

Abstract
There are two known types of microbial two-component flavin-dependent monooxygenases that catalyze oxygenation of p-hydroxyphenylacetate (HPA), and they are distinguished by having structurally distinct reductases and oxygenases. This paper presents a detailed analysis of the properties of the enzyme from Pseudomonas aeruginosa, an example of one group, and compares its properties to those published for the Acinetobacter baumannii enzyme, an example of the alternative group. The reductase and oxygenase from P. aeruginosa were expressed in Escherichia coli. The reductase was purified as a stable C-terminal His-tagged yellow protein containing weakly bound FAD, and the oxygenase was purified as a stable colorless N-terminal His-tagged protein. The reductase catalyzes the reduction of FAD by NADH and releases the FADH− product into solution, but unlike the reductase from A. baumannii, this catalysis is not influenced by HPA. The oxygenase binds the released FADH− and catalyzes the oxygenation of HPA to form 3,4-dihydroxyphenylacetate, after which the FAD dissociates to be re-reduced by the reductase, a common overall pattern for two-component flavin-dependent oxygenases. With this system, it appears that interactions between the reductase and the oxygenase can facillitate the transfer of FADH− to the oxygenase, although they are not required. We show that the P. aeruginosa oxygenase system in complex with FADH− reacts with O2 to form a quasi-stable, unusually high-extinction flavin hydroperoxide species that binds HPA and reacts to form the product. The resultant flavin hydroxide decomposes to FAD and water while still bound to the oxygenase, and then releases product and FAD from the protein. Unlike the enzyme from A. baumannii, during normal catalysis involving both the reductase and oxygenase, the rate-determining step in catalysis is the dissociation of FAD from the oxygenase in a process that is independent of the concentration of HPA. Structures for the reductases and oxygenases from A. baumannii and from Thermus thermophilus (similar to the P. aeruginosa system) form a basis for interpreting the molecular origins of the differences between the two groups of flavin-dependent two-component oxygenases.
Many flavin-dependent two-component oxygenases have been studied over the last decade, and it is now recognized that these enzymes are widely spread throughout the microbial world (1). The enzymes consist of two separate proteins (a reductase and an oxygenase) that combine their activities to achieve the same catalytic outcome as the integrated single-component flavoprotein oxygenases that have been widely studied for more than 40 years (1). Several two-component flavin-dependent systems have been isolated that hydroxylate or epoxidize a wide variety of organic compounds (1). In these systems, the function of the reductase is to reduce a flavin cofactor, which the oxygenase utilizes to react with oxygen and catalyze hydroxylation of a unique substrate. The first such two-component flavin-dependent system studied was bacterial luciferase, which oxygenates long chain fatty aldehydes to the corresponding acids and, in addition, gives off light (2 and references therein). In this paper we focus on a two-component aromatic hydroxylase, p-hydroxyphenylacetate hydroxylase (HPAH), EC 1.14.13.3, which is found in numerous bacteria and by historic precedent has become a model example of two-component hydroxylases. This enzyme is classified by function next to p-hydroxybenzoate hydroxylase (EC 1.14.13.2), the model example of the single-component hydroxylases (3). Single-component flavin-dependent hydroxylases contain a tightly bound flavin and function so that the reduction of the flavin, the subsequent reaction with oxygen, and the hydroxylation reactions all take place on a single protein (1,3). Although the overall reactions catalyzed by both the single– and two-component hydroxylases are almost identical, the proteins responsible are not structurally related. Thus, these enzyme systems provide a fascinating example of evolutionary convergence to carry out the same overall function. Until recently, published work implied that in microorganisms there were at least three structural and functional variants of HPAH. The enzyme system from Pseudomonas putida (the first HPAH studied) was reported to consist of a flavoprotein and a coupling protein (4), the enzyme systems from P. aeruginosa (this study) and Escherichia coli (5) consist of a small reductase and a large oxygenase, and the enzyme from Acinetobacter baumannii consists of a large reductase and an oxygenase that is smaller than those from E. coli and P. aeruginosa (6). We now know that the HPAH from P. putida is functionally very similar to the HPAH from A. baumannii (Durgan, Ballou, and Entsch, unpublished results). The P. putida colorless coupling protein is actually the oxygenase, and the flavoprotein is the reductase. Thus, over a wide range of organisms, there are only two, rather than three distinct forms of HPAH known. In this paper, we demonstrate some of the subtle differences in the functions of these distinct HPAHs that have different, but related structures (7-9). It has also been instructive to compare the reactions catalyzed by these two-component enzymes with those of the one-component enzymes with the same overall function.
Although a number of two-component aromatic hydroxylases have been isolated and studied to some degree, only a few have been investigated at the mechanistic level. Significant examples are HPAH from A. baumannii (6,10,11) and ActVA-ActVB, which participates in the biosynthesis of actinorhodin and is found in Streptomyces coelicolor (12). The HPAH from A. baumannii consists of a large reductase (C1, 35.5 kDa) and a small oxygenase (C2, 47 kDa), while the ActVA-ActVB system consists of a small reductase (Act VB, 18 kDa) and a small oxygenase (Act VA, 40 kDa). The ActVA-ActVB enzyme is more difficult to study in detail because it has a complex, unstable, and expensive substrate, dihydrokalafungin. Moreover, there are no available structures of its two proteins.
Structures have recently been published for members of each of the two types of HPAH mentioned above, that from A. baumannii (7), and that from Thermus thermophilus (8,9). The latter has a small reductase (16.1 kDa) and a larger oxygenase (54.3 kDa) like HPAH from P. aeruginosa and from E. coli (5). Moreover, our preliminary studies with HPAH from P. aeruginosa (13,14) showed that the amino acid sequences are similar to those of HPAH from T. thermophilus. Thus, the crystal structures of the T. thermophilus enzymes are useful models for the P. aeruginosa HPAH to compare mechanistic and structural features with those of the distinctly different HPAH from A. baumannii that catalyzes the same reaction. At present, there are no published mechanistic studies on HPAH from T. thermophilus.
As shown by the structures, both types of reductases have similar core folding patterns (9, P. Chaiyen, personal communication), but C1 has an extra C-terminal, α-helical domain that is not part of the more common smaller reductases found in two-component enzymes (9,15). Given the lack of sequence similarity between the oxygenases (10), it was surprising to find that they have similar core folding patterns (7,8), but rather different strategies for binding flavin and substrate. The distinct binding features are responsible for some of the differences in catalytic behavior. In this paper, we highlight some key properties of the proteins from P. aeruginosa and compare them to our published work on the A. baumannii proteins. Unlike C1, whose activity is greatly stimulated by the binding of p-hydroxyphenylacetate (HPA), the reductase (HpaC) from P. aeruginosa catalyzes the reduction of FAD by NADH without being affected by the presence of HPA. C1 is specific for FMN while the HpaC can reduce either FAD or FMN; both oxygenases can use either reduced flavin, but the P. aeruginosa oxygenase (HpaA) reacts faster and more efficiently with FADH−. However, there are a number of mechanistic differences between the two systems. For example, as described in this manuscript, rather than the presence of HPA controlling the rate of reduction of the flavin by the reductase as a means of regulation, the overall control in the P. aeruginosa system is mainly exerted by the flavin-binding properties of the oxygenase.
Materials and Methods
Materials
The following reagents were purchased from Sigma: 4-hydroxyphenylacetate (HPA), 3,4-dihydroxyphenylacetate (DHPA), NADH, FAD, and riboflavin. Pure FMN was prepared from FAD by hydrolysis with snake venom from Crotalus adamenteus (16).
Concentrations of reagents were determined using the known extinction coefficients at pH 7.0: NADH (ε340 = 6 220 M-1cm-1), FAD (ε450 = 11 300 M-1cm-1), FMN (ε450 = 12 200 M-1 cm-1), riboflavin (ε450 = 12 300 M-1cm1) and HPA (ε277 = 1 500 M-1cm-1). Concentrations of the proteins were determined from the extinction coefficients at 280 nm (apo-HpaA, ε280 = 9 828 M-1cm-1 and HpaC, ε280 = 7 570 M-1cm-1, calculated from the amino acid compositions using the ExPASY tools) or when HpaC is bound to FAD, using the ε450 = 10 500 M-1cm-1.
All plasmids and the His-binding resin used to purify the reductase (see below) were bought from Novagen. The dioxygenase (dihydroxyphenylacetate dioxygenase [DHPAO]) used to measure 3,4-dihydroxyphenylacetate was purified from Pseudomonas sp. by following the reported procedure (17).
Cloning, expression, purification, and characterization of recombinant HpaC and HpaA
The complete gene (phaA) for the oxygenase was amplified by PCR from P. aeruginosa genomic DNA using a proofreading DNA polymerase (from Finnzymes) and the primers below that were derived from the P. aeruginosa genomic sequence.
5′-GGGGAATTCCATATGAAACCCGAAGATTTCCGTGCCTC (coding strand)
5′-CGGGATCCGTCATTGGCGGATGCGATCGAG (complementary strand)
The complete gene for the reductase (phaC) was amplified by PCR in a similar fashion using the primers below.
5′GGGGAATTCCATATGTCCCAGCTCGAACCCAGGCAG (coding strand)
5′-CGGGATCCTTCAGGCCGCCCGCCGGG (complementary strand).
In order to achieve a selective isolation of the proteins in E. coli, which contains genes for its own HPAH, the genes from P. aeruginosa were cloned initially into the expression vector pET-15b (Novagen), which encodes six histidines at the N-terminal of the protein codes. The His-tag modification enabled us to isolate both proteins in large amounts free from their E. coli counterparts.
The expression vectors were transformed into separate colonies of E. coli BL21(DE3), and cells were grown in LB/Ampicillin at 37 °C in flasks. Protein production was induced by adding 0.5 mM IPTG when turbidity at 600 nm reached ∼1.0. After induction, the temperature was lowered to 20 °C and incubation continued overnight with shaking at 250 rpm. The cells were harvested, washed with 50 mM K phosphate buffer, pH 7.0 and stored at −80 °C. Typical yield of cells was 5 g per liter of medium.
