

Abstract
Background
Arsenic is a ubiquitous element that is a potential carcinogen and teratogen and can cause adverse developmental outcomes. Arsenic exerts its toxic effects through the generation of reactive oxygen species (ROS) that include hydrogen peroxide (H2O2), superoxide-derived hydroxyl ion, and peroxyl radicals. However, the molecular mechanisms by which arsenic induces cytotoxicity in murine embryonic maxillary mesenchymal (MEMM) cells are undefined.
Methods
MEMM cells in culture were treated with different concentrations of pentavalent sodium arsenate [As (V)] for 24 or 48 hours and various end points measured.
Results
We show that treatment of MEMM cells with the pentavalent form of inorganic arsenic resulted in caspase-mediated apoptosis, accompanied by generation of ROS and disruption of mitochondrial membrane potential. Treatment with caspase inhibitors markedly blocked apoptosis. In addition, the free radical scavenger N-acetylcysteine dramatically attenuated arsenic-mediated ROS production and apoptosis, and exposure to arsenate increased Bax and decreased Bcl protein levels in MEMM cells.
Conclusions
Taken together, these findings suggest that in MEMM cells, arsenate-mediated oxidative injury acts as an early and upstream initiator of the cell death cascade, triggering cytotoxicity, mitochondrial dysfunction, altered Bcl/Bax protein ratios, and activation of caspase-9.
Keywords: Reactive oxygen species, arsenate, mitochondria, apoptosis, caspases
INTRODUCTION
Arsenic is a ubiquitous element that exists in a variety of oxidation states in nature, with the +3 and +5 states being the most predominant (HSDB, 2001; ATSDR, 2007; Kachinskas et al. 1994). In the United States, arsenic is primarily used for manufacturing pesticides, but is also a by-product of the smelting industry and of the burning of arsenic-contaminated coal. Humans are exposed to arsenic chiefly through inhalation or ingestion of arsenic-contaminated water (Gebel, 1999). Chronic exposure to inorganic arsenical compounds can cause liver toxicity in addition to increased risk of cancers of the lung, bladder, skin and liver (Smith et al., 1992). For these reasons, the US Environmental Protection Agency (EPA) has placed arsenic at the top of its list of hazardous substances (US EPA, 2001). In addition to its potential as a carcinogen, arsenic has also been implicated as a teratogen, causing adverse reproductive outcomes in animal models (Ferm and Carpenter, 1968; Golub et al., 1998; Holson et al., 2000; Spiegelstein et al, 2005; Wang et al., 2006). Prolonged low-level exposure to arsenic has been associated with spontaneous abortion, stillbirth, developmental impairment and congenital malformations (Pastides et al., 1988). The hazard to the developing fetus of arsenic exposure is exacerbated by the fact that arsenic can cross the placenta and accumulate in the fetus during gestation (Lindgren et al., 1984).
In mammals, four main forms of arsenic can be found: arsenite (As III), arsenate (As V), monomethlyarsenic acid (MMA) and dimethlyarsenic acid (DMA) (Vahter et al., 2002). The relative teratogenic potency of these forms is not clear. Arsenic is known to generate various reactive oxygen species (ROS), including hydrogen peroxide (H2O2), superoxide-derived hydroxyl ion (*OH), and peroxyl (ROO*) radicals (Hei et al., 1998; Oya-Ohta et al., 1996). Other studies have reported arsenical compounds causing a dose dependent increase in ROS in cultured human-hamster hybrid cells (Liu et al., 2001) and Chinese hamster ovary (CHO) cells (Wang et al., 1996). Preliminary studies in our laboratory demonstrated significant increases in exencephaly and orofacial dysmorphology in murine embryos following in utero arsenate exposure. The craniofacial region in the developing embryo is one of the most dynamically growing areas, which renders it highly susceptible to various malformations, particularly those induced by exposure to teratogens. Normal development is dependent upon exquisitely tuned events – both morphological and molecular. It thus stands to reason that any alteration in one of these coordinated processes can lead to abnormal development of the craniofacial region. In the United States, common orofacial malformations, such as cleft lip and cleft palate, occur with a frequency of 1 in 700 live births annually (March of Dimes, 2008). A common feature in cases of orofacial clefting, in humans and animal models, is a significant growth insufficiency of the lip, palate, and/or surrounding tissues (Bhattacherjee et al., 2003). The developing mammalian midfacial region, derived primarily from the maxillary processes of the first branchial arch, has proven to be an excellent experimental system for understanding the regulation and interaction of molecular signals during embryogenesis (Dhulipala et al., 2004; Pisano et al., 2003; Warner et al., 2005). Thus, the present study was designed to test the hypothesis that pentavalent arsenate, like trivalent arsenite, causes cell death in primary cultures of murine embryonic maxillary mesenchymal (MEMM) cells via a mechanism involving the generation of reactive oxygen species and subsequent mitochondrial perturbation. We show here that arsenate mediated cytotoxicity involves generation of reactive oxygen species (ROS), changes in the protein ratio of mitochondrial proteins Bcl (anti-apoptotic) and Bax (pro-apoptotic), mitochondrial membrane perturbation and activation of caspases 3 and 9. To our knowledge, this is the first study that describes a mechanism of arsenate-mediated apoptosis in an in vitro system relevant to murine orofacial development.
MATERIALS and METHODS
Materials
Sodium arsenate (99.4% pure), and N-acetylcysteine (NAC) were obtained from Sigma Chemical Company (St. Louis, MO), 5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyaniniodide (JC-1) and MitoTracker Orange were obtained from Molecular Probes (Seattle, WA). CytoTox 96® non-radioactive cytotoxycity assay kit was purchased from Promega (Madison, WI) while membrane permeable caspase inhibitors were purchased from R&D Systems (Minneapolis, MN). Polyclonal antibodies against Bcl, Bax and β-actin were obtained from Santa Cruz (Santa Cruz, CA).
Methods
Animal dosing and primary cell cultures
ICR mice (Harlan, Indianapolis, IN, USA) were housed in a controlled environment at a temperature of 22°C with an alternating light/dark cycle. Mature male and female mice were mated overnight and the presence of a vaginal plug the following morning was taken as evidence of mating (gestational day 0). Pregnant dams were injected IP with 20 mg/kg sodium arsenate or saline on days 7 and 8 of gestation and embryos removed for observation on gd 10 and 17. To establish primary cell cultures, embryos were removed from pregnant dams on gd 13 and embryonic maxillofacial tissue was dissected in sterile, cold phosphate-buffered saline. Cells were dispersed by gentle trypsinization with 0.025% Trypsin/0.27 mM EDTA for 10 minutes at 37°C, and plated at a density of 6 × 103 cells/cm2. These cells are referred to as MEMM (murine embryonic maxillary mesenchyme) cells.
