

Abstract
GluRδ2 is a member of the iGluR family, but despite a prominent role in cerebellar synaptic plasticity, this receptor does not appear to function as an ion channel. Endogenous ligands that modulate the activity of native GluRδ2 in the cerebellum have not been identified, but two candidate modulators are d-serine and extracellular calcium. Taking advantage of known crystal structures and spontaneously active GluRδ2 receptors containing the lurcher mutation (GluRδ2Lc), we investigated the mechanism by which calcium and d-serine regulate the activity of GluRδ2Lc. Our data suggest that calcium binding stabilizes the dimer interface formed between two agonist-binding domains and increases GluRδ2Lc currents. The data further suggest that d-serine binding induces rearrangements at the dimer interface to diminish GluRδ2Lc currents by a mechanism that resembles desensitization at AMPA and kainate receptors. Thus, we propose that calcium and d-serine binding have opposing effects on the stability of the dimer interface. Furthermore, the effects of calcium are observed at concentrations that are within the physiological range, suggesting that the ability of native GluRδ2 to respond to ligand binding may be modulated by extracellular calcium. These findings place GluRδ2 among AMPA and kainate receptors, where the dimer interface is not only a biologically important site for functional regulation, but also an important target for exogenous and endogenous ligands that modulate receptor function.
Keywords: electrophysiological recordings, delta2, structure–function relationship, pharmacology, Xenopus oocytes, disulfide bond
Introduction
The GluRδ2 receptors show weak sequence identity (21–25%) to AMPA, kainate, and NMDA receptors (Araki et al., 1993; Lomeli et al., 1993), which are ionotropic glutamate receptors (iGluRs) that mediate the majority of fast excitatory neurotransmission in the CNS (Dingledine et al., 1999). Although it is assumed that GluRδ2 forms ligand-gated ion channels, none of the iGluR agonists or any other known compound induce measurable current responses from wild-type GluRδ2 (GluRδ2Wt) (Schmid and Hollmann, 2008).
GluRδ2 is localized in dendritic spines of Purkinje cells and their postsynaptic localization suggests that the receptor is involved in synaptic transmission (Mayat et al., 1995; Landsend et al., 1997; Zhao et al., 1998). The majority of experimental data on the functional role of GluRδ2 in Purkinje cells originate from studies on genetically engineered GluRδ2−/− mice and GluRδ2 mutant mice, such as the hotfoot strains with naturally occurring mutations within the GluRδ2 gene (Vogel et al., 2007). These mice have several deficits in cerebellar development and function, including impaired Purkinje cell synaptogenesis and activity-dependent AMPA receptor endocytosis during cerebellar long-term depression (Kashiwabuchi et al., 1995; Matsuda and Yuzaki, 2002).
In the absence of known GluRδ2 agonists, the receptor function of GluRδ2 has been studied by evaluating the properties of GluRδ2 containing the lurcher mutation (A654T) (GluRδ2Lc). GluRδ2Lc receptors display apparent constitutive activity, because the lurcher mutation enables spontaneous activation of the ion channel in the absence of agonist. Spontaneously active GluRδ2Lc channels possess properties that are similar to those of other iGluRs, including Ca2+ permeability that is similar to that found in GluR2-lacking AMPA receptors (Zuo et al., 1997; Kohda et al., 2000; Wollmuth et al., 2000).
Recently, crystallographic structures of the GluRδ2 agonist-binding domain were obtained in the apo-form and in complex with d-serine bound within the agonist-binding pocket (Naur et al., 2007). Comparison of these structures shows that upon d-serine binding, the agonist-binding domain closes around the ligand, which is the initial conformational change that is believed to trigger activation of iGluRs (Armstrong and Gouaux, 2000). Interestingly, d-serine binding leads to a reduction in the spontaneously active GluRδ2Lc current, but does not activate currents at recombinant GluRδ2Wt (Naur et al., 2007). The GluRδ2 agonist-binding domain crystallized as a dimer in the absence of ligand, and this apo structure revealed a dimer interface with bound Ca2+. The information describing a Ca2+-binding site in the dimer interface provides a structural hypothesis with which to examine the previously described potentiation of spontaneously active GluRδ2Lc currents by extracellular Ca2+ (Wollmuth et al., 2000). In the present study, we investigated the functional consequences of d-serine binding to the agonist-binding pocket and Ca2+ binding to the dimer interface of GluRδ2Lc receptors. The data suggest that binding of the endogenous ligands Ca2+ and d-serine have opposing effects on the stability of the dimer interface. This new insight to the structure–function relationship of GluRδ2 provides a conceptual framework by which to interpret future studies on role GluRδ2 in cerebellar synaptic plasticity.
Materials and Methods
DNA constructs, mutagenesis, and cRNA synthesis.
Wild-type rat GluRδ2 (GenBank U08256) was subcloned into a pCI-IRES-bla vector containing a T7 site upstream from the 5′ untranslated region (Hansen et al., 2008). Mutations, including the lurcher mutation and the P528C and L789C mutations in the described previously GluRδ2-S1S2 expression construct (Naur et al., 2007), were introduced using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol and verified by DNA sequencing. For expression in Xenopus oocytes, DNA constructs were linearized by restriction enzymes to produce cRNAs using the mMessage mMachine kit (Ambion). The amino acids are numbered according to the full-length protein including the signal peptide.
Electrophysiological experiments.
Xenopus oocyte preparation and maintenance were performed essentially as described previously (Traynelis et al., 1998). Oocytes were injected with cRNA encoding GluRδ2 with or without mutations the day after surgical excision and collagenase treatment. Oocytes expressing GluRδ2Lc were maintained with 25 μm 1-naphthyl acetyl spermine (NASP) (Sigma-Aldrich) to prevent cytotoxicity. Two-electrode voltage-clamp current recordings were performed 24–72 h after injection at 23°C. Unless otherwise stated, the extracellular solution contained (in mm) 90 NaCl, 3 KCl, 0.5 BaCl2, 10 HEPES, pH 7.6. Solution exchange was computer controlled through an 8-modular valve positioner (Digital MVP Valve; Hamilton Company). Voltage and current electrodes were filled with 0.3 and 3.0 m KCl, respectively, and current responses were recorded at a holding potential of −40 mV. Data acquisition and voltage control were accomplished with a two-electrode voltage-clamp amplifier (OC-725; Warner Instruments). Choline chloride or N-methyl-d-glucamine chloride was used to maintain osmolarity and a constant concentration of Cl− in the extracellular solution during experiments where CaCl2 was applied to the oocytes. Reduction of disulfide bonds was accomplished by soaking the oocytes in 30 mm DTT (Sigma-Aldrich) in extracellular solution for 3 min at ambient temperatures before the recording essentially as described previously (Weston et al., 2006).
Data analysis.
Data were analyzed with GraphPad Prism 5.0 (GraphPad Software). Concentration–response data for individual oocytes were fitted to the Hill equation. Fitted EC50 values and Hill coefficients (nH) from individual oocytes were used to calculate the mean and SEM. For graphical presentation, data sets from individual oocytes were normalized to the maximum current response in the same recording. The averaged data points were then fitted to the Hill equation and plotted together with the resulting curve.
SDS-PAGE and Western blot analysis.