The His-tagged oxygenase could not be purified using a Ni+2 column because it irreversibly precipitated in the column matrix. Instead, a stable homogeneous active enzyme was obtained by precipitating the protein from the crude extract with 1 mM NiSO4, removing the supernatant, and then dialyzing the precipitate extensively with 100 mM EDTA in 50 mM K phosphate, 20% glycerol, pH 7.6 to remove Ni+2 and dissolve the protein. This procedure yielded an enzyme solution that could be stored at −20 °C without any loss in activity. The preparation was estimated to be more than 99 % pure by SDS-PAGE.
Molecular weight of the native enzyme was determined by FPLC using gel filtration column chromatography (Tricorn columns with Superose 6 10/300 GL from Pharmacia). The column was equilibrated with 50 mM K phosphate, 150 mM NaCl, pH 6.8, the proteins were eluted using a flow rate of 0.4 mL min-1. Calibration was done using a gel filtration standard mix from BioRad consisting of molecular weight markers ranging from 1,350 to 670,000 daltons. A calibration curve was constructed by plotting the ratio [elution volume (Ve)] /[void volume (V0)] vs. the logarithm of the known molecular weights of the protein standards. The molecular weight of the His-tagged oxygenase was determined by comparing its Ve/ V0 with that of the protein standards.
The His-tagged reductase was isolated as an active yellow protein by using a Ni2+ column. This apparently pure enzyme was found to be unstable and probably somewhat damaged by peptidase activity, as evidenced from its behavior on SDS-PAGE and in mass spectrometry. In order to obtain an intact protein, an alternative approach of attaching histidines at the C-terminal end was attempted. This was achieved by designing primers for site-directed mutagenesis that incorporated four histidines in phaC immediately before the stop codon. Two histidines were introduced with one set of mutagenic oligonucleotides below:
5′-CCCGGCGGGCGGCCCATCATTGAAGGATCCGGCTG (coding)
5′-CAGCCGGATCCTTCAATGATGGGCCGCCCGCCGGG (complementary).
The mutations were incorporated using the Stratagene QuickChange XL site-directed mutagenesis (SDM) kit. The plasmid was transformed into E. coli XL-10 competent cells and sequenced. Positive clones were used for the introduction of the next two histidines by site-directed mutagenesis using the primers below:
5′ -CGGGCGGCCCATCATCATCATTGAAGGATCCGGCTG (coding)
5′-CAGCCGGATCCTTCAATGATGATGATGGGCCGCCCG (complementary)
The amplified product was transformed into E. coli XL-10 cells as before. The recombinant plasmid was sequenced to confirm clones of phaC-4His.
For the production of active enzyme, the pET11a plasmid (Novagen) containing the His-tagged reductase gene was transformed into E. coli BL21(DE3), and as with the oxygenase, the cells were grown, expression was induced by adding 0.5 mM IPTG, and the cells were harvested. Purification of the C-terminal His4-tagged reductase using a Ni2+ column yielded an active yellow protein (at least 20-fold more active than the N-terminal tagged enzyme) that was stable at 4 °C in 50 mM K phosphate buffer, pH 7.6, containing 10 % glycerol. This reductase with a C-terminal tag was confirmed as a single band on SDS-PAGE and a single species by MALDI-MS. The molecular weight of the native protein was determined by size-exclusion FPLC using Superose 6 (Pharmacia) and TSK-G2000SW (Sigma) as described above for the oxygenase.
Enzyme assays
Reductase activity of HpaC was monitored by following NADH oxidation at 340 nm using various flavins as substrates. Assays were performed aerobically at 25 °C in 25 mM Tris-Cl, pH 8.0, containing 100 μM NADH and 50 μM flavin.
The product of hydroxylation, DHPA, is a substrate for the dioxygenase, DHPAO. The dioxygenase converts DHPA to 5-carboxymethyl-2-hydroxymuconic semialdehyde, which has an absorbance maximum at 380 nm (Δε = 46 000 M-1 cm-1 at pH 9.0, as determined by the complete conversion of a measured amount of DHPA to the semialdehyde before adjustment of pH to 9.0). This reaction was used to monitor the amount of DHPA formed in reactions catalyzed by HPAH.
Spectral studies
UV-visible absorbance spectra were recorded with a Hewlett Packard diode array (HP 8452A), a Cary 3, or a Shimadzu 2501PC spectrophotometers. Fluorescence measurements were made using a Jobin Yvon Fluoromax-3 spectrofluorometer. All of these instruments were equipped with thermostatic cell compartments.
Transient-state kinetics
Rapid reaction studies were performed with a Hi-Tech Model SF-61 stopped-flow instrument in absorbance or fluorescence modes, and double-mixing experiments were performed with a Hi-Tech Model SF-61DX stopped-flow instrument in the absorbance mode. For anaerobic reactions, the system was flushed and incubated overnight with an anaerobic solution of protocatechuate (500 μM) and protocatechuate-3,4-dioxygenase (∼1 μM) to scavenge any oxygen. This solution was replaced with oxygen-free buffer before introducing anaerobic reaction mixtures. The enzyme solution was made anaerobic in a glass tonometer by repeated cycles of evacuation and subsequent equilibration with oxygen-free argon, and finally left under a small positive pressure of argon for use on the stopped-flow apparatus. Buffers and other non-protein reagent solutions were made anaerobic by bubbling with oxygen-free argon in a syringe for 15-20 min.
Kinetic data were analyzed by fitting to exponential equations using the Marquardt algorithm incorporated into Program A (developed by Chung-Jen Chiu, Rong Chang, Joel Dinverno and David P. Ballou, University of Michigan).
Experimental conditions
Unless mentioned otherwise, all stopped-flow experiments were done in 50 mM K phosphate, pH 7.2 at 4 °C.
Results
Preparation of Reductase, HpaC
Initial attempts to express HpaC in E. coli as an N-terminal His-tagged derivative were successful in producing large amounts of soluble protein containing some yellow color. This protein behaved as a flavin reductase but was unstable, and its properties changed with storage. Examination of this preparation by MALDI mass spectrometry and by electrophoresis showed that it contained polypeptides with fragments missing from the C-terminal end. To counter this problem, the reductase was expressed in E. coli as a C-terminal His-tagged derivative (see Materials and Methods). The latter derivative purified as a stable, yellow protein that was judged by mass spectrometry to be a single molecular species with a molecular weight that matched the mass calculated from the sequence of the translated gene (see below). The yellow color was due to FAD (see Fig. 1A), which was present at approximately 1 FAD per polypeptide in some preparations. However, some purified samples had only partial occupation of the active site with FAD, indicating that FAD is not bound tightly (see below). Size-exclusion chromatography showed that the reductase was a monomer in solution. Based upon the amino acid sequence, the monomeric structure (without His-tag, but with FAD) has a molecular weight of 19 400. Thus, HpaC belongs to the family of low molecular weight flavin reductases that are similar in structure. The sequence of HpaC is 32 % identical to the reductase component of HPAH of T. thermophilus (although the latter is shorter at both the N- and C-termini) (9) and also similar to PheA2, the reductase component of phenol hydroxylase from Bacillus thermoglucosidasius A7 (15). HpaC is 58% identical (and higher in similarity) in sequence (with no gaps) to the reductase of HPAH from E. coli. Thus, one would predict that these two reductases should have very similar properties. As detailed below, comparisons with the published results for the E. coli enzyme (18) shows that this is true, except for a large difference in the affinities of the reductases for NADH. The crystal structures of the HPAH reductase from T. thermophilus and of PheA2 show that they are both dimers (15) and although a His-tag at the C-terminus of the HpaC preparation is not likely to interfere with the operation of the active site, because of the proximity of the C-terminal residues in the dimer structures, it is possible that the His-tag could prevent formation of the dimer in solution. Nevertheless, we can note that not all of the known small flavin reductases are homodimers. For example, Fre from E. coli functions in solution as a monomer (2).
Figure 1.
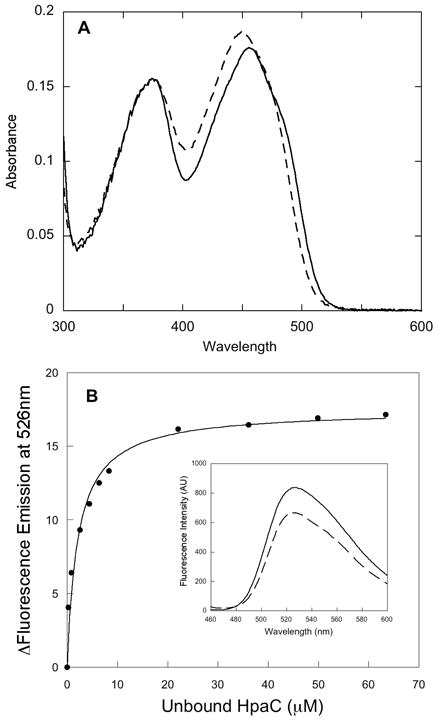
Spectral analysis of the reductase component of HPAH from P. aeruginosa. A - Spectrum of HpaC in buffer (solid line) and after addition of SDS to dissociate FAD from the protein (dashed line). B - Fluorimetric titration of FAD with apo-HpaC at 4 °C. A solution of 4.5 μM FAD in 50 mM K phosphate buffer, pH 7.2 was titrated with aliquots of concentrated apo-HpaC (0.13 mM), and fluorescence emission spectra were recorded with excitation at 450 nm. Free (or unbound) HpaC was measured by taking the difference between measured bound HpaC and total HpaC. Changes in fluorescence emission intensity were plotted against the concentration of unbound HpaC in solution. The data were fitted to a hyperbolic equation (solid line in B) to obtain the Kd for FAD of 3 μM. The inset shows the spectrum of the free FAD (solid line) and that of the final titration point after adding 68 μM HpaC (dashed line).
Properties of reductase
HpaC was usually isolated with some FAD bound, and was found to display reductase activity using NADH as the reductant of the FAD and O2 to reoxidize the FADH− during turnover The reductase has no measureable activity with NADPH as reductant. To measure binding of FAD, a solution of FAD was titrated with apoprotein, and the partial quenching of FAD fluorescence was followed (see Fig. 1B). At 4 °C, the Kd for FAD was determined to be 3 μM. Thus, the affinity for FAD is only in the range of tight substrate binding, not in the nanomolar range that would be typical of a flavin prosthetic group. There is a wide range of affinity for FAD by the different reductases associated with two-component flavin-dependent hydroxylases. For example, the PheA2 reductase for phenol hydroxylase (above) binds FAD as a prosthetic group with a Kd of 9.8 nM (15), while the similar reductase for HPAH from T. thermophilus (9) binds FAD as a substrate, with a Kd in the μM range, like HpaC.