Determination of Cytotoxicity
Arsenate cytotoxicity was determined at different time intervals by colorimetric measurement of cellular lysis-induced release of lactate dehydrogenase (LDH) into culture medium using the CytoTox 96® kit (Promega, Madison, WI). This assay is based on the generation of NADH by reduction of lactate and NADH-dependent conversion of 2-[4- Iodophenyl]-3-[4-nitrophenyl]-5-phenyltetrazolium chloride (INT) by diaphorase. The amount of red formazan product formed at 490 nm is proportional to the number of lysed cells. Maximal LDH activity was assessed by cell lysis with 0.1% Triton X-100. Basal levels of LDH release were determined in medium from untreated cells. Relative values for LDH activity were calculated as follows: (LDH release from treated cells minus basal release) divided by (maximal LDH release minus basal release).
Determination of cell viability by Trypan blue dye exclusion
MEMM cells were grown to ~ 80 % confluence and treated with 1, 10 or 100 μM pentavalent sodium arsenate [As(V)] for 24 or 48 hrs. One set of cells was pretreated for two hours with 10 mM N-acetyl cysteine (NAC) before being treated with arsenate for the same time duration. Post-treatment, cells were analyzed by light microscopy for morphological alterations indicative of apoptosis. The trypan blue dye exclusion assay was performed to quantify the extent of cell viability. In this assay, light microscopy is used to distinguish viable from non-viable cells. Compromised cell membranes of non-viable cells allow trypan blue to enter the cell. Viable cells, with intact membranes, prevent the dye from entering and thus remain unstained.
Detection of apoptosis
Arsenate-induced apoptosis in MEMM cells was assessed by using the Apostat™ membrane permeable dye system (R&D Systems, Minneapolis, MN). Apostat is a cell permeable dye that binds specifically to activated caspases. MEMM cells were grown to ~80% confluence and treated with 1, 10 or 100 μM arsenate for 24 h. One sample was also pretreated for two hours with 10 mM N-acetyl cysteine (NAC) prior to exposure to 100 μM arsenate for the same time duration. Five μl of Apostat dye was included for the final 30 min of the incubation period. Cells were then washed with PBS followed by treatment with the nuclear stain DAPI (200 ng/ml for 5 min). Apostat staining was visualized via fluorescence microscopy using a FITC filter for green Apostat fluorescence, and a UV filter for blue DAPI staining.
Involvement of ROS in Arsenate mediated apoptosis
The involvement of reactive oxygen species (ROS) in arsenate-induced apoptosis was assessed utilizing MitoTracker Orange (CM-H2TMROS) dye (Molecular Probes, WA) as an indicator of arsenate-generated ROS. MitoTracker Orange is a lipophilic molecule which in its reduced form is non-fluorescent. Upon its oxidation by ROS, it emits orange-red fluorescence. MEMM cells, grown to 80% confluence, were treated with increasing concentrations of arsenate (1, 10, 100 μM) for 4 h. As a control, one sample was also pretreated for two hours with 10 mM N-acetyl cysteine (NAC) prior to exposure to 100 μM arsenate for the same time duration. NAC will quench any ROS formed as a result of exposure to arsenate. Thirty minutes before the end of incubation period, all samples received one (1) μl of MitoTracker Orange dye. Subsequently, cells were washed with PBS and visualized via fluorescence microscopy using a Texas red filter. Orange fluorescence was taken as evidence of the presence of ROS-oxidized CM-H2TMROS.
Involvement of caspases in arsenate-induced apoptosis
To examine the involvement of caspases in arsenate-induced apoptosis, synthetic fluoro-methylketone labeled, cell permeable caspase inhibitors were employed. Briefly, MEMM cells were grown to ~80% confluence and pre-treated for 1 hr with 20 μM (final concentration) Z-IETD-FMK (caspase-8 inhibitor), Z-DEVD- FMK (caspase-3 inhibitor), Z-LEHD-FMK (caspase-9 inhibitor) or Z-VAD-FMK (pan caspase inhibitor). Arsenate was added to the culture medium to a final concentration of 100 μM and cells incubated for an additional 24 h. At the end of incubation period, cells were examined for morphological indicators of apoptosis, including membrane blebbing, photographed, using a Nikon TE 2000 inverted microscope, and then collected and assayed for viability using trypan blue dye exclusion. None of the inhibitors utilized had any effect on MEMM cell viability at the concentration used. Further, the caspase inhibitor solvent, DMSO, a known scavenger of ROS, did not interfere with arsenate-induced apoptosis at the concentrations used in the assay (data not shown).
Assessment of arsenate-induced changes in mitochondrial membrane potential
Mitochondrial permeability was determined by labeling cells with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyaniniodide (JC-1) (Molecular Probes, USA) as described (Reers et al., 1995). Cells growing in culture were trypsinized with 0.25% Trypsin/0.27 mM EDTA for 5 minutes at 37°C, collected by centrifugation (500 × g, 4°C for 5 min) and resuspended in phenol red-free DMEM. Cell suspensions containing 1 × 105 cells were then treated with arsenate at various concentrations (1, 10, 100 μM) for 4 h at 37°C and 5% CO2. One sample each was treated with 100 μM As (V) for 8 hrs while another was pretreated with 10 mM NAC for two hours before being treated with 100 μM As (V) for 4h. Fifteen minutes prior to the end of incubation, 100 nM JC-1 was added to the cell suspensions. Subsequently, cells were pelleted, washed with PBS and suspended in Cell Buffer (Cell Fluorescence LabChip® Kit) to give a final concentration of 30,000 cells/10 μl. Decreased fluorescence, due to lower membrane potential, and representative of increased mitochondrial permeability, was quantified using the Cell Fluorescence Module of the Agilent 2100 Bioanalayzer (Agilent Biosystem, Palo Alto, CA). Cells not treated with arsenate served as controls.
