylindoles (AAI), such as WIN55,212-2; and the antagonist/inverse agonists, such as SR141716A (6). Interaction sites, independent of CB1 and CB2 receptors, also appear to be responsible for the anticancer activity of cannabinoids (7–10). There are more than 60 cannabinoids in Cannabis sativa. In addition to Δ9-THC, cannabidiol (CBD), cannabinol (CBN), and cannabigerol (CBG) are also present in the plant (11). CBN has low affinity for CB1 and CB2 receptors, whereas the non-psychotropic cannabinoids, CBD and CBG, have negligible affinity for the cloned receptors (12–14). While CBN and CBG have not been tested for their ability to inhibit human brain cancer, CBD has been reported to inhibit the growth of a human glioblastoma in a xenograft model (7, 15, 16).
There are conflicting reports as to what extent other cannabinoids can modulate the activity of Δ9-THC, and it has been suggested that non-psychoactive cannabinoids can either potentiate or inhibit the actions of Δ9-THC (17–20). Cooperative effects have also been observed with endogenous cannabinoids (21). The potential benefits of using a cannabinoid-based medicine comprised of multiple cannabinoids has been a driving force in recent human clinical trials (20, 22, 23). Investigations have shown that non-pyschoactive cannabinoids can alter the physiological response to Δ9-THC, potentially by altering its metabolism (17–19, 24, 25). However, no investigation to date has provided molecular mechanisms explaining how cannabinoids, acting through distinct pathways, could converge onto a shared pathway resulting in a modulation of activity unique to the combination.
In this study, we sought to determine whether the plant-derived cannabinoid, CBD, would modulate the ability of Δ9-THC to inhibit glioblastoma cell proliferation and survival. We found that CBD enhanced the ability of Δ9-THC to inhibit glioblastoma cell growth and induce apoptosis. The molecular mechanisms associated with these specific effects are presented.
Material and Methods
Cell culture and treatments
The human glioblastoma cell lines used were SF126, U251 and U87. Cell lines were maintained at 37°C and 5% CO2. In all experiments, the different cell populations were first cultured in RPMI media containing 10% fetal bovine serum (FBS). Glioblastoma cells were then seeded into 96-well plates in 10% FBS and on the first day of treatment the media was replaced with vehicle control or drug in RPMI and 0.1% FBS as previously reported (8). The media with the appropriate compounds were replaced every 24 h. Δ9-THC and CBD were obtained from NIH through the National Institute of Drug Abuse.
MTT assay
Assays were performed as previously described (26). % control was calculated as the MTT absorbance of the treated cells/control cells × 100.
Apoptosis analysis
Cells were grown in 6-well culture dishes and treated with the appropriate compounds every 24 h for 3 days. Cells attached to the plate as well as the cells in media were collected, pelleted, washed once with PBS, and processed for labeling with FITC-tagged annexin and propidium iodide (PI) by use of an Apo-Direct apoptosis kit obtained from Phoenix Flow Systems (San Diego, CA). Briefly, the cell pellet was resuspended in 300 µl of the supplied reaction buffer along with 3 µl of both PI and FITC-tagged annexin. After a 15-min incubation period at room temperature, the labeled cells were analyzed by flow cytometry using a FITC detector (FL1) and a PI emission signal detector (FL2). Cell flow cytometry in combination with PI and annexin staining was used to quantify the percentage of cells undergoing apoptosis in control and treatment groups. % control was calculated as annexin positive staining in treated cells/control cells × 100. PI staining was used to distinguish necrotic cells from those undergoing apoptosis.
Cell cycle analysis
U251 cells were grown in Petri dishes (100 mm ×15 mm) and received drug treatments for 2 days. On the third day, the cells were harvested and centrifuged at 1200 rpm for 5 min. The pellet was washed 1X with PBS + 1% BSA, and centrifuged again. The pellet was resuspended in 0.5 ml of 2% paraformaldehyde and fixed overnight at room temperature. The next day the cells were pelleted and resuspended in 0.5 ml 0.3% Triton in PBS and incubated for 5 min at room temperature. The cells were then washed 2 times with PBS + 1% BSA. The cells were finally suspended in PBS (0.1% BSA) with 10 µg/ml Propidium Iodide and 100 µg/ml RNAse. The cells were incubated for 30 min at room temperature before being stored at 4°C. Cell cycle was measured using a FACS Calibur using Cell Quest Pro and Modfit software.
Boyden chamber invasion assay
Assays were performed in modified Boyden Chambers (BD Biosciences, San Diego, CA) as previously described (26). Data were presented as relative invasiveness of the cells through the Matrigel, where the respective controls are set as 100 %.
Reactive oxygen species (ROS) measurements
The production of cellular reactive oxygen species (ROS)/H2O2 was measured using 2’–7’Dichlorodihydrofluorescein (DCFH-DA, Sigma Aldrich). DCFH-DA is deacylated intracellularly into a non-fluorescent product, which reacts with intracellular ROS to produce 2’–7’Dichlorofluorescein, which remains trapped inside the cell, and can be measured quantitatively. Cells were plated onto 6-well dishes and received drug treatments for three days. On the third day, 10 mM DCFH-DA was added to the media (MEM with 0.1% FBS) and the cells were incubated with DCFH-DA overnight. The next day, the cells were trypsinized, washed with PBS, and the fluorescent intensity was measured using a FACS and cell quest pro software.
Western analysis
Western analysis was performed as previously described (26). Anti-phospho-JNK, anti-phospho-p38, anti-phospho-ERK1/2 and anti-ERK1/2 were obtained from Millipore. Anti cleaved caspase 3, 7, 9 and PARP were obtained from Cell Signaling, Boston, MA. Antibodies were added according to the manufactures protocol.
Polymerase Chain Reaction
Total cellular RNA was isolated from glioblastoma cancer cells treated with vehicle control or with CBD. Transcripts for p8 and for β-actin were reverse transcribed using SuperscriptII Reverse TranscriptaseII (Gibco-BRL), and polymerase chain reaction performed. The 5' and 3' PCR primers were GAAGAGAGGCAGGGAAGACA and CTGCCGTGCGTGTCTATTTA for p8; and GCGGGAAATCGTGCGTGACATT and GATGGAGTTGAAGGTAGTTTCGTG for β-actin. PCR was performed in buffer containing 1 µM of each of the 5' and 3' PCR primer and 0.5 U of Taq polymerase using 18 cycles for amplification of p8 and β-actin cDNAs. The cycle conditions were 45 sec denaturation at 94°C, 45 sec annealing at 55°C, and 1 min extension at 72°C.