Membranes from oocytes expressing GluRδ2Lc receptors with and without double cysteine mutations were homogenized in buffer (30 μl/oocyte) containing 20 mm Tris, pH 8.0, 50 mm NaCl, 2 mm N-ethylmaleimide, 1% (w/v) β-dodecylmaltoside, 0.3% (w/v) CHAPS, and complete protease inhibitor mixture (Roche Diagnostics) as described previously (Weston et al., 2006). The homogenates were gently shook for 30 min at 4°C, centrifuged twice for 10 min at 13,000 × g (4°C), and the supernatant containing the solubilized membranes was removed each time. The samples for SDS-PAGE were prepared by mixing 5 μl solubilized membrane fraction with 5 μl 2× Laemmli sample buffer (62.5 mm Tris, pH 6.8, 25% glycerol, 2% SDS, and 0.01% bromophenol blue) with or without 200 mm DTT. The samples (10 μl) were incubated for 15 min at room temperature (23°C) and then subjected to SDS-PAGE using 10% Tris-HCl gels and blotted onto nitrocellulose membranes. Western blots were performed using goat anti-GluRδ2 primary antibody (1:2000 dilution; Santa Cruz Biotechnology; s.c.-26118) and horseradish peroxidase-conjugated donkey anti-goat secondary antibody (1:10,000 dilution; Santa Cruz Biotechnology; s.c.-2033). Blots were visualized using chemiluminescence and scanned.
Results
Extracellular calcium increases spontaneously active GluRδ2Lc currents
Modulation of GluRδ2Lc currents by extracellular Ca2+ has previously been described using heterologous expression in HEK-293 cells (Wollmuth et al., 2000). To investigate the effects of extracellular Ca2+ in the Xenopus oocyte expression system, we initially applied increasing concentrations of Ca2+ to oocytes expressing either GluRδ2Wt or GluRδ2Lc (Fig. 1A). Extracellular Ca2+ markedly potentiated spontaneously active currents (baseline currents measured in the absence of Ca2+) at oocytes expressing GluRδ2Lc (N = 23), whereas baseline currents at oocytes expressing GluRδ2Wt were insensitive to extracellular Ca2+ (N = 15). The potentiation of spontaneously active GluRδ2Lc currents was concentration dependent with an EC50 of 205 ± 7 μm (nH = 1.7) and a maximal fold potentiation of 4.1 ± 0.2 relative to baseline spontaneously active currents (Fig. 1B). These observations are consistent with previously published data from HEK-293 cells (Wollmuth et al., 2000).
Figure 1.

A, Representative two-electrode voltage-clamp recording of current responses to increasing concentrations of extracellular Ca2+ at GluRδ2Wt and GluRδ2Lc expressed in Xenopus oocytes. GluRδ2Wt expressed in oocytes did not show a current response different from uninjected oocytes; Ca2+ did not induce current in GluRδ2Wt (N = 15). In contrast, GluRδ2Lc showed substantial spontaneously active current, which is potentiated by extracellular Ca2+. B, Potentiation of GluRδ2Lc currents by extracellular Ca2+ was concentration dependent. The data were fitted to the Hill equation, which showed that Ca2+ potentiates GluRδ2Lc currents with an EC50 of 205 ± 7 μm (nH = 1.7) and a maximal fold potentiation of 4.1 ± 0.2 relative to baseline spontaneously active currents (N = 23). C, Representative two-electrode voltage-clamp recording of current responses at GluRδ2Wt and GluRδ2Lc, in which Ba2+ in the extracellular solution (0.5 mm, black bar) was exchanged with Mg2+, Zn2+, Mn2+, Sr2+, or Ca2+ (0.5 mm each, white bars). Application of the different divalent cations is indicated by the bars above the current trace. The effects of Zn2+, Mn2+, Sr2+, and Ca2+ on spontaneously active currents were specific for GluRδ2Lc, since no changes were observed at oocytes expressing GluRδ2Wt (N = 12). D, The effect of 0.5 mm of the indicated divalent cation plotted against the effective ionic radius of the cation (N = 11) shows a preferred optimal ionic radius for potentiation that matches that of Ca2+. The effective ionic radius of an element describes how closely the ion approaches another ion in a solid compound, and it roughly depends on the oxidation state and the number of nearest neighbor ions (coordination number). The values used here assume that the ions have an oxidation state of +2 and a coordination number of 6 (Shannon, 1976). The current responses are shown relative to the baseline spontaneously active currents measured in 0.5 mm Ba2+ (dotted line). Exchanging Ba2+ with Mg2+ did not markedly affect the spontaneously active currents, whereas Zn2+ and Sr2+ induced a modest increase. The largest potentiation of the spontaneously active current was observed for Mn2+ and Ca2+. The radius of Ba2+ is well beyond the optimal radius, consistent with the idea that Ba2+ has no effect on spontaneously active currents.
To investigate the ability of other divalent cations to modulate the spontaneously active GluRδ2Lc currents, we measured the effects of exchanging Ba2+ in the extracellular solution with Mg2+, Zn2+, Mn2+, Sr2+, or Ca2+ (0.5 mm each, N = 11) (Fig. 1C). Exchanging Ba2+ with Mg2+ only minimally affected the spontaneously active currents (1.06 ± 0.01 relative to baseline with Ba2+), whereas Zn2+ and Sr2+ induced a modest increase (1.33 ± 0.02 and 1.33 ± 0.05, respectively) (Fig. 1D). The largest potentiation of the spontaneously active current was observed for Mn2+ and Ca2+ (2.66 ± 0.13 and 3.91 ± 0.22, respectively). The effects of Zn2+, Mn2+, Sr2+, and Ca2+ on spontaneously active currents were specific for GluRδ2Lc, since no changes were observed at oocytes expressing GluRδ2Wt (N = 12). The effect of the divalent cations on spontaneously active currents plotted against their effective ionic radius (Shannon, 1976) indicated a preferred optimal ionic radius for potentiation that matches that of Ca2+ (Fig. 1D). In contrast, the radius of Ba2+ is well beyond the optimal radius, consistent with the idea that Ba2+ has no effect on spontaneously active currents. We also investigated whether extracellular Na+ is able to modulate GluRδ2Lc currents by measuring the current–voltage relationship at 0, 30, 60, and 90 mm extracellular Na+ (N = 5 for each curve; data not shown). However, the current–voltage curves in the analyzed voltage range (−60 to + 30 mV) obeyed the Goldman–Hodgkin–Katz flux equation, arguing that extracellular Na+ does not modulate GluRδ2.
d-Serine-induced inhibition of spontaneously active GluRδ2Lc currents is mediated by binding to the agonist-binding domain
Crystallographic structures of the GluRδ2 agonist-binding domain in complex with d-serine have demonstrated that d-serine can bind and induce closure of the agonist-binding domain around the ligand (Naur et al., 2007). Although a similar conformational change triggers activation of AMPA, kainate, and NMDA receptors, d-serine fails to activate currents at recombinant GluRδ2Wt (Naur et al., 2007). Nevertheless, application of d-serine to Xenopus oocytes expressing GluRδ2Lc reduces the spontaneously active current (Naur et al., 2007) (Fig. 2A). This observation is in contrast to findings at the AMPA receptor GluR1 containing the lurcher mutation (GluR1Lc), where increased activity is observed upon application of glutamate (Kohda et al., 2000; Klein and Howe, 2004). Furthermore, the spontaneous activity observed at GluR1Lc was attributed to activation by contaminating glutamate in the extracellular solution (Klein and Howe, 2004). It was further suggested that the lurcher mutation in GluR1 primarily increased the glutamate affinity and reduced desensitization (Klein and Howe, 2004).