Immunoblot analysis of mitochondrial Bax and Bcl proteins
MEMM cells were plated as described above, allowed to grow to ~80% confluence, and subsequently treated with 0, 10 and 100 μM concentrations of pentavalent sodium arsenate for 48 h. One set of cells was pretreated for two hours with 10 mM N-acetyl cysteine (NAC) prior to exposure to 100 μM arsenate for the same time duration Cells were then collected in 200 μl of modified RIPA buffer [ (Biosource, Camarillo, CA; containing 50 mM Tris-Cl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 0.5% Nonidet P-40, 0.25% sodium deoxycholate, 50 mM β-glycerophosphate (pH 7.3), 10 mM NaPP, 30 mM NaF, 1 mM benzamidine, 2 mM EGTA, 1 mM sodium orthovanadate, 1 mM dithiothreitol, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride]. Floating and detached cells were also collected and resuspended in the same lysis buffer. Cells were centrifuged at 2000 × g for 5 min at 4°C and supernatants recovered and used for subsequent immunoblotting. Protein levels were determined using the Bradford assay (Bradford, 1976). Forty μg of cleared lysates were separated on 12 % polyacrylamide gels (Invitrogen) and transferred to polyvinylidene fluoride (PVDF) membranes. Membranes were blocked with 5% (w/v) non-fat dry milk in TBST buffer [50 mM Tris (pH 7.6), 150 mM NaCl, and 0.1% Tween-20] for 1 h at room temperature. Polyclonal anti-Bax antibody (Santa Cruz, CA) was diluted in the same blocking buffer and incubated with the membranes for 60 min at room temperature, washed, and then incubated with anti-mouse horseradish peroxidase conjugated secondary antibody. The ECL-Plus™ chemiluminescent detection system (Amersham Pharmacia Biotech, Arlington, IL, USA) was used to visualize immune complexes as per the manufacturer’s instructions. Blots were stripped and reprobed with polyclonal anti-Bcl antibody (Santa Cruz, CA) in similar fashion. To confirm equal loading of protein, the blots were stripped and reprobed with an antibody against β-actin (Santa Cruz, CA). Densitometric analysis was carried out using NIH Image, version 1.09.
Statistical analysis
All data are presented as mean ± S.E.M of three separate experimental determinations. Statistical analyses were conducted using the Statistical Package for Social Sciences© (SPSS; Chicago, IL) version 13.0. Treatment group and time differences were assessed by a multivariate repeated measures ANOVA. Posthoc analyses of individual differences were carried out using the Turkey’s test for multiple comparisons. Statistical significance was assigned p < 0.05.
RESULTS
Phenotypes generated by in utero arsenate exposure
Our studies indicate that i.p. treatment with pentavalent sodium arsenate resulted in about 25% of the embryos/fetuses demonstrating embryolethality and neural tube defects and oro-facial malformations when compared with the saline-dosed controls (Table 1). Prominent among these are (Figure 1) - isolated exencephaly (b [gd10] and f [gd18]), complete cranial/facial clefting (c [gd10]), exencephaly and midfacial hypoplasia (d [gd17]), acrania (e [gd17]), and gastroschisis (not shown).
Table 1.
Developmental outcomes following in utero exposure to 20 mg/kg As (V) a
| Dose | No. of litters (N) | No. of implants (N) | No. of resorptions (%) b | No. of birth defectsd (%) c | No. of live births (N) |
|---|---|---|---|---|---|
| 0 (vehicle) | 12 | 108 | 3 (2.8) | 1 (0.95) | 102 |
| 20 mg/kg | 11 | 100 | 15 (15)* | 8 (10.2)* | 77 |
Dams were injected intraperitonially on gd 7 and 8 and the fetuses examined on gd 17.5.
Percent of implants.
Percent from the total number of live born fetuses
Birth defects include NTDs, orofacial abnormalities and gastroschisis.
Significantly different when compared to vehicle treated control animals (p< 0.05).
Fig. 1.
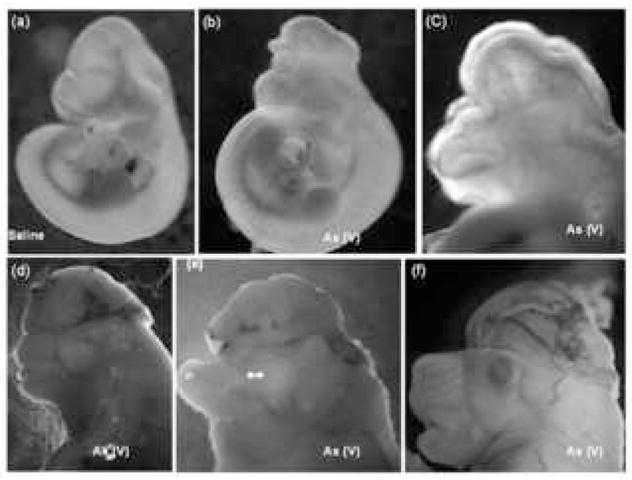
Predominant phenotypes resulting from in utero arsenic exposure of pregnant mice. Dams were dosed i.p. with 20 mg/kg of pentavalent sodium arsenate on gestation days (gd) 7 and 8, euthanized on gd 10 or 17, and embryos/fetuses removed for observation and photography. Prominent phenotypes generated include isolated exencephaly (b [gd10] and f [gd18]), complete cranial/facial clefting (c [gd10]), exencephaly and midfacial hypoplasia (d [gd17]), acrania (e [gd17]), and gastroschisis (not shown). Panel (a) shows a saline control embryo on gd 10.
Arsenate treatment results in dose - and time- dependent cell death
Arsenate cytotoxicity was determined at different time intervals by colorimetric measurement of cellular lysis-induced release of lactate dehydrogenase (LDH). The repeated measures ANOVA revealed a significant effect of arsenic treatment on LDH release from MEMM cells (p<0.05). In addition, there was both an effect of time on arsenic-induced LDH release (p<0.05), as well as a significant treatment by time interaction (p<0.05). Further analysis revealed increases in LDH release from MEMM cells 12 hours after exposure to the 100 μM arsenic concentration and 48 hours following treatment with all of the arsenic concentrations tested (0.1 μM, 10 μM, 100 μM) (Figure 2).
Fig. 2.

Toxicity of pentavalent sodium arsenate (As V) was determined by measuring cellular release of lactate dehydrogenase. MEMM cells were exposed to different concentrations of arsenate and, after the indicated time intervals; aliquots of culture medium were removed and assayed for the presence of LDH as described (see Methods). Absorbance of the red formazan product at 490 nm was directly proportional to the extent of cell death. Each bar in a cluster represents a level of absorbance recorded at a different concentration of arsenate ranging from 0 μM (negative control) to 100 μM. The cluster marked “staur.” represents absorbance values from cells treated with 0.1 μM staurosporine (positive control). Results were obtained from four separate experiments performed in triplicate and mean values ± S.E. are plotted for each treatment condition. * p <0.05.