Pharmacological and statistical analyses
In the proliferation assays, IC50 values with corresponding 95% confidence limits were calculated using non-linear analysis of logged data (GraphPad Prism, San Diego, CA). When just the confidence limits of the IC50 values overlapped significant differences were determined using unpaired Student’s t-test. Significant differences were also determined using one-way ANOVA where suitable. Bonferroni’s multiple comparison post-hoc analyses were conducted when appropriate. P values <0.05 defined statistical significance. Positive and negative aspects of constituent interaction were determined in a 2 × 2 design using two-way ANOVA as described by (27). IC20 and IC80 values were calculated using the equation ICF = (F/100-F)1/H • IC50, where F is the fractional response expressed as a percentage, ICF is the quantity of drug needed to inhibit an F percentage response and H is the hillslope. Treatment groups were divided into 1) no treatment (control), 2) Δ9-THC alone, 3) CBD alone, and 4) Δ9-THC and CBD combined. Data was analyzed using a two-way ANOVA with an interaction term that was used to test for whether the combination of Δ9-THC and CBD differed from the additive effects of each alone (GraphPad Prism, San Diego, CA).
To further test for synergism, the combination index (CI) was calculated where CI< 1, = 1, and >1 indicates synergism, additive effect, and antagonism, respectively (28, 29). Based on the classic isobologram for mutually exclusive effects relative to the end point of measurement, the CI value for × % inhibition is calculated as: CI = (D)1/(Dx)1 + (D)2/(Dx)2 (D)1 THC; (D)2 represents CBD; (Dx)1 and (Dx)2 are the doses for x% growth that can be obtained using the ICF equation described above. (D)1 and (D)2 are the concentrations in the combination that also inhibit cell growth by ×% (29).
A cell cycle analysis program was used to estimate the proportions of cells in each of three compartments: G0/G1, S, and G2/GM. The experiments were conducted on four different dates and there were two replicates for each date. It was noticed that in the control (vehicle) experiments the percentage of cells in each compartment varied significantly from day to day, therefore each treatment compartment percentage estimate was standardized by dividing it by the average percentage for the vehicle on that date. This procedure was carried out for data from each experiment on each day. Multivariate analysis of variance (MANOVA) on the vector of cell cycle compartment standardized ratios (G0/G1, S and G2/GM) was performed to control the multiple comparison type 1 (false positive) error rate. Since this produced a significant result (at p<0.05), it was concluded that there were differences due to treatments. Standardized ratios for each compartment were then tested separately using univariate one-way ANOVA with treatment as the explanatory factor. These tests were also significant at p<0.05 for each compartment except G2/GM. It was concluded that there was evidence for a treatment effect in each of the three cell cycle compartments G0/G1, S and G2/GM. We then tested the ratios for each treatment within a compartment to determine if they significantly differed from 1.0, indicating a treatment effect for that particular treatment. Finally, we tested for a significant interaction of CBD and Δ9-THC, each at their lowest dose by ANOVA with an interaction term. This was carried out after transforming the standardized ratios to logarithms so a test for additive interactions could be performed.
Results
Δ9-THC and CBD inhibit the growth of multiple glioblastoma cell lines
The CB1 and CB2 receptor agonist, Δ9-THC, can inhibit glioblastoma cell proliferation in culture and in vivo and is currently being used in a clinical trial (4). CBD, a cannabinoid constituent with negligible affinity for CB1 and CB2 receptors, can also inhibit the proliferation of glioblastoma in culture and in vivo (7, 16). SF126, U251 and U87 cells were treated for three days with a range of concentrations of either Δ9-THC or CBD. The antiproliferative activity of the compounds was assessed using the MTT assay and the corresponding IC50 values were calculated as previously described (26). The IC50 values for Δ9-THC in SF126, U251 and U87 cells were 2.5 µM (1.8–3.4), 3.3 µM (2.4–4.6) and 3.3 µM (2.3–4.8), respectively. The IC50 values for CBD in SF126, U251 and U87 cells were 1.2 µM (1.1–1.3), 0.6 µM (0.5–1.0), 0.6 µM (0.5–0.7), respectively. CBD was therefore a more potent inhibitor of cell growth than Δ9-THC in the three cell lines studied.
CBD enhances the inhibitory effects of Δ9-THC on glioblastoma cell growth
It has been suggested that non-psychoactive cannabinoid constituents can either potentiate or inhibit the actions of Δ9-THC (11, 17–19). Therefore, the glioblastoma cell lines that were originally used to test the antiproliferative activity of individual cannabinoids were used to determine the effects of combination treatments. The positive and negative aspects of constituent interaction were tested by analyzing the activity of different combinations of Δ9-THC and CBD in a 2 × 2 design (Figure 1). The concentrations used for the treatments were IC80 or IC20 values calculated from the IC50 values as described in the methods. When applied in combination at the predicted IC80 concentration, Δ9-THC and CBD produced a greater than additive inhibition of cell growth in SF126 and U251 cells. This was not observed in U87 cells (data not shown). In SF126 cells, cell viability in presence of Δ9-THC (3.9 µM) was 26 ± 9 %, CBD (1.4 µM) was 40 ± 4 %, and Δ9-THC (3.9 µM) + CBD (1.4 µM) was 8 ± 4 % (Figure 1A). In U251 cells, cell viability in presence of Δ9-THC (5.4 µM) was 55 ± 3 %, CBD (0.9 µM) was 69 ± 4 % and Δ9-THC (5.4 µM) + CBD (0.9 µM) was 2 ± 2 % (Figure 1B). Predicted IC20 concentrations of Δ9-THC and CBD that alone produce only minimal effects on cell growth were combined and further tested in a 2 × 2 factorial design in the positive responding cell lines (SF126 and U251). Again, greater than additive effects were observed in SF126 and U251 cells. In SF126 cells, cell viability in presence of Δ9-THC (1.6 µM) was 90 ± 4 %, CBD (1.1 µM) was 63 ± 4 %, and Δ9-THC (1.6 µM) + CBD (1.1 µM) = 25 ± 6 % (Figure 1C). In U251 cells, cell viability in presence of Δ9-THC (1.7 µM) was 71 ± 4 %, CBD (0.4 µM) was 83 ± 5 % and Δ9-THC (1.7 µM) + CBD (0.4 µM) was 7 ± 4 % (Figure 1D). In the cell lines demonstrating significant interactions (SF126 and U251 cells), we further tested for synergism using the combination index (CI) described in the methods. As shown in Table 1 supplementary, in both U251 and SF126 cells a synergistic increase in the antiproliferative activity of the cannabinoids was observed. A CI value of <1, 1, and >1 indicates synergism, additivity, and antagonism, respectively (29). Synergistic activity was observed at all the concentration ranges tested in U251 cells, therefore, this cell line was used primarily in the remainder of the experiments.
Figure 1. CBD enhances the inhibitory effects of Δ9-THC on glioblastoma cell growth.