Figure 2.

A, Representative two-electrode voltage-clamp recording of d-serine-mediated current responses at GluRδ2Lc expressed in Xenopus oocytes. Applications of saturating concentrations of d-serine (30 mm) and the channel blocker 1-naphthyl acetyl spermine (NASP; 100 μm) are indicated by the bars above the current trace. At the beginning of the recording, the baseline currents are predominantly mediated by the spontaneously active GluRδ2Lc. The spontaneously active GluRδ2Lc currents are inhibited upon application of a maximally effective concentration of d-serine. Application of the channel blocker NASP results in complete inhibition (100%) of the spontaneously active GluRδ2Lc current. B, Representative two-electrode voltage-clamp recording of current responses at GluRδ2Lc R530K. d-Serine at 30 mm minimally reduced spontaneously active currents at GluRδ2Lc R530K (5%). C, d-Serine at 30 mm reduced the spontaneously active current by 66 ± 2% at GluRδ2Lc (N = 7) relative to complete inhibition (100%) by NASP. The d-serine response (i.e., d-serine-induced inhibition) was decreased to 5 ± 2% for R530K (N = 9) and 5 ± 1% for D742A (N = 9) relative to complete inhibition (100%) by NASP. In the structure of the GluRδ2 agonist-binding domain in complex with d-serine, the guanidinium group of R530 interacts with the α-carboxyl group of d-serine, and the carboxylate group of D742 interacts with the α-amino group of d-serine.
If the spontaneous activity observed at GluRδ2Lc is mediated by contaminating d-serine or other unknown agonists in the extracellular solution, then mutations that will abolish agonist binding to GluRδ2Wt should also eliminate the spontaneous activity. In the structure of the GluRδ2 agonist-binding domain in complex with d-serine, the guanidinium group of R530 interacts with the α-carboxyl group of d-serine (Naur et al., 2007). In addition, the carboxylate group of D742 interacts with the α-amino group of d-serine. This binding mode of the amino acid moiety is essentially identical for all agonists so far crystallized in complex with the agonist-binding domains of iGluRs, including AMPA, NMDA, and kainate receptor subunits. Perturbations of this binding mode by mutating the arginine to lysine or the aspartate to alanine in other iGluRs results in an apparent loss or strong decrease in agonist potency (Uchino et al., 1992; Hirai et al., 1996; Williams et al., 1996; Hansen et al., 2005). We therefore mutated R530K and D742A in GluRδ2Lc and measured the ability of d-serine to reduce spontaneously active currents. d-Serine failed to reduce currents at GluRδ2Lc R530K (N = 9) and D742A (N = 9) and only a modest reduction could be observed at a d-serine concentration of 30 mm. However, these agonist-binding site mutants retain similar levels of spontaneously active currents as that of GluRδ2Lc (Fig. 2B). The response to 30 mm d-serine relative to complete inhibition (100%) of the spontaneously active GluRδ2Lc current by 1-naphthyl acetyl spermine (NASP) was 66 ± 2% at GluRδ2Lc (N = 7), whereas the relative d-serine-induced inhibition (hereafter referred to as d-serine response) was decreased to 5 ± 2% for R530K and 5 ± 1% for D742A (Fig. 2C). Since perturbations of the binding pocket by R530K and D742A mutations do not abolish spontaneous activity of GluRδ2Lc, we conclude that contaminating agonist in the extracellular solution is not responsible for the spontaneously active GluRδ2Lc currents. Furthermore, the effect of d-serine on GluRδ2Lc currents is not caused by competitive inhibition by d-serine or an unknown contaminating agonist. We conclude that the ability of d-serine to reduce currents at GluRδ2Lc is caused by binding in the agonist-binding pocket as shown in the crystallographic structure. Thus, the lurcher mutation appears to have different effects in GluR1Lc, where glutamate increases activity, and GluRδ2Lc, where d-serine reduces activity. This difference likely reflects variation in regions of the receptors that are important for gating and desensitization, such as the agonist-binding domain, the transmembrane domain, and the dimer interface.
Extracellular calcium modulates potency of d-serine at GluRδ2Lc
At AMPA and kainate receptors, the stability of the dimer interface is greatly reduced upon agonist binding and closure of the agonist-binding domain around the agonist (Sun et al., 2002; Jin et al., 2003, 2005; Horning and Mayer, 2004; Armstrong et al., 2006; Weston et al., 2006; Hansen et al., 2007). This instability leads to a rearrangement of the dimer interface, followed by repositioning of the transmembrane helices to a nonconducting conformation, where the ion channel is closed and the receptor is desensitized (Armstrong et al., 2006; Mayer, 2006; Hansen et al., 2007). The ability of d-serine to bind in the agonist-binding pocket and induce closure of the GluRδ2 agonist-binding domain suggests that d-serine-mediated reduction of GluRδ2Lc currents may reflect a process similar to desensitization of the other iGluRs that involves rearrangement of the dimer interface. It is conceivable that extracellular Ca2+ potentiates GluRδ2Lc currents by binding at the site between two agonist-binding domains and stabilizing the dimer interface, thereby attenuating structural rearrangements that lead to desensitization. Therefore, we predict that binding of extracellular Ca2+ to GluRδ2Lc should attenuate the ability of d-serine to induce desensitization.
To investigate this hypothesis, we determined the EC50 of d-serine in the absence of extracellular Ca2+ and in the presence of increasing concentrations of extracellular Ca2+ (Fig. 3A,B). Indeed, the EC50 of d-serine increases 8.8-fold when the concentration of extracellular Ca2+ is increased from 0 mm (N = 10) to 3 mm (N = 12) (Fig. 3A, Table 1). The ability of extracellular Ca2+ to reduce d-serine potency (i.e., increase EC50) was concentration dependent with an EC50 of 770 μm (nH = 1.3) (Fig. 3C). This is consistent with the idea that binding of Ca2+ between two agonist-binding domains stabilizes the dimer interface, making d-serine-mediated rearrangement more difficult (Fig. 4A,B). If the binding of extracellular Ca2+ to GluRδ2Lc attenuates the ability of d-serine to induce desensitization, then the binding of d-serine to GluRδ2Lc should also attenuate the ability of extracellular Ca2+ to bind the dimer interface. In keeping with this hypothesis, the EC50 of Ca2+ to potentiate the GluRδ2Lc currents increased from 205 ± 7 μm (N = 23) in the absence of d-serine to 676 ± 13 μm (N = 8) in the presence of 1 mm d-serine (Fig. 3D).
Figure 3.

A, Concentration–response data for d-serine at GluRδ2Lc in the presence of increasing concentrations of extracellular Ca2+. Each curve is generated using data from 8 to 12 oocytes. d-Serine potency decreases (i.e., EC50 increases) with increasing concentrations of extracellular Ca2+. B, Representative two-electrode voltage-clamp recording of d-serine responses at GluRδ2Lc in the absence (top trace) and in the presence of extracellular Ca2+ (bottom trace). Calibration: 100 nA (vertical), 60 s (horizontal). C, Reduction of d-serine potency by extracellular Ca2+ was concentration dependent with an EC50 of 770 μm (nH = 1.3). Each data point is generated using data from 8 to 12 oocytes. D, d-Serine also attenuates the ability of extracellular Ca2+ to potentiate GluRδ2Lc currents. The EC50 of Ca2+ to potentiate the GluRδ2Lc currents increased from 205 ± 7 μm (nH = 1.7, N = 23; open squares) in the absence of d-serine to 676 ± 13 μm (nH = 2.3, N = 8; closed squares) in the presence of 1 mm d-serine.