The reduction of bound FAD by NADH was studied using anaerobic stopped-flow kinetic techniques that measured the absorbance changes of the flavin (usually at 450 nm). The measured rate constant for the reduction of FAD (7 s-1) was independent of the concentration of NADH down to 20 μM, implying that the Kd for NADH is ≤ 1 μM. We note that the initial binding of NADH is much faster than 7 s-1, so that binding does not interfere with measurements of the rate of reduction. In contrast to many similar flavin reductases, for example, Act VB (12), no charge-transfer interactions (with absorbance in the 520-650 nm range) between FADH− and NAD were observed for HpaC. When such interactions are observed, the loss of the charge-transfer absorbance usually indicates the release of NAD (12). Without such interactions with HpaC, the stopped-flow experiments provided no information on the rate of release of products from the enzyme. The rate of FAD reduction was independent of the presence of HPA, demonstrating that, in contrast to C1 (11), the activity of the HPAH reductase is not allosterically regulated by substrate. In the presence of HPA under similar conditions, C1 catalyzes reduction of FMN with a rate constant of 300 s-1 (11). HpaC is isolated with FAD bound and preferentially binds FAD, but it does not discriminate between FAD, FMN, and riboflavin as substrates in its ability to consume NADH during aerobic steady-state turnover.
A critical consideration in the function of this two-component enzyme is the fate of FADH− formed on the reductase. This was examined in the stopped-flow spectrophotometer. HpaC was reduced photochemically under anaerobic conditions in a tonometer and then mixed at 4 °C with either anaerobic or aerobic solutions of dichlorophenolindophenol (DCIP) or menadione. Oxygen had no observable effect upon the reactions with either of the redox dyes. The results with DCIP were most readily interpreted, because the fate of the oxidized dye could be easily followed independently by the loss of absorbance at 600 nm as the blue dye was reduced. With a 2-fold excess of dye over FADH−, approximately 65% of the reaction occurred in the dead time of the instrument. This type of very rapid reaction is typical of reactions of free reduced flavins with quinones and related molecules such as DCIP (19). The remaining 35 % of the FADH– reacted in a complex manner with DCIP that could be fitted to a two-exponential equation, giving rate constants of approximately 17 and 1 s-1 for the two phases that were of nearly equal magnitude. Based upon these observations from multiple experiments, it is clear that ∼ 65% of the FADH– was not bound to protein under the conditions of the experiments. Given the protein concentration used in the reactions (18 to 20 μM), it was estimated that the overall Kd for FADH− is ∼20 μM, which is ∼7-fold greater than that for FAD. This difference in affinities is appropriate for the reductase to transfer the reduced flavin to the oxygenase and then re-bind the oxidized flavin product to promote its re-reduction during catalysis. The overall reaction with DCIP accounted for all the reduced flavin in the reaction, even in the presence of oxygen. Therefore, as expected for a reductase, we conclude that the fraction of the total FADH− that remains bound to the protein does not react rapidly with oxygen. The rate of dissociation of FADH– from the protein could be either of the measured rate constants (above), or both, depending upon the interpretation of the experiment. However, it is clear that the reduced flavin dissociates only at a modest rate from HpaC alone.
Preparation of oxygenase, HpaA
HpaA could be expressed as an N-terminal His-tagged derivative. Inspection of the structure of the related oxygenase from T. thermophilus (referred to as PhaB) (8) shows that a His-tag on the N-terminus of the polypeptide is not expected to interfere with the integrity of the protein, unlike a His-tag on the C-terminus, which is involved in the oligomeric structure essential to oxygenase function. Large amounts of soluble, active HpaA were expressed in E. coli. However, attempts to purify the HpaA with a standard Ni column resulted in irreversible precipitation of the protein on the column. Therefore, a novel method was devised to take advantage of the His-tag. The protein was selectively precipitated from the crude cellular extract by the addition of a solution of NiSO4 (see Materials and Methods). The precipitate was dialyzed against 100 mM EDTA in buffer containing 20% glycerol, which caused the precipitate to slowly dissolve, yielding a solution of active oxygenase that was shown by SDS-PAGE to be pure. HpaA preparations are stable and have been stored at -20 °C for at least three years without loss of activity, provided that the solution contains 20% glycerol. This protein is colorless when pure, and behaves in solution as a homodimer when examined in size-exclusion chromatography experiments (see Materials and Methods). The monomer molecular weight, based upon the amino acid sequence (without the His-tag) is 58,464. The structures of the oxygenases from T. thermophilus (8) and A. baumannii (7) both show a minimum structural unit in the form of a homotetramer. However, the tetrameric oxygenase from T. thermophilus, HpaB, is clearly formed as a dimer of dimers, suggesting that a dimer may be the functional unit. Both published structures show that flavin binding involves residues from two monomers, so that the functional unit must be at least a dimer. No information is available to establish the functional requirement for a tetramer. Thus, our observation of a dimeric solution structure of HpaA from P. aeruginosa is consistent with a dimer being the functional unit without the need for a tetrameric structure. HpaA is similar in sequence to the HpaB, but it is 39 residues larger with additional residues at both the N- and C-termini. The remaining sequences are contiguous (except for deletions of one or two residues) and exhibit 29.2% identity and 45% similarity. In addition, most of the residues interacting with HPA are identical in the two proteins, as are many residues interacting with FAD.
Properties of oxygenase
Titration of FAD with HpaA or addition of FAD to a solution of oxygenase does not cause any detectable perturbation of the absorption spectrum of FAD when working in the range of 0.05 mM of each. This result suggests that HpaA has a low affinity for FAD (Kd ≥ 0.25 mM). For catalysis, HpaA must bind FADH−, but quantification of this affinity presented challenges. Any absorbance changes that might have occurred were too small to measure, especially in view of the fact that the solution tended to develop some turbidity during experimentation. However, it was discovered that when an anaerobic solution of 20 μM FADH− equilibrated with 40 μM oxygenase was mixed with oxygenated buffer, ≤ 10% of the ensuing reaction with oxygen was due to free flavin (which formed oxidized FAD and H2O2); ≥ 90% formed the rather stable C4a-FAD hydroperoxide. Thus, the Kd for FADH− was likely to be ≤ 2 μM. An attempt was made to measure Kd by mixing aerobic solutions of various concentrations of HpaA with an anaerobic solution of FADH− in a stopped-flow spectrophotometer. Flavin that was bound to the oxygenase would react rapidly with O2 to form the quasi-stable C4a-flavin hydroperoxide (see below for details), while the free FADH− would react over the course of ∼ 1 s to form FAD plus H2O2. The amount of C4a-flavin hydroperoxide formed would thus be a measure of the fraction of FADH− that was bound. This method was successful with the C2 oxygenase from A. baumannii (10). Unfortunately, it was found that under the experimental conditions we used, the binding of FADH− to HpaA occurred with an observed rate constant of only ∼ 26 s-1, a rate that was too slow (see below) to accurately determine the equilibrium state between oxygenase and FADH−.
HpaA is not absolutely specific for FAD, although it is specific in any practical sense. Thus, in turnover by coupling HpaA with the FMN-specific C1 from A. baumannii (11), only a small fraction of the NADH used (approximately 10%) resulted in the formation of DHPA. Consistent with this was the observation in stopped-flow kinetic studies (see below), that when 12 μM FMNH− was equilibrated with 30 μM oxygenase and then reacted with oxygen, only a few percent of the flavin formed a complex with the oxygenase, as judged by the formation of the flavin hydroperoxide.
The reaction of oxygen with HpaA that was pre-equilibrated with FADH− in the absence of HPA was also examined. Other oxygenases from two-component systems, for example, C2 from A. Baumannii and ActVA from S. coelicolor (10,12), form metastable flavin hydroperoxides in the absence of their substrates. This contrasts with several of the one-component flavin-dependent oxygenases (3), which only stabilize the hydroperoxide in the presence of substrate, substrate-like effectors, or high concentrations of monovalent anions such as azide (20). We mixed HpaA with FAD, made the solution anaerobic in a tonometer, and reduced the FAD with dithionite. This solution was mixed with an equal volume of oxygenated buffer in the stopped-flow spectrophotometer (Fig. 2). The FADH− spectrum was converted in a second-order reaction with a rate constant of 3 × 105 M-1s-1 to a species with an absorbance maximum at 386 nm, typical of a flavin C4a-hydroperoxide. Thus, HpaA also forms a metastable flavin hydroperoxide. However, the spectrum is distinctly different from the C4a-hydroperoxides of other oxygenases studied, in that it has an extinction coefficient at 386 nm of 12.5 mM-1cm-1 (Fig. 2), compared to values of 8 – 9 mM-1cm-1 for most other flavin-dependent oxygenases (10,12,20). The flavin hydroperoxide of HpaA decomposes slowly, as illustrated in Fig. 2, to form FAD (with a spectrum identical to that of FAD free in solution) with a first-order rate constant of 0.005 s-1. Although the current data are not definitive, it is possible that the decomposition of the flavin hydroperoxide occurs free in solution after slow dissociation of the flavin hydroperoxide from the oxygenase.
Figure 2.

Reaction of oxygen with HpaA in complex with FADH− in the absence of HPA. A solution containing FADH− with oxygenase in the ratio of 1:3 was mixed in a stopped-flow spectrometer with an equal volume of buffer equilibrated with 1.23 mM oxygen at 4 °C. The final solution contained 50 mM K phosphate and 5% glycerol, pH 7.0. Spectra with increasing absorbance at 450 nm are the reduced enzyme before reaction, the flavin-C4a-hydroperoxide (scan taken 30 s after mixing) and at subsequent time intervals to 10 min. The rate of decomposition of the hydroperoxide was measured from these spectra and found to be a first-order process with a rate constant of 0.005 s-1. Inset: Plot of the observed rate constants for formation of the flavin-C4a-hydroperoxide calculated from traces recorded at 390 nm against oxygen concentration in solution. From the plot, a second-order rate constant of 3 × 105 M-1s-1 was obtained for the formation of the hydroperoxide.