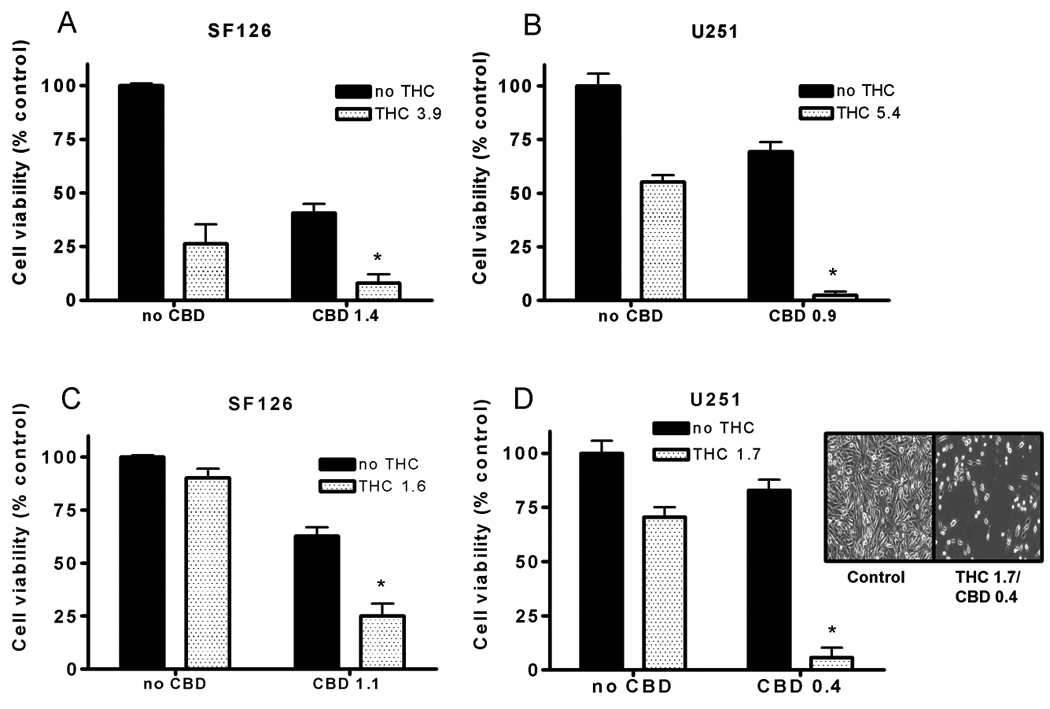
To test for positive and negative interactions a 2 × 2 factorial design using specific µM concentrations of drug were used as described in the methods. Cell proliferation was measured using the MTT assay. A) SF126 and B) U251 cells were treated for three days with vehicle/no drug, Δ9-THC, CBD, or a combination of Δ9-THC and CBD. Concentrations of Δ9-THC and CBD that produce only minimal effects on cell proliferation were also tested in 2 × 2 factorial design in C) SF126 and D) U251 cells. % control was calculated as the MTT product absorbance in the treated cells/control cells × 100. Data are the mean of at least 3 independent experiments; bars, ± SE. Data was analyzed using a two-way ANOVA (GraphPad Prism, San Diego, CA). (*) indicates statistically significant interaction (p<0.01). Inset on D) Representative light microscope image of the effects of the combination treatment on U251 cells (40X).
CBD does not enhance the inhibitory effects of Δ9-THC on glioblastoma cell invasiveness
In addition to uncontrolled cell growth, a hallmark of the aggressive phenotype of glioblastoma cells is their ability to migrate away for the primary tumor of origin and invade into neighboring CNS tissue (30). We, therefore, sought to determine whether the addition of CBD to Δ9-THC would improve the activity of the compound to inhibit migration and invasion through a reconstituted basement membrane in a Boyden chamber assay (Figure 2). Both Δ9-THC and CBD could significantly inhibit the invasiveness of U251 cells. The predicted IC50 values for Δ9-THC and CBD to inhibit U251 cell invasiveness were 85 nM (49–150) and 126 nM (20–796), respectively. Concentrations of 100 nM Δ9-THC and CBD (their approximate EC50 values) were used to test for positive or negative interactions. These concentrations were chosen as compared to the predicted EC80 values to ensure significant increases in cell death would not be produced which could confound the results of the invasion assay, i.e., dead cells will not migrate and invade. Whereas both Δ9-THC and CBD were able to inhibit U251 cell invasiveness, CBD did not enhance the activity of Δ9-THC when the compounds were combined. In U251 cells, invasiveness in presence of Δ9-THC (0.1 µM) was 48 ± 3 %, CBD (0.1 µM) was 72 ± 3 % and Δ9-THC (0.1 µM) + CBD (0.1 µM) was 36 ± 3 %. Since Δ9-THC and CBD acted synergistically to inhibit glioblastoma cell growth, but not to inhibit cell invasiveness, mechanistic experiments were focused on understanding the reduction in cell viability produced by the combination treatment. The 4:1 ratio of Δ9-THC (1.7 µM) and CBD (0.4 µM) (as described above in Figure 1D) was primarily used as the combination treatment for the remainder of the experiments.
Figure 2. Δ9-THC in combination with CBD does not produce a greater overall inhibition of glioma invasiveness.

To test for positive and negative interactions a 2 × 2 factorial design was used as described in the methods. The Boyden chamber invasion assay was used to determine the effects of treatment on the invasiveness of U251 cells. U251 cells were treated for three days with Δ9-THC (0.1 µM), CBD (0.1 µM), or a combination of Δ9-THC (0.1 µM) and CBD (0.1 µM). Data are presented as relative invasiveness of the cells through the Matrigel, where the respective controls are set as 100%. Data are the mean of at least 3 independent experiments; bars, ± SE. (*) indicates statistically significant differences from control (p<0.05).
The combination treatment of Δ9-THC and CBD leads to the modulation of specific mitogen activated kinases (MAPK)
The regulation of ERK, JNK, and p38 MAPK activity plays a critical role in controlling cell growth and apoptosis (31). Modulation of these pathways has been indicated in cannabinoid control of cancer cell growth and survival (9, 32–34). We used U251 cells to determine whether modulation of ERK, JNK, and p38 MAPK activity occurred. Treatment with the combination of cannabinoids led to a substantial down-regulation of pERK, but produced no significant change in total ERK (Figure 3A). Additionally, no inhibition of p38 MAPK or JNK activity was observed. When U251 cells were treated with individual concentration of Δ9-THC and CBD, instead of the combination, no changes in pERK were observed (Figure 3B). These data demonstrate the modulation of pERK was specific for the combination treatment. Down-regulation of pERK in the presence of the combination treatment was first observed after two days treatment in U251 cells (Figure 3C). The down-regulation of pERK was also observed in SF126 cells using a combination treatment of Δ9-THC and CBD (Figure 3D). These data demonstrate the modulation of pERK by the combination treatment of THC and CBD appears to represent a common mechanism shared by different glioblastoma cell lines.