Table 1.
Summary of d-serine concentration–response data and potentiation by extracellular Ca2+ at GluRδ2Lc mutants
| GluRδ2Lc mutant |
d-Serine+0 mm Ca2+ |
d-Serine+3 mm Ca2+ |
Fold shift |
Ca2+ potentiation |
||||
|---|---|---|---|---|---|---|---|---|
| EC50 (μm) | nH | N | EC50 (μm) | nH | N | EC50 3 mm / 0 mm | (relative to baseline) | |
| GluRδ2Lc | 309 ± 8 | 1.0 | 12 | 2720 ± 90 | 2.1 | 10 | 8.8 | 3.8 ± 0.23 |
| R530K | n.d. | n.d. | n.d. | 3.3 ± 0.20 | ||||
| D742A | n.d. | n.d. | n.d. | 3.5 ± 0.11 | ||||
| E531A | 319 ± 10 | 1.1 | 8 | 705 ± 55 | 1.9 | 8 | 2.2 | 1.6 ± 0.08 |
| D535A | 1600 ± 80 | 2.3 | 10 | 1580 ± 240 | 2.2 | 8 | 1.0 | 1.0 ± 0.01 |
| D782A | 278 ± 11 | 1.0 | 12 | 304 ± 18 | 1.1 | 8 | 1.1 | 0.8 ± 0.02 |
| E531A+D535A | 10,200 ± 1000 | 2.2 | 10 | 8770 ± 630 | 2.2 | 12 | 0.9 | 1.0 ± 0.04 |
| E531A+D782A | 8240 ± 180 | 2.3 | 5 | 7540 ± 140 | 2.4 | 6 | 0.9 | 0.9 ± 0.04 |
| D535A+D782A | 345 ± 31 | 1.0 | 6 | 351 ± 6 | 1.1 | 5 | 1.0 | 0.6 ± 0.01 |
| E531C+D782C | 1170 ± 180 | 0.8 | 6 | 897 ± 212 | 0.8 | 6 | 0.8 | 0.9 ± 0.01 |
| D535C+D782C | 472 ± 99 | 0.9 | 6 | 514 ± 90 | 1.0 | 4 | 1.1 | 0.9 ± 0.01 |
| P528C+L789C | 898 ± 192 | 1.1 | 7 | 1070 ± 200 | 0.8 | 4 | 1.2 | 0.9 ± 0.01 |
d-Serine EC50 ± SEM in the presence and absence of extracellular Ca2+ were used to calculate the fold shift in d-serine EC50. N is the number of oocytes used to generate the data; nH is the Hill slope; and n.d. indicates not determined. Ca2+ potentiation is the fold potentiation ± SEM of baseline spontaneously active GluRδ2Lc currents by the application of 3 mm Ca2+.
Figure 4.

A, Side and top view of the dimer formed by two agonist-binding domains of GluRδ2 in the absence of d-serine (apo-form) (PDB code 2V3T) (Naur et al., 2007). Soluble proteins containing the isolated agonist-binding domain of iGluRs have been constructed by deleting the N-terminal domain and replacing the transmembrane domains with a short linker. In these structures, the agonist-binding domain exists in a bilobed clamshell-like arrangement with the agonist-binding pocket located deep within the cleft between two lobes (D1 and D2). The two GluRδ2 subunits (colored yellow and orange) form a dimer that is symmetrical around a pseudo twofold axis. The dimer binds two calcium ions (shown as green spheres) at the top of the dimer interface. Residues at the dimer interface that were mutated in this study are shown as space-filled residues. B, Magnified top view of the solvent-accessible Ca2+-binding site with the backbone of one agonist-binding domain (protomer) shown in yellow and the other shown in orange. Dashed lines indicate potential coordination of Ca2+ by residues in the GluRδ2 dimer interface. The Ca2+-binding site is ideally situated to modulate the stability of the dimer as it is formed by acidic residues from both subunits. More specifically, Ca2+ interacts with E531 (backbone carbonyl and sidechain carboxyl), V534 (backbone carbonyl), and D535 (sidechain carboxyl) from one subunit and D782 (sidechain carboxyl) from the other subunit. The “top” of the Ca2+-binding site (i.e., the opposite side of Ca2+ relative to the sidechain carboxyl of E531) is exposed to the solvent. Residues that were mutated in this study and V534 are shown as gray sticks. The following two pairs of solvent-accessible cysteine mutations were made in GluRδ2Lc: E531C+D782C and D535C+D782C. Residues P528 and L789, which do not participate directly in Ca2+ binding, were also mutated to cysteines (Fig. 5A). C–F, Concentration–response data for d-serine at GluRδ2Lc (C) and the GluRδ2Lc mutations D782A (D), D535A (E), and E531A (F) in the absence (circles) and presence (triangles) of extracellular Ca2+ (3 mm). EC50 values are listed in Table 1. d-Serine EC50 (i.e., potency) at GluRδ2Lc is highly sensitive to extracellular Ca2+, whereas d-serine EC50 at GluRδ2Lc D535A and D782A is unaffected by extracellular Ca2+. The EC50 of d-serine at GluRδ2Lc E531A is less sensitive to extracellular Ca2+. G, Summary of fold shifts in d-serine EC50 in the presence of extracellular Ca2+ (3 mm) relative to d-serine EC50 in the absence of extracellular Ca2+. H, Summary of fold shifts (i.e., Ca2+ potentiation) of spontaneously active currents in the presence of extracellular Ca2+ (3 mm) relative to baseline spontaneously active currents in the absence of extracellular Ca2+. Numerical values are listed in Table 1. Black bars indicate fold shift at nonmutated GluRδ2Lc, white bars indicate GluRδ2Lc with mutations in the d-serine-binding site, striped bars indicate GluRδ2Lc with mutations in the Ca2+-binding site, and n.d. indicates not determined. Fold shifts of d-serine EC50 were not determined at R530K and D742A, since d-serine EC50 could not be determined at these mutants.
The effects of extracellular calcium are caused by binding to the dimer interface
We predict that Ca2+ binding to the site identified at the dimer interface by crystallography mediates the observed effects. To rule out the possibility that Ca2+ potentiation is mediated by binding to sites of GluRδ2 that are different from the binding sites at the dimer interface, we mutated residues E531, D535, and D782 that form the Ca2+ binding sites at the dimer interface (Fig. 4). Mutating D782A in GluRδ2Lc completely abolished both the Ca2+-mediated reduction of d-serine potency and the potentiation of spontaneously active currents. Similar results were observed for GluRδ2Lc D535A; GluRδ2Lc E531A remained partially sensitive to extracellular Ca2+. The double mutants GluRδ2Lc E531A+D535A and GluRδ2Lc E531A+D782A were completely insensitive to extracellular Ca2+. Spontaneously active currents at GluRδ2Lc with mutations in the d-serine-binding site (R530K and D742A) were still potentiated by extracellular Ca2+ (Fig. 4H, Table 1). Based on these results, we conclude that the effects of extracellular Ca2+ on d-serine potency and potentiation of spontaneously active currents are caused by Ca2+ binding within the dimer interface.