We also examined the reaction of oxygen with HpaA in complex with FADH− in the presence of the substrate, HPA. We mixed oxygenase (2-fold in excess over FAD) with FAD and HPA in a tonometer, made the solution anaerobic, and reduced the FAD with dithionite. This solution was mixed with an equal volume of oxygenated buffer in the stopped-flow spectrophotometer. The reaction was followed by absorbance traces collected at many individual wavelengths from 300 to 500 nm, and by fluorescence emission at wavelengths greater than 500 nm. Fluorescence excitation was at 370, 390, and 415 nm, wavelengths useful for detecting the flavin-C4a-hydroperoxide and hydroxide, and at 450 and 470 nm, wavelengths at which the C-4a adducts have very little absorbance, but at which oxidized FAD fluoresces strongly. Upon mixing with oxygen, the bound FADH− reacted rapidly to form the same high extinction flavin hydroperoxide as that shown in Fig. 2. The reaction was monitored at 390 nm using a range of oxygen concentrations, and it was found that it behaved as a second-order process with a rate constant of 3 × 105 M-1s-1, identical to the reaction without HPA present. The full reaction is illustrated by selected absorbance traces in Fig. 3A and fluorescence traces in Fig. 3B. By inspection, it can be seen that the absorbance trace at 415 nm represents a minimum of four phases in the reaction. Only the initial reaction with oxygen is dependent upon oxygen concentration, but at low O2 concentrations, the first two phases were not well separated because the observed processes occur consecutively at similar rates. Thus, only at the highest oxygen concentration used (0.62 mM), which makes the first phase significantly faster than the second, is it possible to clearly resolve all of these phases. The four phases thus observed are consistent with the following processes occurring: 1) the initial reaction with oxygen to form the C4a-flavin hydroperoxide (the first increase at 415 nm up to about 8 ms), 2) hydroxylation of HPA (the small decrease in absorbance at 415 nm from ∼ 8 to 80 ms caused by a blue shift in the spectrum as the C4a-hydroxyflavin adduct is formed), 3) formation of oxidized flavin (the increase in absorbance at 415 and 470 nm and the decrease at 390 nm occurring between 80 ms and 0.5 s), and 4) release of FAD from the oxygenase (the decrease in absorbance at 415 nm between 0.5 and 10 s). The last phase was identified by the fact that the final spectrum at 10 s was that of free FAD rather than the spectrum of the first observed oxidized FAD bound to the enzyme (see below). In addition, a large fluorescence increase appeared at the same rate as the final absorbance change (this was the only change seen when fluorescence was excited at 470 nm [Fig. 3B]), and this is consistent with the formation of the fluorescence due to free FAD. The intermediate phases that can be seen in the absorbance traces were more definitively identified by comparison to the fluorescence trace that was produced from excitation at 390 nm. Because flavin-C4a-hydroxides, but not flavin-C4a-hydroperoxides, are often highly fluorescent in flavoprotein oxygenases (3), the appearance of this fluorescence is a useful signature of the actual step involving hydroxylation. Fluorescence excitation at 390 nm of HpaA revealed an intensely fluorescent transient species (Fig. 3B) that is not seen with excitation at 470 nm. The formation and decay of the transient species excited at 390 nm corresponds well with the second and third phases in the absorbance trace at 415 nm (Fig. 3A). Thus, the second phase seen in the absorbance trace at 415 nm is due to the blue shift of the flavin-C4a-adduct as it converts from the hydroperoxide to the hydroxide, and it can be confidently attributed to the hydroxylation of the substrate that yields the C4a-hydroxyflavin. Decay of this species represents the formation of bound oxidized FAD. The fluorescence traces recorded with excitation near 390 nm could only be properly fitted when 5 exponentials were incorporated in a consecutive reaction, even though only three unique changes in fluorescent amplitude were readily apparent (Fig. 3B). From the analysis of the fluorescence traces, the observed rate constant for hydroxylation was 28 s-1, that for formation of FAD was 18 s-1, and the release of FAD from the oxygenase occurred at 0.5 s-1 under the conditions used in the experiment. No fluorescence change occurred during formation of the flavin hydroperoxide (see Fig. 3B), and even inclusion of the rate constant obtained from absorbance traces for the formation of the hydroperoxide (see above) does not give perfect fits for the substantial lag in the fluorescence traces before the hydroxylation step. Good fitting of this lag requires a fifth exponential, and in fact, absorbance traces obtained between 370 and 410 nm also could not be fitted accurately with four exponentials, but required the same fifth exponential with a value of ∼ 40 s-1. We hypothesized that this extra phase is the interaction (binding) of HPA with the oxygenase. Because the presence of HPA in the reaction had no effect on the initial reaction with oxygen, we surmised that binding occurred after formation of the FAD-C4a-hydroperoxide.
Figure 3.

Reaction of oxygen with HpaA in complex with FADH− and HPA. A – A solution of FADH− (15.5 μM), oxygenase (33 μM) and HPA (0.5 mM) in buffer (50 mM K phosphate, pH 7.2, containing 2.5% glycerol) was mixed with an equal volume of buffer containing oxygen (0.62 mM) in a stopped-flow spectrophotometer at 4 °C. The concentrations given are for the reaction mixtures after mixing in the stopped-flow instrument. The reaction was followed by the change in absorbance at the indicated wavelengths. The wavelengths (390, 415 and 470 nm) selected are those that best characterized the various steps of the reaction. B – The same experiment as in A after reconfiguration of the instrument to record fluorescence emission beyond 500 nm. One trace was from fluorescence excited at 390 nm (to monitor the formation of the C4a-hydroxyflavin), and the other was from fluorescence excited at 470 nm (to monitor the formation of oxidized FAD). C – Selected absorbance traces at 410 and 460 nm from an experiment similar to those in A and B, but with inclusion of 0.1 M KCl in the reaction. The final concentrations in this experiment were 26.8 μM FADH−, 60 μM oxygenase, 1.25 mM HPA, and 0.62 mM oxygen in the same buffer at 4 °C.
The above experimental evidence implies that there are five phases in the reaction, and all make sense when the requirements of the oxygenase are considered. However, there was one more problem arising from the data. Absorbance traces around 450 to 470 nm were fitted to a rate constant (10 – 11 s-1) that appeared to be due to the oxidation of the flavin; this was somewhat slower than the rate of loss of fluorescence due to decay of the flavin-C4a-hydroxide in the same experiment (18 s-1). The following experiments gave a reasonable explanation for this observation. We discovered that the addition of 0.1 M KCl to the reaction had a specific ion effect that slowed the release of FAD from the protein (by ∼3.5-fold). However, all other phases remained unchanged (see Fig. 3C). This experimental perturbation highlighted an additional phase in the overall reaction – a phase with a rate constant of ∼ 2 s-1 that occurs between the actual formation of oxidized FAD at 18 s-1 (the large increase in absorbance in Fig. 3C) and the slow release of FAD at 0.14 s-1 in the presence of Cl− (the final decrease in absorbance in the traces in Fig. 3C). We attribute the sixth phase to the release of product (3,4-DHPA) from the oxygenase. When this phase is incorporated into the original data, the time courses for absorbance at all wavelengths can be accounted for by the six rate constants. The complete reaction observed is summarized in Scheme 1. After 0.35 s from the start of the reaction in the stopped-flow spectrophotometer (see for example the peak in the 415 nm trace in Fig 3A), the FAD is completely oxidized, but only a small fraction has released from the oxygenase. The spectrum at 0.35 s was plotted from individual reaction traces at a series of wavelengths, and it is compared to the final spectrum of released FAD in Fig. 4. The portion of the spectrum from 410 to 550 nm can also be obtained by scanning the spectrum during enzyme-monitored turnover (described in the Combined operation section below). This transient spectrum is a composite of those for FAD in complex with oxygenase alone and for FAD in a complex that also includes the product, DHPA. The individual spectra could be more clearly observed in the reaction inhibited by KCl. However, because when Cl− is bound to the oxygenase it causes further changes in the FAD spectrum, the results are more difficult to quantify. The essential point learned from these observations, however, is that the spectrum of FAD is more resolved before it is released from HpaA, and this is consistent with it being in a non-aqueous more hydrophobic environment provided by specific interactions with the oxygenase. We could not reproduce these interactions with HpaA by simply adding FAD to the oxygenase, because FAD binds only weakly, and the complex can only be seen transiently just before FAD is released into solution. Thus, although this complex is not thermodynamically stable, it may be a protein conformation that has sufficient kinetic stability that it can be resolved experimentally. The conformational change that releases FAD from the oxygenase is sensitive to the composition of the reaction solution. For example, in addition to the effect of KCl, it was also found that the concentration of glycerol in the solution modifies the rate constant for the release of FAD.
Scheme 1.

Reaction mechanism of the oxygenase (PhaA) of HPAH from P. aeruginosa as deduced from the experimental analyses described in this paper.
Rate constants determined in this study are: k1 ≤ 2 × 106 M-1s-1; k2 = 3 × 105 M-1s-1; k3 ≤ 60 s-1; k4 = 28 s-1; k5 = 18 s-1; k6 = 2 s-1; k7 = 0.5 s-1.
Figure 4.

Spectra of FAD bound to HpaA obtained from an experiment under the same conditions as described in the legend to Figure 3A. The spectra of the oxygenase with FADH− and HPA bound are shown as the solid line (note the similarity to the reduced spectrum in Figure 2). Absorbance traces were collected over a range of wavelengths from 320 to 500 nm. The dashed line is the spectrum of FAD released from the oxygenase obtained by scanning 30 s after initiation of the reaction. The spectrum formed by a solid line with circular data points was obtained by plotting the absorbance (relative to the final FAD spectrum) 0.35 s after initiation of the reaction. This spectrum represents oxidized FAD bound to the oxygenase.