Figure 3. The combination treatment of Δ9-THC and CBD specifically inhibits ERK activity.
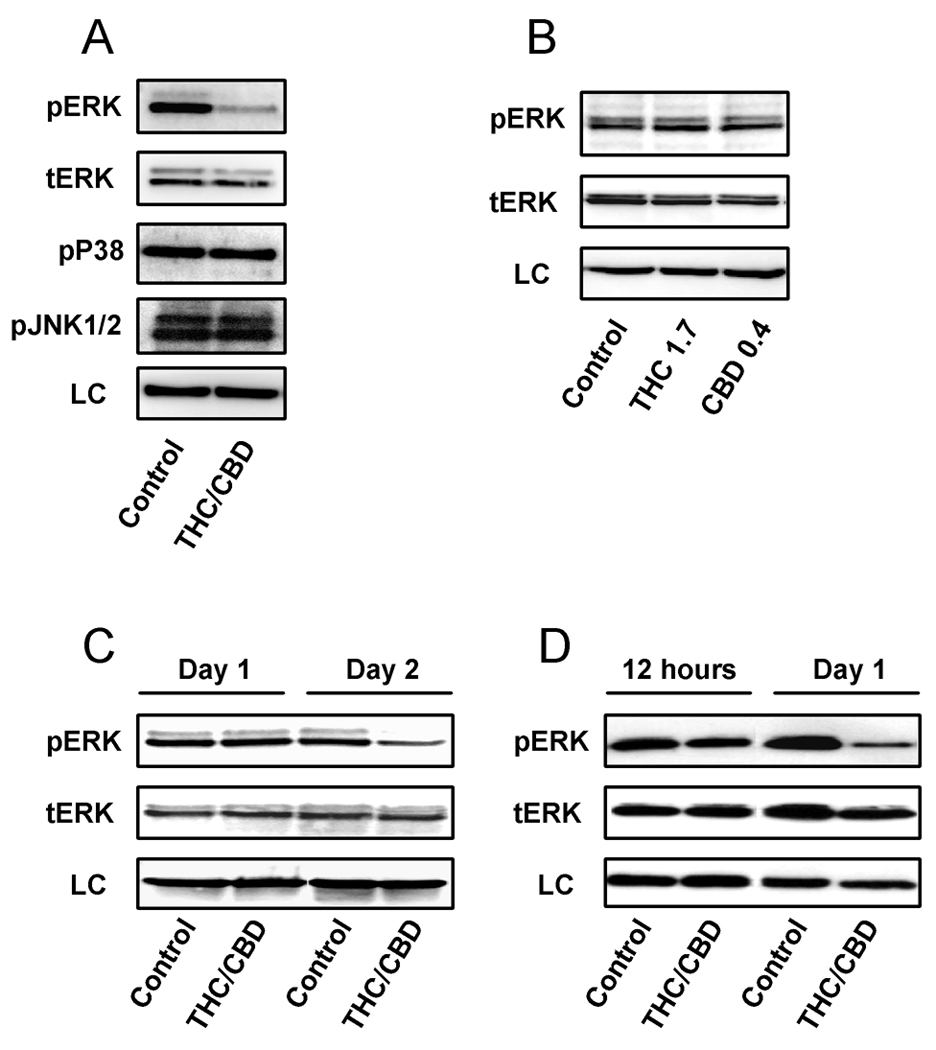
The effects of cannabinoids on kinases activity were analyzed using Western analysis. In A), U251 cells were treated with vehicle or a combination of Δ9-THC (1.7 µM) and CBD (0.4 µM) for three days. Proteins were then extracted and analyzed for pERK, total ERK, pJNK 1/2 and p38 MAPK. In B), U251 cells were treated with Δ9-THC (1.7 µM) or CBD (0.4 µM) alone for three days, and analyzed for pERK and total ERK. In C), U251 cells were treated with vehicle or a combination of Δ9-THC (1.7 µM) and CBD (0.4 µM) for one and two days. In D), SF126 cells were treated with vehicle or a combination of Δ9-THC (1.6 µM) and CBD (1.1 µM) for 12 hours or one day. Either a-tubulin or β-actin was used as a loading control (LC). Blots are representative of at least 3 independent experiments.
The combination treatment of Δ9-THC and CBD inhibits cell cycle and induces apoptosis
Significant reductions in ERK activity have been shown to lead to growth arrest and induction of apoptosis (31). The large reduction in glioblastoma cell viability and ERK activity, observed in the presence of the combination treatment, led us to hypothesize there would be a corresponding modulation of the cell cycle and programmed cell death. Therefore, U251 cells were treated with Δ9-THC and CBD alone or with the combination of the two drugs, and cell cycle was analyzed using cell flow cytometry (Table 1). When administered separately, Δ9-THC and CBD both produced increases in the population of cells in G0/G1 phase, but not in the S and G2/GM phase. The combination of Δ9-THC and CBD produced a greater than additive increase in the population of cells in G0/G1 phase, G2/GM phase and a decrease in cells in S phase.
Table 1. Cannabinoid modulation of cell cycle.
Cell cycle was measured using PI staining and FACS analysis, and Modfit was used to determine the percentage of cell in G0/G1, S and G2/GM phase. U251 cells were treated for three days with CBD (0.4 µM), Δ9-THC (1.7 µM), or a combination of CBD (0.4 µM) and Δ9-THC (1.7 µM). The percentage of cells in each compartment was standardized by dividing it by the average percentage for the vehicle. This procedure was carried out for data from each experiment on each day. Statistical analysis was performed as described in the Material and Methods.
| Treatment | mean(G0/G1) | mean(S) | mean(G2/GM) |
|---|---|---|---|
| CBD 0.4 | 1.08* ± 0.03 | 0.90 ± 0.05 | 0.97 ± 0.28 |
| Δ9-THC 1.7 | 1.12* ± 0.04 | 0.78 ± 0.11 | 1.28 ± 0.37 |
| Δ9-THC 1.7/ CBD 0.4 |
1.23*# ± 0.02 | 0.49*# ± 0.08 | 2.69*# ± 0.56 |
p<0.05
significant interaction
In addition to producing cell cycle arrest, the combination treatment may reduce cell viability through induction of apoptosis. We, therefore, measured apoptosis using annexin staining in combination with cell flow cytometery (Figure 4A). There was minor increase in apoptosis produced with 1.7 µM Δ9-THC, but it was not found to be significantly different from control (n=7). No increase in apoptosis was observed in the presence of 0.4 µM CBD. However, when Δ9-THC and CBD were combined, a greater than additive increase in apoptosis was observed. In a time course analysis studying the induction of apoptosis produced by the combination treatment, we observed only a small increase with the combination treatment after 2 days (Figure 1A and B supplementary), whereas a strong induction of apoptosis was observed by day 3 (Figure 4A).