Cross-linking at the dimer interface by engineered disulfide bonds attenuates d-serine responses and eliminates modulation by calcium
Our data suggest that binding of Ca2+ to the site between two agonist-binding domains attenuates d-serine responses by stabilizing the dimer interface. If d-serine binding to GluRδ2Lc causes desensitization of the receptor by inducing rearrangement of the dimer interface, then stabilizing the dimer interface by other means should attenuate d-serine-mediated reduction of GluRδ2Lc currents. To test this hypothesis, we evaluated whether cross-linking at the dimer interface by engineered disulfide bonds could mimic the effects of extracellular Ca2+.
Initially, cysteine residues were introduced into the dimer interface outside the Ca2+-binding site by site-directed mutagenesis to generate GluRδ2Lc P528C+L789C. When these two cysteine mutations are introduced at the corresponding residues in the AMPA receptor subunit GluR2 and the kainate receptor subunits GluR5, GluR6, and GluR7, the resulting receptors have a cross-linked dimer interface and consequently, these receptors are nondesensitizing (Weston et al., 2006). GluRδ2Lc P528C+L789C was insensitive to extracellular Ca2+ (Table 1), suggesting that the dimer interface is already stabilized by engineered disulfide bonds. Furthermore, the maximal d-serine response at GluRδ2Lc P528C+L789C was small relative to complete inhibition by NASP (9 ± 1%, N = 6). However, the maximal d-serine response did not noticeably increase when the oocytes expressing GluRδ2Lc P528C+L789C were treated with 30 mm DTT for 3 min (12 ± 1%, N = 12). Similarly, the sensitivity to extracellular Ca2+ was not restored when oocytes expressing GluRδ2Lc P528C+L789C were treated with DTT (N = 6; data not shown). These results could suggest that the disulfide bond is inaccessible to DTT or a disulfide bond between P528C and L789C is not formed.
To evaluate whether a disulfide bond is formed between P528C and L789C, we analyzed GluRδ2Lc and GluRδ2Lc P528C+L789C using SDS-PAGE in the presence and absence of DTT in the gel-loading buffer (Fig. 5A). The majority of GluRδ2Lc receptors clearly migrate as monomers under both nonreduced and reduced conditions. In contrast, GluRδ2Lc P528C+L789C receptors migrate as dimers under nonreduced conditions and as monomers under reduced conditions, demonstrating that these receptors are cross-linked by disulfide bonds between P528C and L789C residues. The formation of disulfide bond between P528C and L789C suggests that stabilization of the dimer interface can decrease the effects of d-serine and Ca2+. We speculate that GluRδ2Lc P528C+L789C is resistant to DTT treatment because the disulfide bond is inaccessible to DTT.
Figure 5.
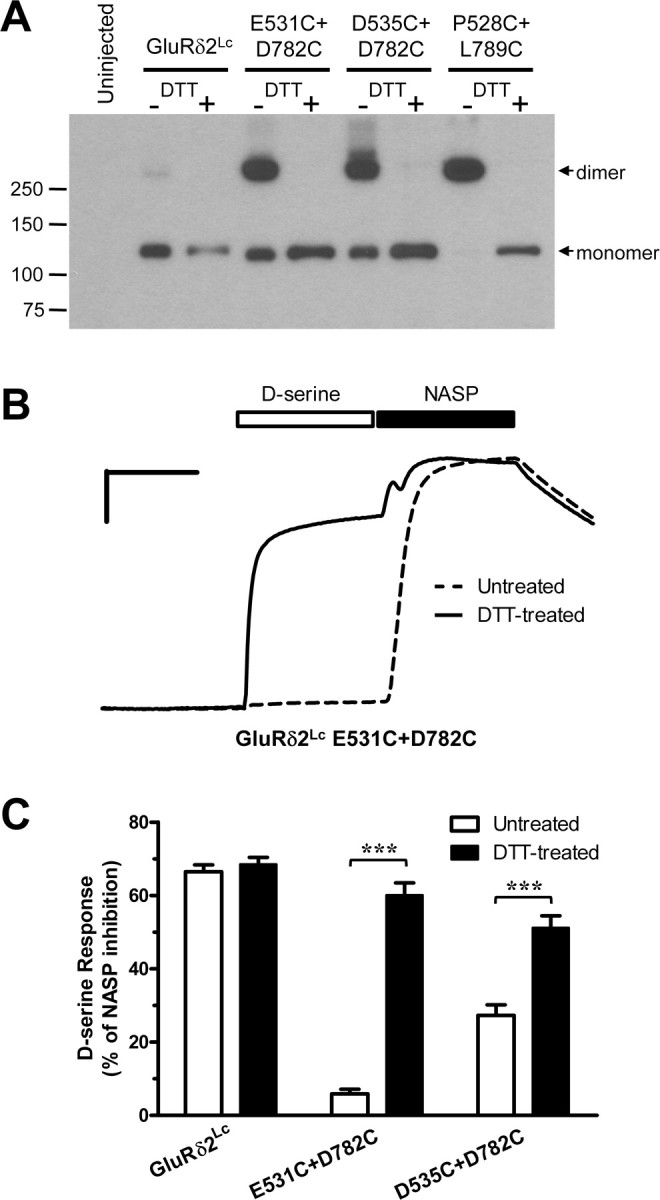
A, Western blot of GluRδ2Lc and double cysteine mutations (E531C+D782C, D535C+D782C, P528C+L789C) expressed in Xenopus oocytes as well as uninjected oocytes. Representative of three experiments. SDS-PAGE was performed without DTT (−DTT) or with 100 mm DTT (+DTT) in the samples. Positions of molecular weight markers (in kDa) are showed to the left. The equivalent of 1/6 an oocyte was loaded per lane. GluRδ2Lc mostly migrates as a monomer, whereas the double cysteine mutants mainly migrate as dimers. These data demonstrate that the double cysteine mutants are cross-linked by disulfide bonds at the dimer interface. B, Representative two-electrode voltage-clamp recording of current responses at GluRδ2Lc E531C+D782C without (dashed line) and with (solid line) DTT treatment. The traces are normalized to better allow comparison. The engineered disulfide bond attenuates the ability of GluRδ2Lc to respond to d-serine binding as the dimer interface is unable to rearrange. However, when the engineered disulfide bond in E531C+D782C is reduced by DTT, the dimer interface is then capable of rearranging and the receptor is able to respond to d-serine binding. Calibration: 20% of NASP response (vertical), 60 s (horizontal). C, Summary of the response to 30 mm d-serine relative to complete inhibition (100%) of the spontaneously active current by 100 μm NASP (d-serine response) before (white) and after (black) treatment with DTT. Treatment of oocytes expressing GluRδ2Lc E531C+D782C and GluRδ2Lc D535C+D782C with DTT significantly increased d-serine responses, whereas GluRδ2Lc was insensitive to DTT treatment (***p < 0.001, t test).
To overcome this obstacle, we took advantage of the structure of the GluRδ2 agonist-binding domain (Naur et al., 2007) to design two additional double cysteine mutants, GluRδ2Lc E531C+D782C and GluRδ2Lc D535C+D782C. In these mutants, the cysteine residues are introduced in the Ca2+-binding site at the surface of the dimer interface, which should be readily accessible to DTT, thereby allowing evaluation of d-serine responses in the presence and absence of the disulfide bonds. In other words, the Ca2+-binding site of these double cysteine mutants was disrupted and replaced by disulfide bonds that should potentially cross-link and stabilize the dimer interface. GluRδ2Lc E531C+D782C and GluRδ2Lc D535C+D782C receptors were also evaluated using SDS-PAGE under nonreduced and reduced conditions, demonstrating that both of these double cysteine mutants are cross-linked by disulfide bonds (Fig. 5A).