In the single-mix stopped-flow spectrophotometric studies described above, it was clear that HPA had no detectable influence on the reaction until the flavin hydroperoxide was formed. This observation is quite different from the case of C2 from A. baumannii (10), where there was clear evidence that HPA could bind to C2 after FADH− was bound. Nevertheless, it is possible that HPA is bound to the P. aeruginosa oxygenase, but does not interact with the active site. Therefore, we tested the response of HpaA to HPA in a double-mixing stopped-flow experiment. In the first mix, an anaerobic solution of oxygenase and FADH− was mixed with aerobic buffer and aged for various time periods (sufficient for the flavin hydroperoxide to be formed completely). Then in the second mix, an aerobic solution of HPA was added to the hydroperoxide. The reaction initiated by the second mix was monitored by both diode array spectrophotometry and single wavelength absorbance changes. Aliquot samples of the reacted mixtures were collected and analyzed for product using the specific DHPAO reaction (see Materials and Methods). These experiments showed that there was approximately one DHPA formed for each FADH– oxidized. Thus, HPA can bind to the flavin hydroperoxide form of the oxygenase to initiate hydroxylation. The rapid scanning diode array spectra showed clearly that mixing oxygenase with HPA caused a small blue shift of the flavin C4a-hydroperoxide spectrum in the first 30 to 50 ms. Unfortunately, later in the time course this reaction mix was sensitive to some photochemical reaction(s) of the flavin when using diode array measurement (caused by the high intensity white light required for the diode array technique), so the entire reaction could not be monitored in this manner.
The interaction between HPA and the bound flavin C4a-hydroperoxide was examined further by single wavelength double-mixing measurements, in which the light intensity is sufficiently low to avoid photochemistry. The absorbance changes at 415 nm (Fig. 5A) show the effect of HPA concentration. These changes are qualitatively the same as those in the 415 nm trace in Fig. 3A. However, because the hydroperoxide had already been formed, the first reaction with oxygen is missing. The concentration of HPA affects the rate of the initial decrease in absorbance (up to ∼ 80 ms). It required 2 mM HPA to saturate this phase. Insufficient data were collected to permit a quantitative analysis of the interaction of HPA with the oxygenase, but at 2 mM HPA, the observed rate constant for the interaction was 50 to 60 s-1. The hydroxylation phase and the phase resulting in formation of oxidized FAD are only marginally slower than the initial interaction with HPA, and thus both are affected by the concentration of HPA. The reactions at different HPA concentrations finally merge in the slow phase due to flavin release (Fig. 5A). Fig. 5B compares the rates of formation of FAD (measured by the absorbance increases at 450 nm) when the flavin hydroperoxide species was mixed with 500, 8 and 0 μM HPA. With approximately equimolar HPA (8 μM trace), it is clear that the rate of formation of FAD is dominated by the nature of the interaction between HPA and the oxygenase. At this concentration of HPA, the limiting process could be an equilibrium that has only a fraction of HPA bound, or a slow rate of a conformational or other change to form the productive active site complex with the oxygenase. However, when the traces for 8 and 0 μM HPA are compared (Fig. 5B), it is clear that because of the long half-life of the hydroperoxide, the flavin hydroperoxide form of the oxygenase is still capable of forming product even at low substrate concentrations.
Figure 5.

Kinetic traces observed upon reaction of HPA with the oxygenase in the form of the flavin-C4a-hydroperoxide. This experiment was performed with a double-mix stopped-flow spectrophotometer. In the first mix, HpaA with FADH− was reacted with oxygen under the same conditions as described in the legend to Fig. 2 to form the flavin-C4a-hydroperoxide. Then, in the second mix, this component was reacted with different concentrations of HPA. There were no observable differences due to using aging times ranging from 50 ms to 10 s. The reaction after the second mix contained 7.5 μM FAD, 20 μM oxygenase, 440 μM oxygen, and various concentrations of HPA in 50 mM K phosphate buffer containing 2.5% glycerol at pH 7.2 and 4° C. A – Reaction traces recorded at 415 nm using the concentrations of HPA indicated on the figure. Note the similarity to the reaction trace at 415 nm in figure 3A. Three phases showed some dependence on HPA concentration (see text). B – Reaction traces recorded at 450 nm. The reaction with 0.5 mM HPA has similar kinetics to the traces in A. The reaction with an approximately stoichiometric concentration of HPA (8 μM) shows that at this concentration of HPA, binding is comparatively slow and limits the reaction rate for forming oxidized FAD.
Combined operation of reductase and oxygenase
To conduct efficient catalysis in the cellular environment, two-component flavin-dependent enzymes must transfer a reduced flavin cofactor from the reductase to the oxygenase without losing valuable reducing equivalents via side reactions with oxygen or other oxidizing agents, processes that could generate potentially dangerous reactive chemical species. In addition, after carrying out the oxygenation, the oxidized flavin must be efficiently returned to the reductase to be re-reduced for additional rounds of catalysis. Two previous studies with different two-component enzymes (including HPAH from A. baumannii) have found that the flavin bound to the reductase is transferred between the proteins during catalysis by diffusion without requiring formation of a complex between them (6,12). We used stopped-flow experiments to test this concept with HPAH from P. aeruginosa. An aerobic solution of reductase plus oxygenase was mixed with an aerobic solution of NADH without HPA. The result is shown in Fig. 6. The absorbance spectrum of the proteins before the reaction was that of FAD bound to the reductase, as expected based upon a consideration of the Kd values for FAD with each protein individually; similar observations have been made with other systems (6,12). Upon mixing, the absorbance trace at 460 nm had a short lag due to binding of NADH to reductase (this lag is not visible on the long time scale of figure 6), followed by reduction of the FAD (absorbance decrease) with a rate constant of 7 s-1, which is exactly as observed with reductase alone (above). At 400 nm, there is again a small fast phase in which the absorbance decreases, followed by an increase in absorbance with a rate constant of 6.5 s-1. At the end of the second phase, the spectrum of the solution was recorded to confirm that the FAD was now on the oxygenase as the flavin C4a-hydroperoxide (data not shown). This reaction was repeated with the NADH solution saturated with 100% oxygen, making the final oxygen concentration 3-fold higher than in the first reaction. The only difference was a slight increase in the rate of formation of the flavin hydroperoxide, as monitored at 400 nm (6.8 vs. 6.5 s-1); thus, although the step that forms the C4a-flavin hydroperoxide is undoubtedly faster at the higher oxygen concentration, it is limited by the rate of reduction of the FAD on the reductase. These results are consistent with fast dissociation of FADH− from the reductase and fast binding to the oxygenase; they are not consistent with the apparent rate of dissociation of FADH− from the reductase as measured by its reaction with DCIP (17 s-1, as reported above) and the rate of its binding to the oxygenase in a separate experiment (26 s-1, as reported above) under similar circumstances. These separately measured rate constants predict a maximum rate of formation of the flavin hydroperoxide of ∼ 4 s-1 (1/7 + 1/17 + 1/26 = 1/k). It is even possible that DCIP can react with the reduced flavin before it fully dissociates from HpaC, making the value 17 s-1 greater than the true value for dissociation. However, full dissociation is obviously required for the oxygenase to bind the FADH− and react it with O2 to form the FAD-C4a-hydroperoxide. Thus, we must consider that some specific protein/protein interactions may occur in the overall reaction of this HPAH.
Figure 6.

FADH− transfer between the reductase and the oxygenase. This process was observed using a stopped-flow instrument. A solution containing reductase and oxygenase was mixed with an equal volume of a solution of NADH, and the reaction was followed by the absorbance at 400 and 460 nm. The final composition in the reaction mixture was 11 μM HpaC (determined by the absorbance a 450 nm due to its bound FAD), 15 μM HpaA, and 50 μM NADH in aerobic buffer (50 mM K phosphate containing 1% glycerol at pH 7.2 at 4 °C). At 460 nm, the trace represents the rapid binding of NADH to the HpaC, followed by the large decrease of absorbance over ∼ 0.5 s upon the reduction of FAD bound to the reductase. At 400 nm, the trace represents the same phases on the reductase initially, followed by the increase in absorbance up to 0.5 s due to the formation of the high extinction flavin-C4a-hydroperoxide on the oxygenase.
An important property of all oxygenases is the degree of coupling achieved in catalysis. That is, in the case of this enzyme, the ratio of hydroxylated product formed per NADH consumed. A perfect oxygenase has a ratio of one; i.e., no reactive oxygen species are formed. Many oxygenases do not achieve this ideal goal, and even the most efficient can form reactive oxygen species under a variety of reaction conditions. We used the following experiment to measure coupling with this HPAH. First, a reaction was set up to follow turnover in the combined system by monitoring the oxidation of NADH with limiting HPA present. When HPA is exhausted, the consumption of NADH should nearly cease in an effective system because almost all of the FAD will be trapped on the oxygenase as the quasi-stable C4a-hydroperoxide. If the HPA concentration is known, the amount of NADH consumed to process the HPA can be measured from the kinetic traces. With a molar ratio of 1 to 2, reductase (5 μM) to oxygenase (10 μM), it was found that coupling was 0.90, and the residual rate of consumption of NADH was low, but finite. Product formation was measured by coupling with DHPAO, which converts the product into an intensely absorbing chromophore that can be quantified (Materials and Methods). When NADH was the limiting substrate, product analysis also showed 0.90 coupling as above.