Figure 4. The effects of the combination treatment are the result of CB2 activation.
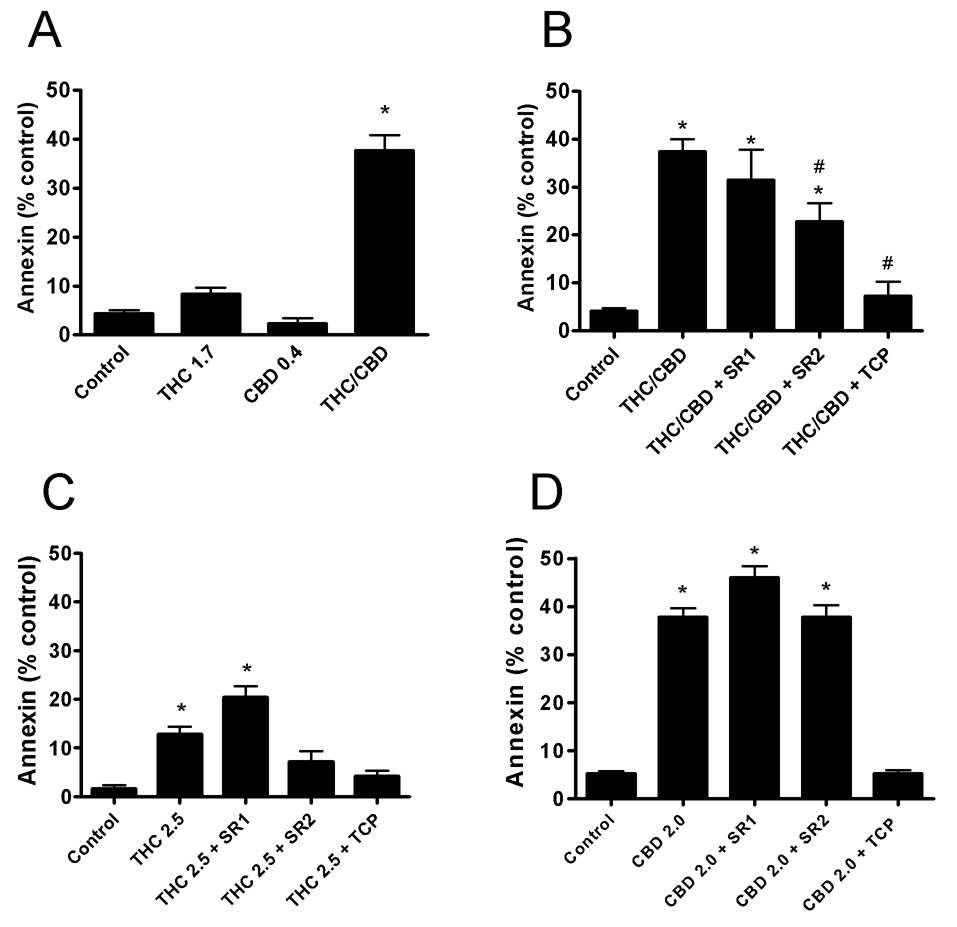
The number of U251 cells positive for annexin (apoptosis) staining after three days treatment were measured using FACS analysis. Cells were treated with: A) Δ9-THC (1.7 µM), CBD (0.4 µM), or a combination of Δ9-THC (1.7 µM) and CBD (0.4 µM) denoted (THC/CBD); B) a combination of Δ9-THC (1.7 µM) and CBD (0.4 µM) denoted (THC/CBD); C) 2.5 µM Δ9-THC; and D) 2.0 µM CBD. In B), C), and D), cells were also treated in the presence of 0.5 µM of the CB1 antagonist, SR141716A (SR1), 0.5 µM of the CB2 antagonist, SR144528 (SR2) or 20 µM α-tocopherol (TCP). % control was calculated as positive annexin staining of the treated cells minus control cells. Data are the mean of at least 3 independent experiments; bars, ± SE. Data were compared using a one-way ANOVA with a Bonferroni’s multiple comparison post-hoc analyses. (*) indicates statistically significant differences from control (p<0.05). (#) indicates statistically significant differences from the combination treatment of THC/CBD (p<0.05).
The inhibitory effects of the combination treatment are the result of CB2 receptor activation and production of ROS
Depending on the glioblastoma cell line used, studies have linked the inhibitory activity of plant derived cannabinoids to activation of CB1 and/or CB2, modulation of MAPKs and induction of cellular stress through increases in ROS and additional stress related proteins, leading to activation of caspases (15, 32, 35). We used the measure of apoptosis to investigate mechanisms by which CBD enhanced the activity of Δ9-THC.
Apoptosis produced by the combination of Δ9-THC and CBD was partially blocked by the CB2 receptor antagonist, SR144528 (SR2), but almost complete reversal was observed in the presence of the anti-oxidant, α-tocopherol (TCP) (Figure 4B). The cannabinoid receptor antagonists and TCP had no effect on apoptosis on their own at 0.5 µM and 20 µM, respectively (data not shown). As predicted by α-tocopherol blockade, the combination of Δ9-THC and CBD produced a significant increase in the formation of ROS as assessed by DCDHF-DA oxidation using FACS analysis (Figure 1C and D supplementary). A small increase in ROS was observed after 1day of treatment with a major induction observed by day 2, preceding the majority of the observed apoptotic cell death.
In order to attempt to match levels of apoptosis produced by the combination treatment, the concentrations of the individual cannabinoids (Δ9-THC and CBD) were next increased. The purpose of these experiments was to determine whether the compounds alone recruited similar pathways as compared to the combination of Δ9-THC and CBD. When U251 cells were treated with Δ9-THC alone, the induction of apoptosis was almost completely blocked by α-tocopherol (TCP) and partially blocked by the CB2 antagonist SR144528 (SR2) (Figure 4C). However, Δ9-THC alone could not produce the level of apoptosis observed with the combination treatment (Figure 4B and C). This finding was not simply an issue of the treatment concentration used since application of Δ9-THC up to 5 µM did not produce a greater induction of apoptosis (data not shown). When U251 cells were treated with CBD alone, the induction of apoptosis was blocked by α-tocopherol (TCP) but no reversal was observed with SR144528 (SR2) (Figure 4D). This result was expected since CBD does not interact efficiently with either CB1 or CB2 receptors.