As expected, GluRδ2Lc E531C+D782C and GluRδ2Lc D535C+D782C did not show any sensitivity to extracellular Ca2+, meaning that these double cysteine mutants did not display any Ca2+-mediated reduction of d-serine potency or potentiation of spontaneously active currents (Table 1). The maximal d-serine response (i.e., d-serine-induced inhibition) relative to complete inhibition of the spontaneously active current by NASP was reduced from 66 ± 2% at GluRδ2Lc control (N = 7) to 6 ± 1% for E531C+D782C (N = 6) and 27 ± 3% for D535C+D782C (N = 8) (Fig. 5B,C). However, when oocytes expressing GluRδ2Lc E531C+D782C and GluRδ2Lc D535C+D782C were treated with 30 mm DTT for 3 min before the recordings, the d-serine response increased to levels similar to GluRδ2Lc lacking engineered disulfide bonds. d-Serine responses were 60 ± 3% for DTT-treated E531C+D782C (N = 9) and 51 ± 3% for DTT-treated D535C+D782C (N = 8), whereas d-serine response at DTT-treated GluRδ2Lc was 69 ± 2% (N = 8). The effects of DTT at GluRδ2Lc E531C+D782C and GluRδ2Lc D535C+D782C could be reversed by incubation with the oxidizing agent DTNB (500 μm for 3 min; N = 8 and N = 8, respectively; data not shown). Treatment of GluRδ2Lc E531C+D782C and GluRδ2Lc D535C+D782C with DTT did not restore sensitivity to extracellular Ca2+ (N = 6 each; data not shown), since their Ca2+-binding sites were disrupted. Similarly, DTT treatment of GluRδ2Lc did not change Ca2+ sensitivity (N = 6), consistent with the idea that GluRδ2Lc does not contain any solvent accessible disulfide bonds that alter function.
Thus, engineered disulfide bonds that cross-link the dimer interface are formed between E531C and D782C as well as D535C and D782C. We conclude that these engineered disulfide bonds markedly attenuate the ability of GluRδ2Lc to respond to d-serine binding because the dimer interface is unable to rearrange, thereby mimicking the effects of extracellular Ca2+. However, when the engineered disulfide bonds are reduced by DTT, the dimer interface is then capable of rearranging and the receptor is able to respond to d-serine binding. In conclusion, the engineered disulfide bonds are able to mimic the effects of extracellular Ca2+ on d-serine responses, thereby demonstrating that the primary effect of Ca2+ binding to GluRδ2Lc is to stabilize the dimer interface and prevent a rearrangement that would otherwise lead to desensitization of the receptor.
Cross-linking of the dimer interface and reducing conformational constraints in the agonist-binding domain convert d-serine from an inhibitor to an activator
A wealth of previously published data for AMPA receptors suggest that agonist binding creates strain on the dimer interface that can be relieved either by rearrangement of the dimer interface to produce desensitized receptors or by rearrangement of the transmembrane helices to open the channel (Sun et al., 2002; Jin et al., 2003, 2005; Horning and Mayer, 2004; Armstrong et al., 2006; Weston et al., 2006; Hansen et al., 2007). Our data with GluRδ2Lc are consistent with this idea in that cross-linking of the dimer interface can reduce the ability of d-serine to trigger dimer interface rearrangement and closure of spontaneously active GluRδ2Lc channels. To examine whether we can obtain any evidence of d-serine binding enhancing channel activity, we hypothesized that other constraints within GluRδ2 might prevent the d-serine bound receptor from rearranging to the open state. One candidate for such constraints is a disulfide bond formed by a pair of cysteine residues located in D2 near the junction of the agonist-binding domain to the transmembrane helices. This pair of cysteines (C756 and C811 in GluRδ2) is conserved across the entire iGluR family and has previously been shown to influence glutamate receptor function. In NMDA receptors, this disulfide bond within the NR1 subunit can be readily reduced by DTT, resulting in pronounced potentiation of receptor activity (Sullivan et al., 1994; Choi et al., 2001). In AMPA receptors, however, this disulfide bond is inaccessible to DTT, but mutating the cysteines increases agonist affinity and potency (Watase et al., 1997; Abele et al., 1998). Because these cysteine residues were cross-linked in the crystal structure of the GluRδ2 agonist-binding domain (supplemental figure, available at www.jneurosci.org as supplemental material), we introduced a serine at position 811 (C811S), which should prevent formation of a disulfide bond and thereby allow larger conformational changes during agonist binding (Oh et al., 1993; Kaye et al., 2007). Neither the C811S mutation nor the triple P528C+L789C+C811S mutation resulted in measurable responses to d-serine in GluRδ2Wt (data not shown, N = 10 and N = 10, respectively). Figure 6 summarizes the effect of C811S introduced into GluRδ2Lc with and without cross-linked dimer interface (P528C+L789C). As we predicted, breakage of the disulfide bond (between C756 and C811) altered the response to d-serine compared with GluRδ2Lc. Furthermore, when agonist-induced dimer interface rearrangement is prevented by cross-linking the dimer interface, breakage of the C756–C811 disulfide bond through mutagenesis (C811S) converted the effects of d-serine from inhibition to potentiation. This result, predicted by our working hypothesis, confirms that conformational changes introduced by d-serine binding to GluRδ2Lc are capable of initiating a set of intraprotein actions (dimer interface rearrangement, pore opening) that can occur in all functional glutamate receptors.
Figure 6.

Representative two-electrode voltage-clamp recording of current responses at GluRδ2Lc mutants. Top left, Responses to d-serine and NASP at GluRδ2Lc without cross-linking of the dimer interface and with conformational constraints created by the naturally occurring C756–C811 disulfide bond (d-serine response was 66 ± 2%, N = 7 relative to NASP response; also shown in Fig. 3). Top right, Responses at GluRδ2Lc P528C+L789C with cross-linked dimer interface and with intact C756–C811 disulfide bond (relative d-serine response 9 ± 1%, N = 6). Bottom left, Responses at GluRδ2Lc C811S without cross-linked dimer interface and without conformational constraints from a C756–C811 disulfide bond (relative d-serine response −17 ± 13%, N = 6). Bottom right, Responses at GluRδ2Lc P528C+L789C+C811S with cross-linked dimer interface and without conformational constraints from a C756–C811 disulfide bond (relative d-serine response −150 ± 70%, N = 7). Mutating C811S converts d-serine from an inhibitor to an activator of spontaneously active GluRδ2Lc currents, consistent with the idea that breakage of the C756–C811 disulfide bond will allow larger conformational changes during agonist binding. Activation of spontaneously active currents at GluRδ2Lc C811S by d-serine is further promoted by cross-linking of the dimer interface using P528C+L789C mutations.
Discussion
In the present study, we define the binding sites for divalent cations at GluRδ2 that mediate potentiation of spontaneously active currents by extracellular Ca2+. Furthermore, we demonstrate that the binding of extracellular Ca2+ in the physiologically relevant concentration range modulates the ability of GluRδ2Lc to respond to d-serine binding in the agonist-binding pocket and vice versa. Experiments using cross-linking of the dimer interface indicate that d-serine binding induces a rearrangement of the dimer interface, whereas Ca2+ binding stabilizes this dimer interface. Thus, d-serine and extracellular Ca2+ exert opposing actions on the dimer interface. These results suggest that the dimer interface controls the function of GluRδ2 and that this feature is shared across the iGluR family (Sun et al., 2002; Jin et al., 2005; Weston et al., 2006; Plested and Mayer, 2007; Plested et al., 2008) (Fig. 7A).