To examine the kinetics of the complete reaction, the technique of enzyme-monitored turnover (EMT) was used (21). With this method, the response of the enzyme in steady state is followed with time from the absorbance due to an intermediate state of the flavin. In some cases, this approach can lead to new insights into the function of the enzyme [see e.g. (22)]. It was established above that FADH− is transferred from the reductase to the oxygenase. With this HPAH complex, there was no indication of product inhibition, a situation ideal for EMT. The reactions were conducted in a stopped-flow spectrophotometer. One syringe contained an aerobic solution of HpaC (which provided the only FAD for the reaction) and HpaA, and the other contained aerobic solution of HPA plus NADH in excess. When the solutions were mixed, oxygen was 0.25 mM and was the limiting substrate in the reaction. Catalysis was followed by absorbance at selected wavelengths (see Fig. 7A). The initial phase observed (up to 0.25 s at 450 nm) was due to the reduction of FAD on the reductase, consistent with observations of the reductase alone. From 0.25 to 2 s, the reaction proceeded through an evolving mixture of enzyme forms in pre-steady-state, before attaining a long period (from 2 to about 50 s) of nearly constant composition of enzyme species that was consistent with steady-state turnover at saturating substrate concentrations. In the final phase, the reaction becomes dependent upon the concentration of oxygen until all the oxygen is consumed, and the reaction stops (shown by the decrease in absorbance at all wavelengths as all of the flavin is converted to FADH−). The reaction mixture contained essentially saturating substrate concentrations for the enzyme, implying nearly Vmax conditions. The number of turnovers occurring during the steady-state period (∼2–50 s) could be calculated by dividing the concentration of the limiting reagent, oxygen, by the enzyme concentration, and the turnover rate (∼Vmax) could thereby be estimated by inspection to be ∼ 0.54 s-1. This value is in excellent agreement with the transient kinetic analyses discussed above. With both reductase and oxygenase studied separately, the outstanding slow step was found to be the release of FAD from the oxygenase with a rate constant of 0.5 s-1. Consistent with these rate constants, it was found that approximately 85 % of the enzyme during steady state was present as FAD (based upon the absorbance at 450 nm), not as FADH− or as C4a-adducts. Furthermore, most of the FAD in steady state was bound to the oxygenase, as can be estimated from the absorbance at 480 nm (note the large difference in absorbance between bound and free FAD at this wavelength – see Fig. 4). From the last phase of turnover (at times > 50 s), where the composition of enzyme species was dependent upon oxygen concentration, the Km for O2 was estimated to be 4 μM using standard EMT methods (21).
Figure 7.

Enzyme-monitored turnover (EMT) analysis of HPAH under conditions where substrate concentrations are saturating over most of the time period of turnover. A – In this experiment, a solution containing reductase and a slight excess of oxygenase was mixed in a stopped-flow spectrophotometer with an equal volume of a solution of NADH and HPA such that oxygen was the limiting substrate. The final reaction mixture in the stopped-flow instrument contained reductase with 10 μM FAD, 15 μM oxygenase, 0.75 mM NADH, 0.75 mM HPA and 0.26 mM oxygen in 50 mM K phosphate buffer containing 2.5% glycerol at pH 7.2 at 4 °C. Selected traces are shown. The pre-steady-state phase lasted for ∼ 2 s, before the mixture of enzyme species attained a steady-state concentration that lasted for about 50 s. Finally, the FAD became reduced as oxygen was depleted in the last segment of the reaction. B – A similar experiment to A in which the concentration of HPA was limiting in the reaction. The final reaction contained the same components as in A, but the amount of HPA was reduced to 0.15 mM. The 415 nm trace is quite different from that in A; when the reaction was complete the absorbance was high compared to that in A because the FAD was now bound to the oxygenase as the flavin-C4a-hydroperoxide rather than as FADH−.
EMT was repeated using the same conditions as above, except HPA was made the limiting substrate by using it at an initial concentration after mixing of 0.15 mM. The absorbance traces at 450 and 415 nm are shown in Fig. 7B. If the traces were printed in linear time, it would be clear that there was a significant dependence on HPA concentration. Quantitative EMT analysis of the HPA dependence gave a Km for HPA of 8 μM and a turnover number as 0.45 s-1. When HPA was exhausted, the absorbance at 450 nm (Fig. 7B) suggested that the enzyme was reduced (i.e., it had the same absorbance as the experiment with oxygen limiting). However, the trace at 415 nm shows that after HPA was exhausted, the enzyme absorbance is characteristic of the flavin hydroperoxide (nearly isosbestic with oxidized FAD) not the reduced flavin, and quite different from the case when oxygen was limiting (compare Figs. 7A and 7B). In cells, HPA is likely to be the limiting substrate from time to time (not oxygen or NADH). This result demonstrates that the “resting” form of the enzyme in a cell most likely contains the available FAD as the hydroperoxide on the oxygenase. Because the oxygenase is likely to be at a greater concentration [e.g., approximately 120 μM in E. coli (5)] than the free FAD in the cell [under strict control with a likely upper limit in bacteria for FAD ∼ 10 μM (23)], this property results in an effective trap for free flavin, preventing the reductase from producing FADH− that otherwise would react with oxygen to form reactive oxygen species.
We were able to demonstrate the above principle by EMT analysis in the presence of both the oxygenase and the reductase. When FAD was present in excess of the concentration of the oxygenase active sites (we tried 2-fold excess of FAD for illustration), and HPA was the limiting substrate, the depletion of HPA failed to limit the rapid consumption of NADH; instead, NADH consumption continued until all the oxygen was used up. Thus, conditions such as these, with FAD in excess of the oxygenase, result in the formation of reactive oxygen species because during turnover there is a supply of free FADH− in solution that cannot be trapped by the oxygenase. This situation probably rarely occurs in a cell, since there is negligible free FAD available for turnover with the reductase and O2.
Discussion
Flavin-dependent hydroxylases generally function by first reducing the flavin by NAD(P)H, reacting the reduced flavin with O2 to form a C4a-flavin hydroperoxide, and then hydroxylating the substrate. The order of addition of substrates can vary with individual enzymes. The reductive reactions and the oxygenative reactions have quite different catalytic requirements. In PHBH and in phenol hydroxylase, which are prototypes of the single component hydroxylases, significant movement of the flavin within the protein structure provides the distinct environments required for the reduction and hydroxylation reactions in the overall catalysis (3). In contrast, these two roles occur on separate proteins in the two-component hydroxylases. Using separate proteins may facilitate adaptation to new substrates more quickly than with single component systems that must coordinate the oxygenase and the reductase functions.
In this paper, we have presented results from experiments that describe the properties and basic mechanism of HPAH from P. aeruginosa to illuminate some similarities and differences between the function of HPAH from this source and that from A. baumannii. The structures of the components of HPAH from A. baumannii (7 and P. Chaiyen, personal communication) and of the components of the HPAH system from T. thermophilus (8,9) will be referred to in this discussion. As mentioned in the Introduction and Results, the essential features of the published structure of the T. thermophilus HPAH are very likely present also in the P. aeruginosa enzyme, and many of these features are different from those of the A. baumannii enzyme. Although these comparisons are useful for developing an understanding of various mechanisms of activating oxygen and regulating these hydroxylations, we note that there are many two-component flavin-dependent hydroxylases that are likely to be considerably different than those discussed here. These include the bacterial luciferases (2) and the halogenases (24), which contain completely different oxygenase structures and therefore may have very different mechanisms.
The larger flavin reductase, C1 from A. baumannii, contains, in addition to the N-terminal catalytic flavin reductase domain found in all of the reductases, a C-terminal regulatory domain that binds HPA (P. Chaiyen, personal communication). The sequence of the N-terminal domain of C1 is similar to the complete sequence of the smaller reductases of the alternative group of two-component enzymes, including that of the P. aeruginosa reductase, HpaC (25). C1 has a tightly bound FMN that is reduced by NADH two orders of magnitude faster when HPA is bound to the C-terminal domain of the reductase than when it is absent (11). This mode of regulation is analogous to that in one-component hydroxylases, such as PHBH, that do not reduce flavin rapidly unless the substrate is bound (3). In the absence of substrate, this property prevents rapid formation of reduced flavin, which would react with oxygen to waste energy and produce H2O2 and O2− •. By contrast, HpaC from P. aeruginosa is fully capable of using NADH to reduce FAD, even at micromolar concentrations, and is not regulated by HPA. As is discussed below, regulation with the P. aeruginosa HPAH occcurs via the oxygenase, HpaA. Both classes of reductase have the common property that they bind oxidized flavin more tightly than reduced flavin, and this property facilitates the delivery of reduced flavin to the partner oxygenases and the rebinding of the oxidized flavin products for further rounds of catalysis. A similar scenario exists in the ActVA-ActVB system that participates in the biosynthesis of actinorhodin (12). The transfer of reduced flavin to the oxygenase is much faster than the overall catalytic rate with both the A. baumannii (6) and the P. aeruginosa enzymes (see Fig. 6). Our data from experiments with the P. aeruginosa HPAH suggest that there is some form of protein-to-protein interaction that facilitates transfer of FADH−, while no such requirement exists for transfer of FMNH− in the A. baumannii enzyme (6) or in the ActVA-ActVB system (12).
The oxygenase C2 from A. baumannii is a homotetramer with subunits of 422 amino acids, while the HpaA oxygenase from P. aeruginosa is a dimer of dimers with each subunit containing 520 amino acids. The amino acid sequences show possible distant evolutionary relationships, but gaps are required in the A. baumannii sequence to permit alignment, and there is little identity to the P. aeruginosa sequence. Nevertheless, as is mentioned in the Introduction, structures of both oxygenases show similar patterns of folding, indicating that they are likely to have derived from a common origin. The important differences in the structures occur in the details of the binding of the flavin cofactor, the binding of HPA, and the residues in the active site. Three prominent loops in the central domain between β-sheet strands in the T. thermophilus oxygenase PhaB (8) are directly involved in the structural differences (Fig. 8). The P. aeruginosa oxygenase PhaA sequence suggests that it has similar loops, but these loops do not exist in the C2 A. baumannii oxygenase. The isoalloxazine ring of the flavin is bound in a similar slot in both PhaB and C2 (between the central β-sheet domain and the α-helical C-terminal domain). However, in PhaB, the AMP portion of FAD has a binding pocket partly formed by loop A in Fig. 8 (scarlet) that is not found in the A. baumannii oxygenase. This loop has several residues that interact with the AMP moiety, including R151 and Q148 (R164 and Q157 in HpaA) that H-bond to the phosphate, to provide the specificity for FAD that has also been found in the P. aeruginosa oxygenase (see Results). However, in C2, the ribityl phosphate is exposed to solvent (7), which explains why the protein is equally active with FMNH− or FADH− (The specificity for FMN in the A. baumannii HPAH is derived from the reductase, C1). Therefore, if C2 is coupled with an FAD reductase, it will readily catalyze the formation of DHPA (10).
Figure 8.