The ability of the higher concentrations of Δ9-THC (2.5 µM) and CBD (2.0 µM) to inhibit pERK were also studied and compared to the combination treatment (Figure 2 supplementary). Again, the combination treatment produced a substantial down regulation of pERK. However, the higher concentration of Δ9-THC alone had no effect on pERK. The higher concentration of CBD produced only a small inhibition of pERK. Taken together, this data suggests that a unique pathway was activated by the combined administration of Δ9-THC and CBD, which led to the down-regulation of pERK.
Cannabinoid mediates apoptosis through p8 and caspases
The induction of the stress associated gene, p8, has been shown to be a specific event in THC-induced apoptosis, but its involvement in CBD induced apoptosis has not been determined (35, 36). This pathway was evaluated to determine the role p8 played in the observed increase in apoptosis during cannabinoid treatments (Figure 5A and B). Treatment of U251 cells with CBD lead to a small reduction in p8 expression compared to control, however, this change was not statistically significant (n=4). Treatment with Δ9-THC alone or the combination of Δ9-THC and CBD led to an up-regulation of p8 expression. The magnitude of the effect was similar between treatment groups. This data demonstrates the modulation of p8 was not specific for the combination treatment.
Figure 5. When combined, Δ9-THC and CBD produce an increase in activation of p8 and multiple caspases.
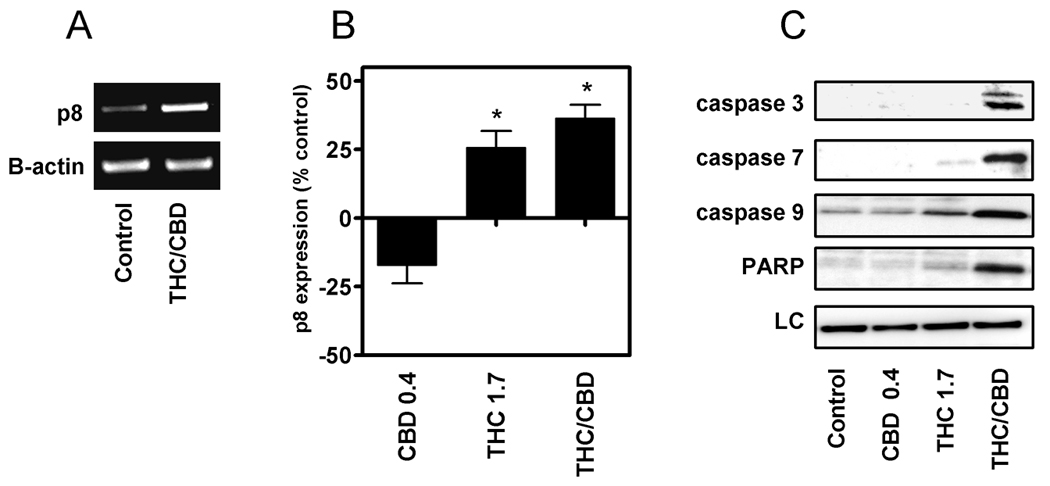
The effects of cannabinoids on p8 and caspase expression were analyzed using semi-quantitative RT-PCR and Western analysis, respectively. RNA and protein were collected from U251 cells treated for three days with CBD (0.4 µM), Δ9-THC (1.7 µM), or a combination of Δ9-THC (1.7 µM) and CBD (0.4 µM). A) RT-PCR was run on RNA extracted from control-treated and Δ9-THC/CBD-treated samples. Expression of the β-actin gene product was used as a control for equal loading. B) Data are represented as percentage p8 expression of the treated cells/control cells ×100 and all values were normalized against β-actin. Blots and PCR reactions are representative of at least 3 independent experiments. Data were compared using a one-way ANOVA with a Dunnett’s multiple comparison post-hoc analyses. (*) indicates statistically significant differences from control (p<0.05). C) Proteins were extracted from treated cells and analyzed for cleaved caspase 3, 7, 9 and PARP expression.
Multiple caspase pathways were next evaluated to determine additional mechanisms by which the combination treatment increased apoptosis (Figure 5C). In the presence of CBD alone, no significant changes in caspase activity were observed. Small increases in the activity of caspase 7, caspase 9 and PARP, but not caspase 3, were observed when U251 cells were treated with the Δ9-THC alone. Treatment with the combination of Δ9-THC and CBD led to a substantial up-regulation of caspase 3, 7, and 9 activities as well as an increase in PARP expression. These data demonstrate that a unique modulation of caspase activity is produced when glioblastoma cells are treated with the combination of Δ9-THC and CBD as opposed to the individual cannabinoids.
Discussion
We observed that plant derived cannabinoids inhibit the proliferation of human glioblastoma cell lines. Compared to Δ9-THC, CBD was significantly more potent than Δ9-THC at inhibiting cancer cell growth. This finding is in agreement with studies using models of aggressive breast cancers (26, 37). Δ9-THC is currently being used in a clinical trial for treatment of recurrent glioblastoma (5). Past studies have suggested that non-psychoactive cannabinoids can modulate the actions of Δ9-THC (17–20). We hypothesized that the cannabinoid therapy utilizing Δ9-THC alone could be improved using a strategy of combination treatments.
We discovered that CBD enhanced the ability of Δ9-THC to inhibit cell proliferation and induce cell cycle arrest and apoptosis. This activity occurred in two of three glioblastoma cell lines tested. Treatment of U251 cells with the combination led to a substantial down-regulation of ERK activity, but not p38 MAPK and JNK1/2. The reduction in ERK activity was specific for the combination treatment and occurred in more than one glioblastoma cell line. Importantly, continuing to increase the concentration of Δ9-THC alone did not result in inhibition of ERK activity. This data indicates that the enhanced effects observed were not solely due to an increase in potency of Δ9-THC in U251 cells upon co-application with CBD. Further support for this conclusion was observed when studying the activity of Δ9-THC and CBD on U251 cell invasiveness. Both compounds were effective at inhibiting the invasiveness of U251 cells, however, there was no evidence that CBD improved the activity of Δ9-THC upon co-application.
In human glioblastoma cells, the ability of Δ9-THC to inhibit growth and induce apoptosis has been linked to the initial activation of CB1 and CB2 receptors (35). Similar effects produced by CBD have been linked in part to CB2 receptor activation, but the initial interaction site for the additional activity of CBD remains to be clarified (7). We observed that increases in apoptosis produced by Δ9-THC alone, or the combination of Δ9-THC and CBD, were partially dependent on CB2 receptor activation. Apoptosis produced by CBD alone was not dependent on CB2 receptor activation. Importantly, the induction of apoptosis in the presence of the combination treatment was significantly greater than that observed with Δ9-THC alone. Apoptosis produced by the combination of Δ9-THC and CBD was dependent on the production of oxidative stress and resulted in a unique activation of both intrinsic and extrinsic caspases.