Figure 7.

A, The results from the present study suggest that the dimer interface controls the function of GluRδ2, and stability of this interface is influenced by Ca2+ (green spheres). This feature is shared across the iGluR family. Desensitization of AMPA receptors is inhibited by binding of positive modulators at the dimer interface, such as cyclothiazide binding to GluR2 (shown as spacefill, PDB code 1LBC). Desensitization of kainate receptors is regulated by binding of anions and cations at the dimer interface, such as two sodium (purple spheres) and one chloride (green) binding to GluR5 (PDB code 3C32). Mutations that stabilize the dimer interface attenuate desensitization of both AMPA and kainate receptors. B, Hypothetical model that illustrates the opposing effects of d-serine and Ca2+ on GluRδ2Lc (lurcher mutation indicated by red stars). d-Serine binding in the agonist-binding pocket between D1 and D2 and subsequent domain closure can result in a rearrangement of the dimer interface formed by two agonist-binding domains. d-Serine binding to spontaneously active GluRδ2Lc results in a rearrangement of the dimer interface. This rearrangement at the dimer interface results in desensitization by repositioning the transmembrane helices to a nonconducting conformation, where the ion channel is closed. However, binding of Ca2+ attenuates d-serine-induced desensitization by stabilizing the dimer interface in a conformation that permits opening of the ion channel. The Ca2+-stabilized conformation of the dimer interface permits increased opening of the ion channel and thus results in potentiation of the spontaneously active current.
d-Serine binding and calcium binding have opposing effects on dimer interface stability
A hypothetical model that illustrates the effects of d-serine and Ca2+ on GluRδ2Lc is proposed in Figure 7B. d-Serine binding in the agonist-binding pocket induces domain closure, which could potentially result in either rearrangement of the dimer interface or reorientation of the transmembrane helices and opening of the ion channel. The reason that d-serine responses at GluRδ2Wt cannot be detected using two-electrode voltage-clamp recordings could be the relatively slow temporal resolution of this technique. However, activation and rapid desensitization of GluRδ2Wt expressed in HEK-293 cells were not observed in response to fast application (<1 ms) of d-serine in the absence (0 mm) or presence of extracellular Ca2+ (10 mm) (Naur et al., 2007). Thus, we know that binding of d-serine to GluRδ2Wt does not result in opening of the ion channel and that binding of d-serine to spontaneously active GluRδ2Lc does not increase the opening of the ion channel. We predict that d-serine binding to GluRδ2Lc, and likely also to GluRδ2Wt, results in a rearrangement of the dimer interface to produce desensitization by repositioning the transmembrane helices to a nonconducting conformation. However, binding of Ca2+ attenuates d-serine-induced desensitization by stabilizing the dimer interface in a conformation that permits opening of the ion channel. At GluRδ2Lc, the Ca2+-stabilized conformation of the dimer interface results in potentiation of the spontaneously active current relative to the conformation without bound Ca2+. Thus, d-serine and Ca2+ act at different sites of GluRδ2 with opposing actions on a common downstream substrate: the dimer interface.
Desensitization and dimer interface stability in AMPA and kainate receptors
This model for the opposing actions of d-serine and Ca2+ on GluRδ2 is similar to the proposed actions of agonists and ions on the agonist-binding domains of kainate receptors (Wong et al., 2006, 2007; Plested and Mayer, 2007; Plested et al., 2008). Na+ and Cl− bind at dimer interface of GluR5 with a 2:1 stoichiometry and their binding is required to maintain kainate receptors in the active conformation. Specifically, these ions stabilize the dimer interface and thus attenuate agonist-induced desensitization. Accordingly, the ion sensitivity of kainate receptors is lost when desensitization is prevented by cross-linking the dimer interface at residues corresponding to P528 and L789 in GluRδ2 (Plested and Mayer, 2007; Plested et al., 2008). The residues that form the Ca2+-binding site of GluRδ2 are conserved in GluR5, except for V534 that is an isoleucine in GluR5. However, the spatial orientation of these residues is different in GluR5 and GluRδ2. In GluR5, the residues of one agonist-binding domain form a Na+-binding site that bridges the dimer interface through solvent-mediated hydrogen bonds to residues of the other agonist-binding domain, including D761 that corresponds to D782 in GluRδ2. This means that in kainate receptors, all of the contacts with Na+ are made by one side of the dimer interface (Plested et al., 2008). The Ca2+-binding sites in GluRδ2 are also largely, but not entirely, formed by a single subunit: the other subunit only contributes with the sidechain of D782 (Fig. 4B). Moreover, the crystallographic structure of the GluRδ2 agonist-binding domain in complex with d-serine is a monomer and the sites contain Na+ instead of Ca2+ (Naur et al., 2007). This is most likely due to the loss of D782 that stabilizes the dimer interface, thereby leaving sites that resemble the Na+-binding sites of kainate receptors.
In the AMPA receptor subunit GluR2, the residues corresponding to those that participate in coordination of Ca2+ in GluRδ2 are also conserved with the exception that D782 in GluRδ2 is replaced by an asparagine in GluR2 (Fig. 5B). Consequently, divalent and monovalent cations are unable to bridge the dimer interface of AMPA receptors directly or through solvent mediated hydrogen bonds, which likely explains why the function of these receptors is unaffected by cations (Bowie, 2002; Plested et al., 2008). Instead, E486 of GluR2, which corresponds to E531 in GluRδ2, participates in an intermolecular salt-bridge with K493 (GluR2) of the opposite agonist-binding domain that stabilizes the dimer interface (Armstrong and Gouaux, 2000). Disruption of this salt-bridge accelerates desensitization of AMPA receptors (Horning and Mayer, 2004). E509 in GluR5, which corresponds to E531 in GluRδ2, makes up part of the Na+-binding site and, similar to GluR2, participates in a salt-bridge across the dimer interface with K516 of the opposite agonist-binding domain (Plested et al., 2008). K516 in GluR5 also interacts with anions bound at the dimer interface, thereby enabling coupling between cation and anion-binding sites at the dimer interface (Plested and Mayer, 2007; Plested et al., 2008). The lysine of GluR2 (K493) and GluR5 (K516) is replaced by a threonine in GluRδ2 (T538), which thereby has lost a key interaction that stabilizes the dimer interface.
Physiological relevance of modulation of GluRδ2 by extracellular calcium
The ability of extracellular Ca2+ to modulate d-serine potency at GluRδ2Lc has an EC50 of ∼0.8 mm. Extracellular Ca2+ surrounding the cerebellar synapses is depleted during high-frequency stimulations (Nicholson et al., 1977, 1978), and simulations have shown that the concentration of extracellular Ca2+ in the cleft can decrease by 50% or more (Vassilev et al., 1997; Rusakov, 2001). At parallel fiber to Purkinje cells synapses, where GluRδ2 is highly expressed (Mayat et al., 1995; Landsend et al., 1997), the concentration of extrasynaptic Ca2+ can decrease from a baseline of ∼1.2 to ∼0.8 mm during repetitive stimulation and to ∼0.12 mm during spreading depression (Nicholson et al., 1977, 1978). The Ca2+ EC50 measured in the present study (∼0.8 mm) is therefore well within the physiological range of extracellular Ca2+ concentrations. Although the potencies and efficacies by which d-serine and extracellular Ca2+ modulate GluRδ2Lc are likely different at GluRδ2Wt due to their different unliganded resting states, we predict that native GluRδ2 receptors may be able to sense depletion of extracellular Ca2+ and that this could modulate the ability of native GluRδ2 to respond to ligand binding.