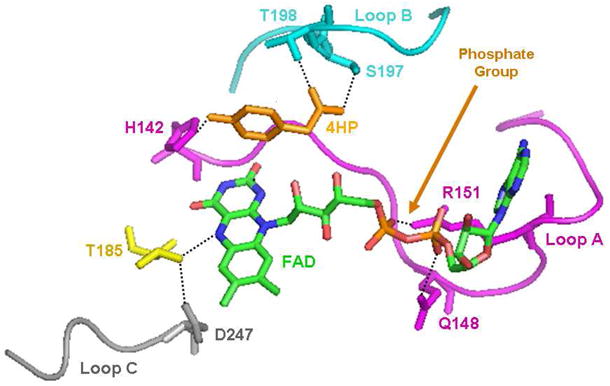
Illustration of three large loops in the structure of the oxygenase from T. thermophilus (adapted from the structure 2YYJ in the PDB) that are not found in the structure of the oxygenase from A. baumannii (7). FAD and HPA bound to the protein are shown in CPK colors, loop A (between strands β5 and β6 and interacting with part of FAD) is shown in scarlet, loop B (between strands β8 and β9 and interacting with HPA) is shown in cyan, and loop C (between strands β10 and β11 and possibly interacting with the flavin hydroperoxide) is shown in grey. Some potential hydrogen bonds are shown as dotted line.
In HpaB significant movements of two loops accompany the binding of FAD, causing the formation of the binding site for HPA and leaving the pyrimidine portion of the isoalloxazine exposed to solvent (8). On binding of HPA, further movement of the loops occurs, which blocks access of the active site to solvent. This led to the suggestion that in catalysis HPA binds to the complex of HpaB with its reduced flavin, as happens with C2 (10). This speculation does not correspond to our experimental results. We only see binding of HPA after the C4a-hydroperoxide has formed. We note that the crystals for the structures were obtained with a very high concentration of FAD rather than FADH– (8).
Both of the two-component HPAH enzymes bind HPA over the re face of the isoalloxazine in similar orientations that are suitable for hydroxylating the 3-position of the substrate (Fig. 8). The orientation of substrate is different from that in the one-component flavoenzymes (26), but the relative orientation of the substrate for attack by the flavin hydroperoxide is probably not very different. The residues that bind HPA are quite different for the two classes of two-component oxygenases. With HpaB, movement of loop B (Fig. 8, cyan) causes it to close over and cover the active site, and also to provide interactions with HPA via S197 and T198 (S197 aligns with S210 in HpaA and T198 is not conserved, but Q212 in HpaA may serve a similar role); loop B does not exist in C2. Binding of FMNH− and HPA by C2 involves only very minor changes in the orientation of a small number of protein residues, whereas binding of HPA and FADH− by HpaB involves significant conformational changes in loops A and B, as mentioned above [Fig. 8 and (8)]. These features in the structures provide a foundation for understanding the differences in the kinetic mechanisms of the two different oxygenases and provide some insight into the way that protein evolution has achieved the same chemical catalysis with one- and two-component hydroxylases.
In both types of two-component HPAH systems, the first step in the oxygenation reaction is to bind reduced flavin to the oxygenase. Under similar experimental conditions and with the flavin and oxygenase at ∼ 10 μM, this process for C2 is very fast (> 500 s-1) (10), but with HpaA was easily measured in the stopped-flow spectrophotometer (26 s-1 - see Results). The ∼ 20-fold difference in rate for the two oxygenases may be partially due to the conformational changes that are required in the T. thermophilus-type (8), but not in the A. baumannii type. Both C2 and HpaB provide a cavity (H2O is present in the structure of HpaB) near the C4a-position that can accommodate oxygen to form the hydroperoxide. With reduced flavin bound, both C2 and HpaA react rapidly with oxygen to form the flavin C4a-hydroperoxide in the absence of HPA. C2 can also react with oxygen when HPA is bound, but the reaction is ∼ 10-fold slower than in its absence (10). We found no evidence for HpaA binding HPA before reacting with oxygen (see Results), but evidence was clear for interactions with HPA after formation of the flavin-C4a-hydroperoxide.
HpaA and C2 form remarkably stable hydroperoxides in the absence of HPA. This is most likely due to several factors. First, both restrict solvent access to the C4a and N5 positions. The one-component aromatic hydroxylases like PHBH, which do not form stable hydroperoxides, only isolate the C4a and N5 positions of the flavin from solvent when 4-hydroxybenzoate is bound and the flavin is reduced; stabilization of the C4a-hydroperoxide is not important in the absence of substrate because due to slow reduction of the flavin, very little forms (26). In addition, both C2 and HpaB (and presumably HpaA) appear to form H-bonds to N5 of the isoalloxazine ring that may be significant in the stabilization of the flavin hydroperoxide. In HpaB, T185 coupled to D247 forms a hydrogen bond to the N5 proton of the reduced isoalloxazine ring (Fig. 8). In general, an H-bond to N5 is not found in those one-component hydroxylases that do not stabilize the hydroperoxide in the absence of substrate (e.g., PHBH). In contrast, the recent structure of a one-component flavin monooxygenase that does stabilize the hydroperoxide without substrate clearly has an H-bond to N5, and it is formed by the essential nicotinamide from NADP that is bound in the active site (27). It is notable that the hydroperoxide of HpaA is more stable (7-fold longer half-life) than that for the C2 oxygenase. Perhaps HpaA has a stronger H-bond to N5 of the flavin hydroperoxide than does C2. Such a stronger H-bond may even be responsible for the considerably larger extinction coefficient for the FAD-C4a-hydroperoxide formed in HpaA compared to those for other flavoprotein hydroxylases (see Fig. 2). Arginine-100 of HpaB is likely to form a hydrogen bond with the C4a-hydroperoxide, and thus also may contribute to its stability. Another possibility for the increased stability of the C4a-hydroperoxide in HpaA is that loop B (Fig. 8), even in the absence of HPA, makes contact of the flavin with solvent less probable. Unfortunately, crystal structures of static equilibrium conditions are not sufficient to resolve these possibilities.
The two model HPAH oxygenases display very comparable hydroxylation kinetics under similar experimental conditions. The rate constant for hydroxylation by HpaA is 28 s-1 compared to 20 s-1 for C2 under the same conditions (10). These reactions are only 2- to 3-fold slower than those of the one-component model enzyme, PHBH. However, both oxygenases prefer to bind HPA to the flavin hydroperoxide state of the oxygenase, a catalytic strategy quite different from that of many of the one-component enzymes, which usually bind substrate before forming the flavin hydroperoxide. (Note, however, that the microsomal flavin monooxygenase and the Baeyer-Villiger oxygenase, cyclohexanone monooxygenase, bind substrate after the hydroperoxide has formed (3,13)). The structures show that the sensitive C4a, N5 region of the isoalloxazine can remain solvent free, while HPA can bind over the re face of the flavin by the rearrangement of residues on that face of the flavin. It would be fascinating to know the configuration of the hydroperoxide group in the protein cavity during the transient process of HPA binding.
Kinetic analysis of C2 (10) showed a two-step binding process for HPA, with the initial interaction being quite weak (Kd of 0.35 mM). With HpaA, we found that it took 1 to 2 mM of HPA to reach the limiting rate of 50 – 60 s-1 for the binding of HPA (Fig. 5A). Given that this rate reaches a limiting value, the binding process for this oxygenase must also involve at least two steps, with the first equilibrium characterized by a large Kd. In the processes following hydroxylation, the kinetics of the two enzymes are significantly different. C2 releases the product with a rate constant of ∼ 8 s-1, and subsequently releases FMN (or possibly, the flavin hydroxide) with a rate constant of 8.3 s-1. This order of events makes C2 sensitive to inhibition by HPA (KI of 41 μM), because this substrate forms a dead-end complex with the flavin hydroxide (10), a process that also occurs with many of the one-component enzymes, as originally described for PHBH (28). HpaA has an active site that permits loss of water from the flavin hydroxide with a rate constant of 18 s-1 (by some as yet unknown mechanism), followed by product dissociation at 2 s-1, and finally, dissociation of FAD from the non-aqueous oxygenase active site at 0.5 s-1 (see Results). Although the last steps (presumably involving the reversal of the protein conformational changes occurring with FADH− and HPA binding to start the reaction) are comparatively slower with the P. aeruginosa enzyme, this enzyme is not sensitive to HPA inhibition. This observation reinforces the earlier conclusion that HPA mainly binds only to the flavin hydroperoxide form of the enzyme, whereas C2 can bind HPA before reacting with oxygen.
Finally, how do these HPAHs compare when operating as a two-component enzyme with reductase and oxygenase at approximately the same concentrations? In the detailed study of the A. baumannii enzyme (6), it was found that the turnover number of the reaction was 2 s-1 at 4 °C, with a coupling ratio of 0.86, and that both the reductase and oxygenase activities were regulated by HPA concentration. Under the high HPA concentrations conditions generally used for assays, the last step in the oxygenase reaction (loss of water from the flavin hydroxide) was inhibited by HPA, and this governed the overall rate. It was clearly established for the A. baumannii HPAH system that the highest degree of coupling NADH and oxygen consumption to product formation occurred when the FMN concentration was ≤ the concentration of the oxygenase, C2. With the P. aeruginosa enzyme, the turnover number for the reaction was ∼ 0.54 s-1 at 4 °C (Fig. 7), with a coupling ratio of at least 0.90. Neither reductase nor oxygenase activities were regulated by the HPA concentration. The release of FAD from the oxygenase was the rate-determining step in catalysis, and this process was sensitive mostly to reaction conditions (such as inclusion of glycerol and/or various concentrations of salt) unrelated to substrates. The reductase had to be present in concentrations similar to those of the oxygenase to avoid any influence of the rate of FAD reduction on the overall turnover number of the system (results not shown). With the P. aeruginosa enzyme, it was clear that effective catalysis (i.e., with little or no uncoupling in which NADH consumption produced H2O2) could proceed provided that the FAD concentration in the reaction remained less than the concentration of active sites in the oxygenase (see Results). This FAD could come simply from the reductase in the mixture, or could be added separately, whether the reductase was initially saturated with FAD or not. In the environment of the cell, where ample NADH is usually present, when HPA is at low concentrations this HPAH probably contains FAD largely as the quasi-stable C4a-hydroperoxide bound to the oxygenase (Fig. 7B) and also depends upon the stabilization of the flavin hydroperoxide as a means to regulate catalysis and avoid producing reactive oxygen species. In the case of the A. baumannii enzyme in a cell that contains NADH but no HPA, catalysis by the reductase and the oxygenase, as well as transfer of FMNH− between the two proteins, become very slow compared to the reactions with HPA present (6). Thus, some of the available FMN probably resides on both the reductase and the oxygenase; this hypothesis has not been tested. Successful product formation by the two-component enzymes depends on the capture of the HPA by the oxygenase before the hydroperoxide decomposes. Presumably, evolutionary pressure has resulted in rather stable C4a-flavin hydroperoxides to assure that good coupling occurs.