Arsenate-induced cell death is primarily via the apoptotic pathway
As the LDH assay does not distinguish between apoptotic, necrotic, and other mechanisms of cell death, Apostat, a cell permeable dye that binds specifically to activated caspases, was utilized in order to determine if As (V)-induced cell death was mediated via apoptosis. Exposure of MEMM cells to 100 μM As (V) for 48 hr resulted in green fluorescence (Fig. 3A), indicative of Apostat binding to activated caspases, a feature unique to apoptosis. Pretreatment with NAC resulted in almost complete quenching of the green fluorescence, indicating the possible involvement of ROS in arsenate-mediated apoptosis. Each image is a representative from a set of three independent experiments. These data suggest that in response to exposure to As (V), MEMM cells exhibiting signs of cell death are undergoing apoptosis. Membrane blebbing, cell shrinkage and formation of membrane apoptotic bodies are some of the characteristic features of apoptosis that distinguish it from necrosis. MEMM cells exposed to As (V) demonstrate such morphology, which was reduced significantly in cells pretreated with NAC (Fig. 3B, lower panel). Utilizing dye exclusion as a marker for cell viability, As (V)-induced apoptosis was further verified by demonstrating that exposure of MEMM cells for either 24 or 48 hr to 100 μM arsenate dramatically decreased the number of viable cells (p<0.05 for both time points) (Fig. 4). It is interesting to observe that cells pretreated with N-acetylcysteine (NAC), the N-acetyl derivative of L-cysteine, which elevates the antioxidant glutathione levels, are protected from As (V)-induced apoptosis. After 48 h of arsenate exposure (100 μM), nearly 70% of MEMM cells remained viable, compared to 12–15% of the As (V)-treated cells that did not receive NAC pretreatment (p<0.05). As NAC is also a known scavenger of free radicals, including reactive oxygen species ROS (Mayer and Noble, 1994), this observation prompted exploration of the possible involvement of ROS in arsenate-induced apoptosis of MEMM cells.
Fig. 3.


(A) Apostat ™ labeling of MEMM cells undergoing As (V) mediated apoptosis. Apostat, a FITC-conjugated, membrane permeable dye, was utilized for visualization of apoptotic cells. MEMM cells were exposed to different concentrations of As (V) for 48 hrs. Apostat was added 15 minutes before the end of the incubation period; cells were washed and co-labeled with the nuclear stain DAPI before being examined for FITC and DAPI using a Nikon TE 2000 inverted microscope equipped with FITC and UV filters. Apoptotic cells are green in the field. The photographs are representative of three identical, separate experiments. (B) Morphological assessment of MEMM cell death following treatment with 100 μM As (V). Cells were examined for membrane blebbing (→), cell condensation (↔) and formation of apoptotic bodies (*) after 48 h of treatment. Lower panel shows the effect of pretreatment with NAC. Representative photographs from three separate experiments with similar results are shown.
Fig. 4.

Assessment of cell viability subsequent to arsenate-induced apoptosis in MEMM cells. Trypan blue dye exclusion was used to assess viability of harvested MEMM cells 24 or 48 h after treatment with different concentrations of As (V). Viable cells retain their membrane integrity and thus exclude the dye. Non-viable cells appeared blue and were quantified using a hemocytometer. The percentage of viable cells was plotted against arsenate concentration. Cells pretreated with N-acetylcysteine (NAC) are protected from As (V)-induced apoptosis. Nearly 70% of MEMM cells remained viable after 48 h of treatment with 100 μM As (V), compared to 12–15% of similarly treated cells that did not receive NAC pretreatment. In each case, mean values ± S.E. from three separate experiments with similar results were plotted. *p<0.05.
Arsenate generates reactive oxygen species
The lipophilic MitoTracker Orange dye (CM-H2TMROS) (Molecular Probes, WA) was utilized to detect ROS. In its reduced form, MitoTracker Orange is non-fluorescent, while its oxidized form emits orange-red fluorescence. Generation of ROS will enhance oxidation, thereby converting the dye to its fluorescent form. MEMM cells exposed to 10 and 100 μM As (V) exhibited an increase in ROS levels (Fig. 5). Figure 5 is a representative of three separate experiments that yielded similar results. Pretreatment with the ROS scavenger N-acetylcysteine reduced the amount of arsenate-induced ROS generated by MEMM cells as evidenced by significantly reduced fluorescence.
Fig. 5.
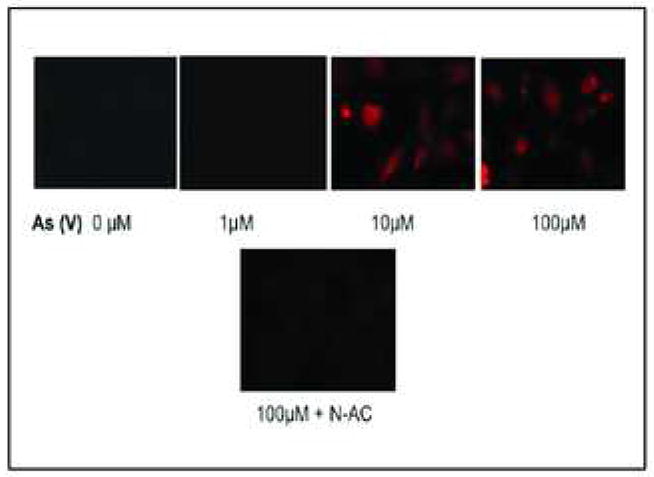
ROS generation in MEMM cells exposed to As (V). MEMM cells were exposed to different concentrations (1–100 μM) of As (V) for 4 h. For the final 30 min. of the incubation period, MitoTracker Orange (CM-H2TMROS), a membrane permeable dye that only fluoresces in its oxidized form, was added to the culture medium. As (V) induced generation of ROS was thus detected by ROS-induced oxidation of MitoTracker Orange. Pretreatment with the ROS scavenger N-acetylcysteine significantly reduced MitoTracker Orange fluorescence, confirming that fluorescence was the result of ROS generation. Photographs are representative of three similar, separate experiments.