Studies have shown that the inhibitory activity of cannabinoids in glioblastoma is dependent on activation of CB1 and CB2 receptors, modulation of MAPKs and induction of multiple types of cellular stresses leading to apoptosis (32, 35, 38, 39). In the case of Δ9-THC, up-regulation of p8 appears to be a specific event which leads to apoptosis in multiple types of cancers (35, 36). Treatment of U251 cells with the combination of Δ9-THC and CBD led to an up-regulation of p8 expression, but similar activity was seen with Δ9-THC alone. This is in contrast to what we observed when studying modulation of caspase activity, and suggests that the enhanced apoptotic activity produced by the combination treatment was not the results of an interaction with the p8 pathway.
The ability of CBD to inhibit growth and induce apoptosis in glioblastoma and additional cancers has been primarily associated with the up-regulation of ROS and multiple caspases, and has been linked to alterations in NADPH oxidases (9, 15). A link between ROS production and modulation of the LOX pathway has been hypothesized as a potential mechanism of antitumor activity of CBD in glioblastoma (16). In this study, an initial increase in ROS was clearly linked to a latter induction of apoptosis. Individually, both Δ9-THC and CBD could increase apoptosis through the production of ROS, however, Δ9-THC was significantly less efficient at inducing this process as a single agent as compared to when it was used in combination with CBD. Even though the concentration of CBD used in the combination treatment did not significantly stimulate ROS, it may have primed this pathway for Δ9-THC through a convergence on shared signal transduction pathways. A similar hypothesis could explain the unique down-regulation of phosphorylated ERK that was produced by the combination treatment. Alternatively, CBD may have potentiated the activity of Δ9-THC by inhibiting pathways that impart drug resistance in glioblastoma. For instance, a recent study showed that amphiregulin expression was associated with increased ERK activation, which mediated resistance to THC-induced apoptosis in gliomas (40). Therefore, CBD may have potentiated the activity of THC-induced apoptosis by inhibiting amphiregulin regulated increases in ERK activation. Future studies will be needed in order to elucidate the detailed mechanism associated with the unique effects of the Δ9-THC and CBD combination treatment.
Individually, Δ9-THC and CBD can activate distinct pathways in glioblastoma cells that ultimately culminate in inhibition of cancer cell growth and invasion as well as induction of cell death (2, 4, 41). We hypothesized that, if the individual agents were combined, a convergence on shared pathways may ensue leading to an enhanced ability of the combination treatment to inhibit certain cancer cell phenotypes. We found this to be true in this investigation. Cannabidiol significantly improved the inhibitory effects of Δ9-tetrahydrocannabinol on glioblastoma cell proliferation and survival, but not on cell invasiveness. The data suggests that the improved activity observed with the combination treatment is the result of a specific modulation of ERK and ROS activity leading to inhibition of cell cycle and induction of apoptosis.
Combinations, compared to individual drug treatments with specific cannabinoid-based compounds, may represent an improvement for the treatment of patients with glioblastoma and perhaps additional cancers. It is also possible that other constituents of Cannabis sativa which are not structurally related to cannabinoids could improve antitumor activity when combined. An important next step will be to perform studies testing for synergistic antitumor activity of cannabinoids in additional preclinical models of glioblastoma. Even if synergism is not evident, combination treatments may allow for increased dosing due to non-overlapping toxicities and decrease development of resistance to the activity of Δ9-THC or CBD when administered alone. With the growing evidence demonstrating cannabinoids are effective inhibitors of multiple types of cancer, it is likely that additional clinical trials will be carried out. Combination treatments with cannabinoids may improve overall efficacy in these future clinical trials.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (grants CA102412 and DA09978), and the SETH Group.
Abbreviations
- CB
cannabinoid
- CBD
cannabidiol
- CBG
cannabigerol
- CBN
cannabinol
- Δ9-THC
Δ9-tetrahydrocannabinol
- ERK
extracellular signal regulated kinases
- ROS
reactive oxygen species
References
- 1.Bifulco M, Di Marzo V. Targeting the endocannabinoid system in cancer therapy: a call for further research. Nat Med. 2002;8:547–550. doi: 10.1038/nm0602-547. [DOI] [PubMed] [Google Scholar]
- 2.Blazquez C, Carracedo A, Salazar M, et al. Down-regulation of tissue inhibitor of metalloproteinases-1 in gliomas: a new marker of cannabinoid antitumoral activity? Neuropharmacology. 2008;54:235–243. doi: 10.1016/j.neuropharm.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 3.Preet A, Ganju RK, Groopman JE. Delta(9)-Tetrahydrocannabinol inhibits epithelial growth factor-induced lung cancer cell migration in vitro as well as its growth and metastasis in vivo. Oncogene. 2007 doi: 10.1038/sj.onc.1210641. [DOI] [PubMed] [Google Scholar]
- 4.Velasco G, Carracedo A, Blazquez C, et al. Cannabinoids and gliomas. Mol Neurobiol. 2007;36:60–67. doi: 10.1007/s12035-007-0002-5. [DOI] [PubMed] [Google Scholar]
- 5.Guzman M, Duarte MJ, Blazquez C, et al. A pilot clinical study of Delta9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197–203. doi: 10.1038/sj.bjc.6603236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacological Therapeutics. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 7.Massi P, Vaccani A, Ceruti S, Colombo A, Abbracchio MP, Parolaro D. Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J Pharmacol Exp Ther. 2004;308:838–845. doi: 10.1124/jpet.103.061002. [DOI] [PubMed] [Google Scholar]
- 8.McAllister SD, Chan C, Taft RJ, et al. Cannabinoids selectively inhibit proliferation and induce death of cultured human glioblastoma multiforme cells. J Neurooncol. 2005;74:31–40. doi: 10.1007/s11060-004-5950-2. [DOI] [PubMed] [Google Scholar]
- 9.McKallip RJ, Jia W, Schlomer J, Warren JW, Nagarkatti PS, Nagarkatti M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol Pharmacol. 2006;70:897–908. doi: 10.1124/mol.106.023937. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz L, Miguel A, Diaz-Laviada I. Delta9-tetrahydrocannabinol induces apoptosis in human prostate PC-3 cells via a receptor-independent mechanism. FEBS Lett. 1999;458:400–404. doi: 10.1016/s0014-5793(99)01073-x. [DOI] [PubMed] [Google Scholar]