The details of how these two-component flavin-dependent hydroxylases activate their substrates and carry out aromatic hydroxylations are not understood to the extent that we know about the single-component hydroxylases such as PHBH. Some specific questions for the two-component systems that remain are: From where does the proton come that protonates the flavin peroxide formed in the reaction between reduced flavin and oxygen? How is the aromatic ring of HPA activated towards electrophilic attack from the hydroperoxide? Is the flavin hydroperoxide activated as occurs in PHBH? These questions may have different answers in each type of HPAH, given that they have different residues in the active site.
Acknowledgments
We would like to thank Dr. Michael Tarasev for help in constructing Figure 8.
Funding Support. This work was supported by the National Institutes of Health GM64711 to D.P.B.
Abbreviations
- HPAH
4-hydroxyphenylacetate hydroxylase
- HPA
4-hydroxyphenylacetate
- DHPA
3,4-dihydroxyphenylacetate
- DHPAO
3,4-dihydroxyphenylacetate dioxygenase
- C1
reductase of HPAH from Acinetobacter baumannii
- C2
oxygenase of HPAH from Acinetobacter baumannii
- HpaC
reductase of HPAH from Pseudomonas aeruginosa
- HpaA
oxygenase of HPAH from Pseudomonas aruginosa
- HpaB
oxygenase of HPAH from Thermus thermophilus
- PHBH
p-hydroxybenzoate hydroxylase
- DCIP
dichlorophenolindophenol
- EMT
enzyme-monitored turnover
- LB
Luria Broth
- MALDI-MS
Matrix assisted laser desorption/ionization-Mass Spectrometry
References
- 1.Van Berkel WJ, Kamerbeek NM, Fraaije MW. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J Biotechnol. 2006;124:670–689. doi: 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- 2.Campbell ZT, Baldwin TO. Fre is the major flavin reductase supporting bioluminescence from Vibrio harveyi luciferase in Escherichia coli. J Biol Chem. 2009;284:8322–8328. doi: 10.1074/jbc.M808977200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Entsch B, Cole LJ, Ballou DP. Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Archiv Biochem Biophys. 2005;433:297–311. doi: 10.1016/j.abb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 4.Arunachalam U, Massey V, Miller SM. Mechanism of p-hydroxyphenylacetate 3-hydroxylase, a two-protein enzyme. J Biol Chem. 1994;269:150–155. [PubMed] [Google Scholar]
- 5.Louie TM, Xie XS, Xun L. Coordinated production and utilization of FADH2 by NAD(P)H-flavin oxidoreductase and 4-hydroxyphenylacetate 3-monooxygenase. Biochemistry. 2003;42:7509–7517. doi: 10.1021/bi034092r. [DOI] [PubMed] [Google Scholar]
- 6.Sucharitakul J, Phongsak T, Entsch B, Svasti J, Chaiyen P, Ballou DP. Kinetics of a two-component p-hydroxyphenylacetate hydroxylase explain how reduced flavin is transferred from the reductase to the oxygenase. Biochemistry. 2007;46:8611–8623. doi: 10.1021/bi7006614. [DOI] [PubMed] [Google Scholar]
- 7.Alfieri A, Fersini F, Ruangchan N, Prongjit M, Chaiyen P, Mattevi A. Structure of the monooxygenase component of a two-component flavoprotein monooxygenase. Proc Natl Acad Sci USA. 2007;104:1177–1182. doi: 10.1073/pnas.0608381104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim SH, Hisano T, Takeda K, Iwasaki W, Ebihara A, Miki K. Crystal structure of the oxygenase component (HpaB) of the 4-hydroxyphenylacetate 3-monooxygenase from Thermus thermophilus HB8. J Biol Chem. 2007;282:33107–33117. doi: 10.1074/jbc.M703440200. [DOI] [PubMed] [Google Scholar]
- 9.Kim SH, Hisano T, Iwasaki W, Ebihara A, Miki K. Crystal structure of the flavin reductase component (HpaC) of 4-hydroxyphenylacetate 3-monooxygenase from Thermus thermophilus HB8: structural basis for the flavin affinity. Proteins. 2008;70:718–730. doi: 10.1002/prot.21534. [DOI] [PubMed] [Google Scholar]
- 10.Sucharitakul J, Chaiyen P, Entsch B, Ballou DP. Kinetic mechanisms of the oxygenase from a two-component enzyme, p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii. J Biol Chem. 2006;281:17044–17053. doi: 10.1074/jbc.M512385200. [DOI] [PubMed] [Google Scholar]
- 11.Sucharitakul J, Chaiyen P, Entsch B, Ballou DP. The reductase of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii requires p-hydroxyphenylacetate for effective catalysis. Biochemistry. 2005;44:10434–10442. doi: 10.1021/bi050615e. [DOI] [PubMed] [Google Scholar]
- 12.Valton J, Mathevon C, Fontecave M, Nivière V, Ballou DP. Mechanism and regulation of the two-component FMN-dependent monooxygenase ActVA-ActVB from Streptomyces coelicolor. J Biol Chem. 2008;283:10287–10296. doi: 10.1074/jbc.M709730200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Entsch B, Cole LJ, Ballou DP. Protein dynamics in catalysis by flavoprotein hydroxylases. In: Nishino T, Miura R, Tanokura M, Fukui K, editors. Flavins and Flavoproteins 2005. ARchiTect; Tokyo: 2005. pp. 143–154. [Google Scholar]
- 14.Chakraborty S, Ortiz-Maldonado M, Eschenburg K, Entsch B, Ballou DP. p-hydroxyphenylacetate 3-hydroxylase from Pseudomonas aeruginosa. In: Nishino T, Miura R, Tanokura M, Fukui K, editors. Flavins and Flavoproteins 2005. ARchiTect; Tokyo: 2005. pp. 161–166. [Google Scholar]
- 15.Van den Heuvel RHH, Westphal AH, Hecks AJR, Walsh MA, Rovida S, van Berkel WJH, Mattevi A. Structural studies on flavin reductase PheA2 reveal binding of NAD in an unusual folded conformation and support novel mechanism of action. J Biol Chem. 2004;279:12860–12867. doi: 10.1074/jbc.M313765200. [DOI] [PubMed] [Google Scholar]
- 16.Duch DS, Laskowski M., Sr A sensitive method for the determination of RNA in DNA and vice versa. Anal Biochem. 1971;44:42–48. doi: 10.1016/0003-2697(71)90343-5. [DOI] [PubMed] [Google Scholar]
- 17.Arunachalam U, Massey V, Vaidyanathan CS. p-Hydroxyphenylacetate-3-hydroxylase. A two-protein component enzyme. J Biol Chem. 1992;267:25848–25855. [PubMed] [Google Scholar]
- 18.Galán B, Díaz E, Prieto MA, García JL. Functional analysis of the small component of the 4-hydroxyphenylacetate 3-monooxygenase of Escherichia coli W: a prototype of a new flavin:NAD(P)H reductase subfamily. J Bacteriol. 2000;182:627–636. doi: 10.1128/jb.182.3.627-636.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibian MJ, Rynd JA. Oxidation of reduced flavins by quinones. Biochem Biophys Res Commun. 1969;34:594–599. doi: 10.1016/0006-291x(69)90779-7. [DOI] [PubMed] [Google Scholar]
- 20.Entsch B, Ballou DP, Massey V. Flavin-oxygen derivatives involved in hydroxylation by p-hydroxybenzoate hydroxylase. J Biol Chem. 1976;251:2550–2563. [PubMed] [Google Scholar]
- 21.Gibson QH, Swoboda BE, Massey V. Kinetics and mechanism of action of glucose oxidase. J Biol Chem. 1964;239:3927–3934. [PubMed] [Google Scholar]
- 22.Moran GR, Entsch B, Palfey BA, Ballou DP. Evidence for flavin movement in the function of p-hydroxybenzoate hydroxylase from studies of the mutant Arg220Lys. Biochemistry. 1996;35:9278–9285. doi: 10.1021/bi960360s. [DOI] [PubMed] [Google Scholar]
- 23.Fischer M, Bacher A. Biosynthesis of flavocoenzymes. Nat Prod Rep. 2005;22:324–350. doi: 10.1039/b210142b. [DOI] [PubMed] [Google Scholar]
- 24.Entsch B, Ballou DP. Flavin-mediated hydroxylation reactions. In: Dagley TP, editor. Wiley Encyclopedia of Chemical Biology. Vol. 2. Wiley & Sons; New Jersey: 2009. pp. 8–17. [Google Scholar]
- 25.Thatsaporn K, Sucharitakul J, Wongratana J, Suadee C, Chaiyen P. Cloning and expression of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii : evidence of the divergence of enzymes in the class of two-protein component aromatic hydroxylases. Biochim Biophys Acta. 2004;1680:60–66. doi: 10.1016/j.bbaexp.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 26.Schreuder HA, Prick PA, Wieringa RK, Vriend G, Wilson KS, Hol WG, Drenth J. Crystal structure of the p-hydroxybenzoate hydroxylase –substrate complex refined at 1.9 Å resolution. Analysis of the enzyme-substrate and enzyme-product complexes. J Mol Biol. 1989;208:679–696. doi: 10.1016/0022-2836(89)90158-7. [DOI] [PubMed] [Google Scholar]
- 27.Alfieri A, Malito E, Orru R, Fraaije MW, Mattevi A. Revealing the moonlighting role of NADP in the structure of a flavin-containing monooxygenase. Proc Natl Acad Sci USA. 2008;105:6572–6577. doi: 10.1073/pnas.0800859105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Entsch B, Ballou DP, Husain M, Massey V. Catalytic mechanism of p-hydroxybenzoate hydroxylase with p-mercaptobenzoate as substrate. J Biol Chem. 1976;251:7367–7379. [PubMed] [Google Scholar]