Apoptosis mediated by arsenate involves activation of caspases
In order to determine if exposure to arsenate activates the ‘initiator’ caspases 8 and/or 9, MEMM cells were pretreated with 20 μM of various caspase inhibitors [Z-IETD-FMK (caspase-8 inhibitor), Z-DEVD-FMK (caspase-3 inhibitor), Z-LEHD-FMK (caspase-9 inhibitor), Z-VAD-FMK (pan caspase inhibitor)] prior to exposure to 100 μM arsenate for 24 h. Figure 6 illustrates that the pan-caspase inhibitor Z-VAD significantly inhibited cell death to the greatest extent [~80%] (p<0.05) while pre-treatment with the caspase-3 inhibitor Z-DEVD was nearly as effective (~75%, p<0.05). Pretreatment with the caspase-9 inhibitor Z-LEHD ensured that approximately 70% of the cells in culture remained viable (p<0.05). The caspase-8 inhibitor (Z-IETD-FMK) was least protective against arsenate-induced apoptosis, suggesting that caspase-8 plays little role in arsenate-mediated cytotoxicity. This observation suggests that exposure to arsenate, as is the case with many toxicants, leads to apoptosis in MEMM cells via the intrinsic mitochondrial pathway by utilizing the initiator caspase-9.
Fig. 6.

Effect of caspase inhibitors on As (V) induced cell death. MEMM cells were pretreated with various caspase inhibitors [Z-IETD-FMK (caspase-8 inhibitor), Z-DEVD-FMK (caspase-3 inhibitor), Z-LEHD-FMK (caspase-9 inhibitor), Z-VAD-FMK (pan caspase inhibitor)] followed by exposure to 100 μM As (V) for 24 h. Cells were harvested and viability assessed by trypan blue dye exclusion. For each sample, mean cell viabilities from three separate, independent experiments are plotted. *p<0.05.
Arsenate induces loss of mitochondrial membrane potential (δψm)
Mitochondria are now recognized as playing a central role in cell death. In the mitochondrial pathway of apoptosis, caspase activation is closely linked to mitochondrial outer membrane permeabilization. The loss of mitochondrial membrane potential is a hallmark for apoptosis. In order to determine if exposure to arsenate results in the loss of mitochondrial membrane potential, MEMM cells were labeled with the cationic dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′ tetraethylbenzimidazolylcarbocyanineiodide). In non-apoptotic cells, JC-1 accumulates as aggregates in the mitochondria, resulting in red fluorescence which is proportional to membrane potential. As the mitochondrial membrane potential collapses in apoptotic and necrotic cells, there is a shift towards monomeric forms, resulting in the loss of red fluorescence and an increase in green fluorescence. Exposure of MEMM cells in suspension culture to As (V) (1, 10 and 100 μM) for 4 hrs resulted in a dose-dependent increase in green fluorescence that was accompanied by a concomitant decrease in red fluorescence. This effect was even more apparent when the cells were treated with 100 μM As (V) for 8 hrs (* p<0.05) (Fig. 7). Pretreatment of the cells with NAC partially prevented the collapse of mitochondrial membrane potential elicited by As (V) exposure, as indicated by a small but significant change in the levels of both red (increased) and green (decreased) fluorescence in cells treated with both NAC and 100 μM As when compared to cells exposed only to 100uM As for 8 hrs (* p<0.05). However, no statistically significant change in mitochondrial membrane potential in MeMM cells was apparent when comparing the effect of exposure of the cells to both NAC with 100 μM As, to the effect of exposure only to 100 uM As for 4 hrs.
Fig. 7.

Analysis of mitochondrial membrane potential (δψm) in MEMM cells treated with As (V). MEMM cells in suspension were treated with 0, 10 or 100 μM As (V) for 4 hours or with with 100 μM As (V) for 8 hours (positive control), before being labeled with 100 nM of mitochondria selective dye JC-1 (see Methods). One set of cells in suspension was pretreated with NAC prior to treatment with 100 μM As (V). Fluorescence intensity was analyzed utilizing the cell fluorescence module of the Agilent 2100 Bioanalyzer. Mean values ± S.E. from three separate experiments were plotted. A significant (*p<0.05) dose-dependent increase in green fluorescence and decrease in red fluorescence was apparent and most prominent in cells treated with 100 μM As (V) for 8 hrs. Alterations in red and green fluorescence in NAC + As (V) treated cells were significantly different from cells treated with As (V) for 8 hrs (*p<0.05) but were not significantly different from cells treated with As (V) for 4 hrs.
Exposure to arsenate increases Bax and decreases Bcl protein levels in MEMM cells
Bcl and Bax are mitochondria-associated proteins (Jia et al. 2001; Kim et al, 2003). Bcl localizes to mitochondrial membranes, providing a stabilizing, anti-apoptotic influence. Bax also localizes to mitochondria, but its action is pro-apoptotic, inducing release of cytochrome c into the cytosol (Krajewski et al., 1993). MEMM cells were exposed to increasing concentrations of arsenate (0, 10 and 100) for 48 hours. Cells, including floaters, were then collected, lysed, quantified, separated by SDS-PAGE and immunoblotted for Bax and Bcl. Exposure to As (V) induced an increase in Bax expression and a concomitant decrease in Bcl expression while pretreatment with NAC resulted in reduced levels of Bax and a restoration in the levels of Bcl (Fig. 8).
Fig. 8.
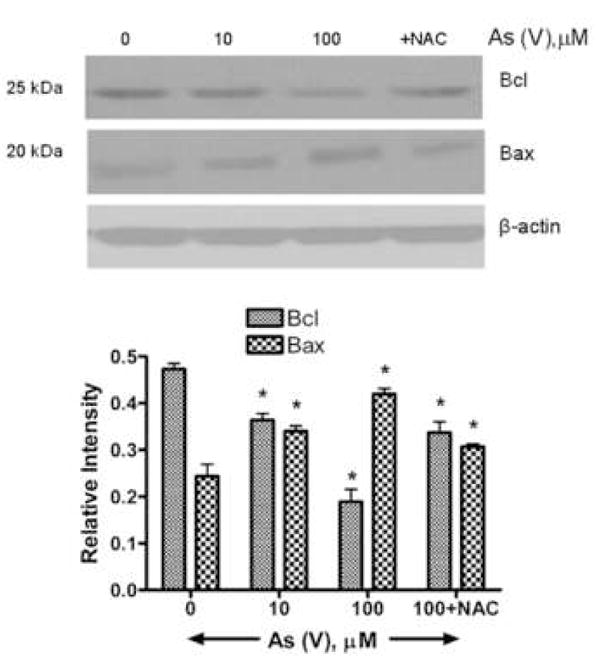
Western blot analysis of Bax and Bcl proteins in MEMM cells exposed to As (V). Cells were exposed to different concentrations of As (V) for 48 h, harvested, and lysed in a modified RIPA buffer (see Methods). One set of cells was pretreated with NAC prior to treatment with 100 μM As (V). 40 μg of total protein lysates were separated by SDS-PAGE, transferred to a PVDF membrane and immunoblotted for Bax and Bcl proteins. (A) Bax, a proapoptotic protein, has a molecular weight of 25 kDa while the antiapoptotic Bcl has a molecular weight of 20 kDa. While the levels of Bcl decrease sharply after 48 hours of 100 μM As (V) treatment, Bax levels show an increase. A change in ratio of Bcl and Bax in favor of Bax is indicative of cellular apoptosis. (B) Densitometric analysis of Western blots from three different experiments. For each treatment condition, the relative intensity of Bcl and Bax protein band was calculated by dividing its numerical intensity by that of corresponding beta-actin band.