- 11.McPartland JM, Russo EB. Cannabis and cannabis extract: greater than the sum of the parts? J Cannabis Therapeut. 2001;1:103–132. [Google Scholar]
- 12.Howlett AC. Cannabinoid inhibition of adenylate cyclase: relative activity of the constituents and metabolites of marijuana. Neuropharmacology. 1987;26:507–512. doi: 10.1016/0028-3908(87)90035-9. [DOI] [PubMed] [Google Scholar]
- 13.Devane WA, Dysarz IFA, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- 14.Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): Identification of cannabinoid receptor subtype selective ligands. The Journal of Pharmacology and Experimental Therapeutics. 1996;278:989–999. [PubMed] [Google Scholar]
- 15.Massi P, Vaccani A, Bianchessi S, Costa B, Macchi P, Parolaro D. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell Mol Life Sci. 2006;63:2057–2066. doi: 10.1007/s00018-006-6156-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Massi P, Valenti M, Vaccani A, et al. 5-Lipoxygenase and anandamide hydrolase (FAAH) mediate the antitumor activity of cannabidiol, a non-psychoactive cannabinoid. J Neurochem. 2008;104:1091–1100. doi: 10.1111/j.1471-4159.2007.05073.x. [DOI] [PubMed] [Google Scholar]
- 17.Jones G, Pertwee RG. A metabolic interaction in vivo between cannabidiol and 1 - tetrahydrocannabinol. Br J Pharmacol. 1972;45:375–377. doi: 10.1111/j.1476-5381.1972.tb08092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krantz JC, Berger HJ, Welch BL. Blockade of (−)-trans-delta-9-tetrahydrocannabinol depressant effect by cannabinol in mice. Amer J Phar. 1971;143:149–152. [PubMed] [Google Scholar]
- 19.Poddar MK, Bhattacharya KC, Ghosh JJ. Potentiating effects of cannabidiol on delta-9-tetrahydrocannabinol-induced changes in hepatic enzymes. Biochem Pharmacol. 1974;23:758–759. doi: 10.1016/0006-2952(74)90643-1. [DOI] [PubMed] [Google Scholar]
- 20.Russo E, Guy GW. A tale of two cannabinoids: the therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med Hypotheses. 2006;66:234–246. doi: 10.1016/j.mehy.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Shabat S, Fride E, Sheskin T, et al. An entourage effect: inactive endogenous fatty acid glycerol esters enhance 2-arachidonoyl-glycerol cannabinoid activity. Eur J Pharmacol. 1998;353:23–31. doi: 10.1016/s0014-2999(98)00392-6. [DOI] [PubMed] [Google Scholar]
- 22.Nurmikko TJ, Serpell MG, Hoggart B, Toomey PJ, Morlion BJ, Haines D. Sativex successfully treats neuropathic pain characterised by allodynia: a randomised, double-blind, placebo-controlled clinical trial. Pain. 2007;133:210–220. doi: 10.1016/j.pain.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 23.Rog DJ, Nurmikko TJ, Young CA. Oromucosal delta9-tetrahydrocannabinol/cannabidiol for neuropathic pain associated with multiple sclerosis: an uncontrolled, open-label, 2-year extension trial. Clin Ther. 2007;29:2068–2079. doi: 10.1016/j.clinthera.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Bornheim LM, Grillo MP. Characterization of cytochrome P450 3A inactivation by cannabidiol: possible involvement of cannabidiol-hydroxyquinone as a P450 inactivator. Chem Res Toxicol. 1998;11:1209–1216. doi: 10.1021/tx9800598. [DOI] [PubMed] [Google Scholar]
- 25.Bornheim LM, Kim KY, Li J, Perotti BY, Benet LZ. Effect of cannabidiol pretreatment on the kinetics of tetrahydrocannabinol metabolites in mouse brain. Drug Metab Dispos. 1995;23:825–831. [PubMed] [Google Scholar]
- 26.McAllister SD, Christian RT, Horowitz MP, Garcia A, Desprez PY. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol Cancer Ther. 2007;6:2921–2927. doi: 10.1158/1535-7163.MCT-07-0371. [DOI] [PubMed] [Google Scholar]
- 27.Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30:723–731. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 28.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 29.Chou TC, Tan QH, Sirotnak FM. Quantitation of the synergistic interaction of edatrexate and cisplatin in vitro. Cancer Chemother Pharmacol. 1993;31:259–264. doi: 10.1007/BF00685668. [DOI] [PubMed] [Google Scholar]
- 30.Visted T, Enger PO, Lund-Johansen M, Bjerkvig R. Mechanisms of tumor cell invasion and angiogenesis in the central nervous system. Front Biosci. 2003;8:e289–e304. doi: 10.2741/1026. [DOI] [PubMed] [Google Scholar]
- 31.Chang F, Steelman LS, Lee JT, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17:1263–1293. doi: 10.1038/sj.leu.2402945. [DOI] [PubMed] [Google Scholar]
- 32.Galve-Roperh I, Sanchez C, Cortes ML, del Pulgar TG, Izquierdo M, Guzman M. Anti-tumoral action of cannabinoids: involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat Med. 2000;6:313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- 33.Gustafsson K, Christensson B, Sander B, Flygare J. Cannabinoid receptor-mediated apoptosis induced by R(+)-methanandamide and Win55,212-2 is associated with ceramide accumulation and p38 activation in mantle cell lymphoma. Mol Pharmacol. 2006;70:1612–1620. doi: 10.1124/mol.106.025981. [DOI] [PubMed] [Google Scholar]
- 34.Melck D, Rueda D, Galve-Roperh I, De Petrocellis L, Guzman M, Di Marzo V. Involvement of the cAMP/protein kinase A pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999;463:235–240. doi: 10.1016/s0014-5793(99)01639-7. [DOI] [PubMed] [Google Scholar]
- 35.Carracedo A, Lorente M, Egia A, et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell. 2006;9:301–312. doi: 10.1016/j.ccr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Carracedo A, Gironella M, Lorente M, et al. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006;66:6748–6755. doi: 10.1158/0008-5472.CAN-06-0169. [DOI] [PubMed] [Google Scholar]
- 37.Ligresti A, Moriello AS, Starowicz K, et al. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J Pharmacol Exp Ther. 2006;318:1375–1387. doi: 10.1124/jpet.106.105247. [DOI] [PubMed] [Google Scholar]
- 38.Jacobsson SO, Wallin T, Fowler CJ. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J Pharmacol Exp Ther. 2001;299:951–959. [PubMed] [Google Scholar]
- 39.Sanchez C, Galve-Roperh I, Canova C, Brachet P, Guzman M. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998;436:6–10. doi: 10.1016/s0014-5793(98)01085-0. [DOI] [PubMed] [Google Scholar]
- 40.Lorente M, Carracedo A, Torres S, et al. Amphiregulin is a factor for resistance of glioma cells to cannabinoid-induced apoptosis. Glia. 2009;57:1374–1385. doi: 10.1002/glia.20856. [DOI] [PubMed] [Google Scholar]
- 41.Parolaro D, Massi P. Cannabinoids as potential new therapy for the treatment of gliomas. Expert Rev Neurother. 2008;8:37–49. doi: 10.1586/14737175.8.1.37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.