DISCUSSION
A known human carcinogen (Abernathy et al., 1999), arsenic has also been implicated in potential deleterious pregnancy outcomes (Rahman et al., 2007; Tabocova et al., 1996). Drinking water contaminated with arsenic has created a serious health hazard in many regions of the world (Chen et al., 2005). Arsenic ingestion through drinking contaminated water during pregnancy has been reported to increase the incidence of abortion (Aschengrau et al., 1989), and maternal arsenic exposure resulted in chromosomal aberrations in fetal cells (Nagymajtenyi et al., 1985).
Of the two major forms of naturally occurring arsenic, arsenite (As III) has been shown to cause cytotoxicity and death in various cell types (Yamanaka et al., 1991; Applegate et al. 1991; Wang et al., 1996; Hengartner, 2000). In contrast, few studies have focused on the equally abundant pentavalent form, arsenate (As V). We report here that arsenate-mediated oxidative injury in MEMM cells acts as an early and upstream initiator of the cell death cascade, triggering cytotoxicity, mitochondrial dysfunction, altered Bcl/Bax protein ratios, and activation of caspase-9. There are two major mechanisms of cell death - apoptosis and necrosis. Unlike necrosis, apoptosis is a highly ordered, complex process with distinct morphological features and biochemical markers. Some of these include internucleosomal DNA fragmentation, membrane blebbing, caspase activation, mitochondrial alterations and the final formation of membrane bound apoptotic bodies (Hengartner, 2000). We have shown that at the arsenate concentrations used in this study, MEMM cells undergo cell death chiefly via the ordered route of apoptosis. This is evident from our observation that exposure of MEMM cells to arsenate results in generation of typical morphological changes associated with apoptosis. Furthermore, we were able to demonstrate that this cell death involved caspases, a family of cysteine proteases, known to be central executioners of apoptosis (Martin and Green, 1995).
There is evidence indicating that reactive oxygen species (ROS), produced by mitochondria, are involved in mediating cell death (Kitchin, 2001). Further, arsenic may also exert its toxic effects through the generation of various ROS which includes hydrogen peroxide (H2O2), superoxide-derived hydroxyl ion (*OH), and peroxyl (ROO*) radicals (Hei et al., 1998; Oya-Ohta et al., 1996). Indeed, arsenic has been shown to cause a dose dependent increase in ROS in cultured human-hamster hybrid cells, and this increase in ROS generation could be quenched by DMSO, a known free radical scavenger (Liu et al., 2001). Antioxidants, such as N-acetylcysteine, suppress apoptosis by scavenging ROS, and their actions provide evidence that ROS act as signaling molecules to initiate apoptosis. Moreover, inorganic arsenicals have been shown to enhance the production of heme-oxygenase, an indicator of oxidative stress (Liu et al., 2001), and arsenic, in its pentavalent state, can cause the uncoupling of oxidative phosphorylation (Chen et al., 1998). Our results show that exposure of MEMM cells to arsenate resulted in generation of ROS. Further, preincubation of cells with the antioxidant NAC, ablated the generation of ROS and abrogated cell death. Inhibition of apoptosis by anti-apoptotic proteins such as Bcl-2 and Bcl-xL is associated with protection against ROS and/or a shift of the cellular redox potential to a more reduced state.
Two major pathways, extrinsic and intrinsic, for the induction of apoptosis, have been identified in recent years (Fulda et al., 2001; Danial and Korsmeyer, 2004; Debatin, 2004). The extrinsic pathway is triggered via ligand binding to tumor necrosis factor family receptors resulting in activation of initiator (i.e. caspase-8) and effecter (i.e. caspase-3) proteolytic enzymes. The intrinsic pathway is activated by agents that target the mitochondria directly. Upon activation, the intrinsic pathway causes the release of cytochrome c and activation of the initiator caspase-9 with subsequent activation of the effecter caspase-3. Our results, using specific caspase inhibitors, have conclusively demonstrated that in MEMM cells, caspase-9 is the primary/initiator caspase in As (V)-induced apoptosis, confirming that arsenate mediated MEMM cell apoptosis proceeds through the intrinsic pathway and involves ROS. Caspase-3 is the common downstream point where both the above-mentioned pathways meet. A variety of key events in apoptosis focus on mitochondria including altered mitochondrial oxidation– reduction, and loss of mitochondrial membrane potential (δψm). Oxidative stress can cause mitochondrial dysfunction, thereby causing cell death (Haga et al., 2005; Ling et al., 2003). JC-1 staining of mitochondria in MEMM cells demonstrated that arsenate can cause depolarization of the membrane, resulting in a process that will ultimately lead to apoptosis. Another significant observation from the present study is that arsenate caused early depolarization of the mitochondrial membrane (~4 h), and this corresponded to the time period during which ROS generation was observed. These data suggest that early generation of ROS may be an important determinant in As (V)-induced apoptosis in MEMM cells.
Bcl-2 is located in the mitochondria, a site where many ROS are generated, as well as a site of action of free radicals. This protein can act as an anti-apoptotic molecule, possibly by scavenging free radicals. Bax, on the other hand, is known to be translocated to the mitochondria, where it causes the release of cytochrome c and subsequent cell death (Lin et al., 2006; Kowaltowski et al., 2001; Jou et al., 2002). Thus, in many cases, ROS-mediated apoptosis involves these proteins at an early stage. Our studies demonstrate that exposure to arsenate caused the levels of Bcl-2 in MEMM cells to fall, while the levels of Bax were found to increase. This suggests the possible involvement of the Bcl family proteins in arsenate-mediated apoptosis in this system.
To understand the potential mechanism of such arsenate-induced cell death in the developing embryo, we have utilized a primary cell culture model utilizing cells derived from the developing orofacial region. The developing secondary palate is a valuable model system to both gain insight into the etiology of palatal clefts and to understand the interplay between various signals governing cellular processes such as proliferation, differentiation, and cell death during embryogenesis. There is abundant data supporting the view that the behavior of MEMM cells in primary culture mimics the in vivo cellular responses of maxillary mesenchymal cells from which they are derived (Greene et al., 1991; Nugent et al. 2001; Potchinsky et al., 1998). Further studies are required to clarify the precise cellular and molecular mechanisms by which various arsenical compounds adversely affect the development of the oro/craniofacial region of mammalian embryos.
Acknowledgments
The authors would like to thank Dr. Kristin Horn for help with statistical data analysis.
This work was supported in part through NIH grants HD053509, DE018215, ES11775, P20-RR017702 from the COBRE program of the National Center for Research Resources, by the Kentucky Science and Engineering Foundation, and by the Commonwealth of Kentucky Research Challenge Trust Fund.
References
- Abernathy CO, Liu YP, Longfellow D, et al. Arsenic: health effects, mechanisms of actions, and research issues. Environ Health Perspect. 1999;107:593–597. doi: 10.1289/ehp.99107593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agency for Toxic Substances and Disease Registry ATSDR. Toxicological Profile for Arsenic. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service; 2007. p. 356. [Google Scholar]
- Applegate LA, Luscher P, Tyrrell RM. Induction of heme oxygenase: a general response to oxidant stress in cultured mammalian cells. Cancer Res. 1991;51:974–978. [PubMed] [Google Scholar]
- Aschengrau A, Zierler S, Cohen A. Quality of community drinking water and the occurrence of spontaneous abortion. Arch Environ Health. 1989;44:283–290. doi: 10.1080/00039896.1989.9935895. [DOI] [PubMed] [Google Scholar]
- Bhatacherjee V, Greene RM, Pisano MM. Divergence of EGF/TGFβ signaling in embryonic orofacial tissue. In Vitro Cell Devel Biol. 2003;39:257–261. doi: 10.1290/1543-706X(2003)039<0257:DOEGFG>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Chen PH, Lan Chen-Che E, Chiou MH, et al. Effects of arsenic and UVB on normal human cultured keratinocytes: impact on apoptosis and implication on photocarcinogenesis. Chem Res Toxicol. 2005;18:139–144. doi: 10.1021/tx049834b. [DOI] [PubMed] [Google Scholar]
- Chen YC, Lin-Shiau SY, Lin JK. Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J Cell Physiol. 1998;177:324–333. doi: 10.1002/(SICI)1097-4652(199811)177:2<324::AID-JCP14>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Debatin KM. Apoptosis pathways in cancer and cancer therapy. Cancer Immunol Immunother. 2004;53:153–159. doi: 10.1007/s00262-003-0474-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhulipala VC, Hanumegowda UM, Balasubramanian G, et al. Relevance of the palatal protein kinase A pathway to the pathogenesis of cleft palate by secalonic acid D in mice. Toxicol Appl Pharmacol. 2004;194:270–279. doi: 10.1016/j.taap.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Ferm VH, Carpenter SJ. Malformations induced by sodium arsenate. J Reprod Fertil. 1968;17:199–201. doi: 10.1530/jrf.0.0170199. [DOI] [PubMed] [Google Scholar]
- Fulda S, Meyer E, Friesen C, et al. Cell type specific involvement of death receptor and mitochondrial pathways in drug-induced apoptosis. Oncogene. 2001;20:1063–1075. doi: 10.1038/sj.onc.1204141. [DOI] [PubMed] [Google Scholar]
- Gebel TW. Arsenic and drinking water contamination. Science. 1999;283:1458–1459. doi: 10.1126/science.283.5407.1455e. [DOI] [PubMed] [Google Scholar]
- Golub MS, Macintosh MS, Baumrind N. Developmental and reproductive toxicity of inorganic arsenic: animal studies and human concerns. J Toxicol Environ Health B Crit Rev. 1998;1(3):199–241. doi: 10.1080/10937409809524552. [DOI] [PubMed] [Google Scholar]
- Greene RM, Linask KK, Pisano MM, et al. Transmembrane and intracellular signal transduction during palatal ontogeny. Genet Dev Biol. 1991;11:262–276. [PubMed] [Google Scholar]
- Haga N, Fujita N, Tsuruo T. Involvement of mitochondrial aggregation in arsenic trioxide (As2O3)-induced apoptosis in human glioblastoma cells. Cancer Sci. 2005;96:825–833. doi: 10.1111/j.1349-7006.2005.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hei TK, Liu SX, Waldren C. Mutagenicity of arsenic in mammalian cells: role of reactive oxygen species. Proc Natl Acad Sci USA. 1998;95:8103–8107. doi: 10.1073/pnas.95.14.8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Holson JF, Stump DG, Clevidence KJ, Knapp JF, Farr CH. Evaluation of the prenatal developmental toxicity of orally administered arsenic trioxide in rats. Food Chem Toxicol. 2000;38:459–66. doi: 10.1016/s0278-6915(00)00015-6. [DOI] [PubMed] [Google Scholar]
- HSDB. Hazardous Substances Data Base. National Library of Medicine; 2001. http://toxnet.nlm.nih.gov/ [Google Scholar]
- Jia L, Patwari Y, Srinivasula SM, et al. Bax translocation is crucial for the sensitivity of leukaemic cells to etoposide-induced apoptosis. Oncogene. 2001;20:4817–4826. doi: 10.1038/sj.onc.1204628. [DOI] [PubMed] [Google Scholar]
- Jou MJ, Jou SB, Chen HM, et al. Critical role of mitochondrial reactive oxygen species formation in visible laser irradiation-induced apoptosis in rat brain astrocytes (RBA-1) J Biomed Sci. 2002;9:507–516. doi: 10.1159/000064723. [DOI] [PubMed] [Google Scholar]
- Kachinskas DJ, Phillipis MA, Qin Q, et al. Arsenate perturbation of human keratinocyte differentiation. Cell Growth Differen. 1994;5:1235–1241. [PubMed] [Google Scholar]
- Kim R, Tanabe K, Uchida Y, et al. Effect of Bcl-2 antisense oligonucleotide on drug-sensitivity in association with apoptosis in undifferentiated thyroid carcinoma. Int J Mol Med. 2003;11:799–804. [PubMed] [Google Scholar]
- Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996;378:150–152. doi: 10.1016/0014-5793(95)01449-7. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, et al. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Lin Y, Kokontis J, Tang F, et al. Androgen and its receptor promote Bax-mediated apoptosis. Mol Cell Biol. 2006;26:1908–1916. doi: 10.1128/MCB.26.5.1908-1916.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren A, Danielsson BR, Dencker L, et al. Embryotoxicity of arsenite and arsenate: distribution in pregnant mice and monkeys and effects on embryonic cells in vitro. Acta Pharmacol Toxicol (Copenh) 1984;54:311–320. doi: 10.1111/j.1600-0773.1984.tb01936.x. [DOI] [PubMed] [Google Scholar]
- Ling YH, Liebes L, Zou Y, et al. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- Liu J, Xie Y, Cooper R, et al. Transplacental exposure to inorganic arsenic at a hepatocarcinogenic dose induces fetal gene expression changes in mice indicative of aberrant estrogen signaling and disrupted steroid metabolism. Toxicol Appl Pharmacol. 2007;220:284–291. doi: 10.1016/j.taap.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SX, Athar M, Lippai I, et al. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc Natl Acad Sci USA. 2001;98:1643–1648. doi: 10.1073/pnas.031482998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo G, Cassady G, Palmisano P. Acute maternal arsenic intoxication with neonatal death. Am J Dis Child. 1969;117:328–330. doi: 10.1001/archpedi.1969.02100030330015. [DOI] [PubMed] [Google Scholar]
- March of Dimes. 2008 www.marchofdimes.com.
- Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- Mayer M, Noble M. N-acetyl-L-cysteine is a pluripotent protector against cell death and enhancer of trophic factor-mediated cell survival in vitro. Proc Natl Acad Sci USA. 1994;91:7496–7500. doi: 10.1073/pnas.91.16.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagymajtenyi L, Selypes A, Berencsi G. Chromosomal aberrations and fetotoxic effects of atmospheric arsenic exposure in mice. J Appl Toxicol. 1985;5:61–63. doi: 10.1002/jat.2550050204. [DOI] [PubMed] [Google Scholar]
- Nugent P, Kusek JC, Pisano MM, et al. Convergence of cAMP, TGF-beta and retinoic acid signaling pathways in cells of the embryonic palate. Life Sci. 2001;69(18):2091–2102. doi: 10.1016/s0024-3205(01)01289-9. [DOI] [PubMed] [Google Scholar]
- Oya-Ohta Y, Daise T, Ochi T. Induction of chromosomal aberrations in cultured human fibroblasts by inorganic and organic arsenic compounds and the different roles of glutathione in such induction. Mutat Res. 1996;357:123–129. doi: 10.1016/0027-5107(96)00092-9. [DOI] [PubMed] [Google Scholar]
- Pastides H, Calabrese EJ, Hosmer DW, et al. Spontaneous abortion and general illness symptoms among semiconductor manufacturers. J Occup Med. 1988;30:543–551. [PubMed] [Google Scholar]
- Pisano MM, Mukhopadhyay P, Greene RM. Molecular fingerprinting of TGFbeta-treated embryonic maxillary mesenchymal cells. Orthod Craniofac Res. 2003;6:194–209. doi: 10.1034/j.1600-0544.2003.00264.x. [DOI] [PubMed] [Google Scholar]
- Potchinsky MB, Lloyd MR, Weston WM, et al. Selective modulation of MAP kinase in embryonic palate cells. J Cell Physiol. 1998;76:266–280. doi: 10.1002/(SICI)1097-4652(199808)176:2<266::AID-JCP5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Rahman A, Vahter M, Ekström EC, et al. Association of arsenic exposure during pregnancy with fetal loss and infant death: a cohort study in Bangladesh. Am J Epidemiol. 2007;165:1389–1396. doi: 10.1093/aje/kwm025. [DOI] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola-Hartshorn C, et al. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–17. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- Smith AH, Hopenhayn-Rich C, Bates MN, et al. Cancer risks from arsenic in drinking water. Environ Health Perspect. 1992;7:259–267. doi: 10.1289/ehp.9297259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelstein O, Gould A, Wlodarczyk B, Tsie M, Lu X, Le C, et al. Developmental consequences of in utero sodium arsenate exposure in mice with folate transport deficiencies. Toxicol Appl Pharmacol. 2005;203:18–26. doi: 10.1016/j.taap.2004.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabacova S, Hunter ES, 3rd, Gladen BC. Developmental toxicity of inorganic arsenic in whole embryo: culture oxidation state, dose, time, and gestational age dependence. Toxicol Appl Pharmacol. 1996;138:298–307. doi: 10.1006/taap.1996.0128. [DOI] [PubMed] [Google Scholar]
- U.S. EPA. 2001. EPA-815-Z-01-001. National Primary Drinking Water Regulations; Arsenic and Clarifications to Compliance and New Source Contaminants Monitoring; Final Rule Federal Register 66:6976.
- Vahter M. Mechanisms of arsenic biotransformation. Toxicology. 2002:181–182. 211–217. doi: 10.1016/s0300-483x(02)00285-8. [DOI] [PubMed] [Google Scholar]
- Wang TS, Kuo CF, Jan KY, et al. Arsenite induces apoptosis in Chinese hamster ovary cells by generation of reactive oxygen species. Cell Physiol. 1996;169:256–268. doi: 10.1002/(SICI)1097-4652(199611)169:2<256::AID-JCP5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Wang A, Holladay SD, Wolf DC, et al. Reproductive and developmental toxicity of arsenic in rodents: a review. Int J Toxicol. 2006;25:319–331. doi: 10.1080/10915810600840776. [DOI] [PubMed] [Google Scholar]
- Warner DR, Greene RM, Pisano MM. Interaction between Smad 3 and Dishevelled in murine embryonic craniofacial mesenchymal cells. FEBS Lett. 2005;579:3539–3546. doi: 10.1111/j.1601-6343.2005.00319.x. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Hasegawa A, Sawamura R, et al. Cellular response to oxidative damage in lung induced by the administration of dimethylarsinic acid, a major metabolite of inorganic arsenics in mice. Toxicol Appl Pharmacol. 1991;108:205–213. doi: 10.1016/0041-008x(91)90111-q. [DOI] [PubMed] [Google Scholar]