The pharmacology and structure–function relationship of GluRδ2 has remained elusive for the past 15 years. Here, we have shown that GluRδ2Lc can undergo conformational changes similar to AMPA and kainate receptors, including agonist-induced closure of the agonist-binding domain and structural rearrangement of the dimer interface, which are the hallmarks of channel activation and desensitization at iGluRs, respectively. In addition, introduction of constraints at the interface and removal of constraints within the agonist-binding domain allows d-serine binding to trigger opening of GluRδ2Lc. Thus, not only does GluRδ2 share sequence similarity with iGluRs, but GluRδ2Lc also shows glutamate receptor-like properties with respect to conformational changes and the importance of the dimer interface, placing the structure–function relationship of GluRδ2 among those of the iGluRs. Nonetheless, recent transgenic experiments showed that insertion of a mutant GluRδ2 into GluRδ2−/− mice can rescue these mice from neurological deficits even when the inserted GluRδ2 has a mutation in the ion channel pore that either disrupts Ca2+ permeability (Kakegawa et al., 2007) or abolishes current flow through the ion channel (Kakegawa and Yuzaki, 2005). These recent data suggest that GluRδ2 does not influence cerebellar function through actions as a ligand-gated ion channel. Our data are compatible with the possibility that GluRδ2 could function through actions independent of current flow through the ion channel, since conformational changes in the agonist-binding domain and the dimer interface may also impact the positions of transmembrane helices and intracellular domains independent of ion flux through the channel pore. Such transmembrane intraprotein rearrangements could affect interactions with intracellular proteins and mediate intracellular signals (Rodríguez-Moreno and Lerma, 1998; Vissel et al., 2001; Nong et al., 2003). Future studies that elucidate the interplay between ligand and Ca2+ binding to native receptors will provide valuable insight to the role GluRδ2 in cerebellar synaptic plasticity.
Footnotes
This work was supported by the National Institutes of Health (Grants NS36654 and NS062204 to S.F.T.), the Michael J. Fox Foundation (S.F.T.), the National Alliance for Research on Schizophrenia and Depression (S.F.T.), the Danish Medical Research Council (P.N., M.G., J.S.K.), the Alfred Benzon Foundation (K.B.H., A.S.K), the Lundbeck Foundation (K.B.H.), Fonden af 17-12-1981 (K.B.H.), and the Villum Kann Rasmussen Foundation (K.B.H.). We thank Dr. Jim Boulter for sharing the GluRδ2 cDNA. We also thank Dr. Lonnie P. Wollmuth and Dr. Shashank M. Dravid for helpful discussions and commentary on this project.
References
- Abele R, Lampinen M, Keinänen K, Madden DR. Disulfide bonding and cysteine accessibility in the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit GluRD. Implications for redox modulation of glutamate receptors. J Biol Chem. 1998;273:25132–25138. doi: 10.1074/jbc.273.39.25132. [DOI] [PubMed] [Google Scholar]
- Araki K, Meguro H, Kushiya E, Takayama C, Inoue Y, Mishina M. Selective expression of the glutamate receptor channel delta 2 subunit in cerebellar Purkinje cells. Biochem Biophys Res Commun. 1993;197:1267–1276. doi: 10.1006/bbrc.1993.2614. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Jasti J, Beich-Frandsen M, Gouaux E. Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell. 2006;127:85–97. doi: 10.1016/j.cell.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Bowie D. External anions and cations distinguish between AMPA and kainate receptor gating mechanisms. J Physiol. 2002;539:725–733. doi: 10.1113/jphysiol.2001.013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chen HV, Lipton SA. Three pairs of cysteine residues mediate both redox and Zn2+ modulation of the NMDA receptor. J Neurosci. 2001;21:392–400. doi: 10.1523/JNEUROSCI.21-02-00392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Hansen KB, Clausen RP, Bjerrum EJ, Bechmann C, Greenwood JR, Christensen C, Kristensen JL, Egebjerg J, Bräuner-Osborne H. Tweaking agonist efficacy at N-methyl-d-aspartate receptors by site-directed mutagenesis. Mol Pharmacol. 2005;68:1510–1523. doi: 10.1124/mol.105.014795. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Yuan H, Traynelis SF. Structural aspects of AMPA receptor activation, desensitization and deactivation. Curr Opin Neurobiol. 2007;17:281–288. doi: 10.1016/j.conb.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Hansen KB, Bräuner-Osborne H, Egebjerg J. Pharmacological characterization of ligands at recombinant NMDA receptor subtypes by electrophysiological recordings and intracellular calcium measurements. Comb Chem High Throughput Screen. 2008;11:304–315. doi: 10.2174/138620708784246040. [DOI] [PubMed] [Google Scholar]
- Hirai H, Kirsch J, Laube B, Betz H, Kuhse J. The glycine binding site of the N-methyl-d-aspartate receptor subunit NR1: identification of novel determinants of co-agonist potentiation in the extracellular M3–M4 loop region. Proc Natl Acad Sci U S A. 1996;93:6031–6036. doi: 10.1073/pnas.93.12.6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horning MS, Mayer ML. Regulation of AMPA receptor gating by ligand binding core dimers. Neuron. 2004;41:379–388. doi: 10.1016/s0896-6273(04)00018-2. [DOI] [PubMed] [Google Scholar]
- Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E. Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci. 2003;6:803–810. doi: 10.1038/nn1091. [DOI] [PubMed] [Google Scholar]
- Jin R, Clark S, Weeks AM, Dudman JT, Gouaux E, Partin KM. Mechanism of positive allosteric modulators acting on AMPA receptors. J Neurosci. 2005;25:9027–9036. doi: 10.1523/JNEUROSCI.2567-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Yuzaki M. A mechanism underlying AMPA receptor trafficking during cerebellar long-term potentiation. Proc Natl Acad Sci U S A. 2005;102:17846–17851. doi: 10.1073/pnas.0508910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Miyazaki T, Hirai H, Motohashi J, Mishina M, Watanabe M, Yuzaki M. Ca2+ permeability of the channel pore is not essential for the delta2 glutamate receptor to regulate synaptic plasticity and motor coordination. J Physiol. 2007;579:729–735. doi: 10.1113/jphysiol.2006.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluR delta 2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- Kaye SL, Sansom MS, Biggin PC. In silico mutation of cysteine residues in the ligand-binding domain of an N-methyl-d-aspartate receptor. Biochemistry. 2007;46:2136–2145. doi: 10.1021/bi061462d. [DOI] [PubMed] [Google Scholar]
- Klein RM, Howe JR. Effects of the lurcher mutation on GluR1 desensitization and activation kinetics. J Neurosci. 2004;24:4941–4951. doi: 10.1523/JNEUROSCI.0660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohda K, Wang Y, Yuzaki M. Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci. 2000;3:315–322. doi: 10.1038/73877. [DOI] [PubMed] [Google Scholar]
- Landsend AS, Amiry-Moghaddam M, Matsubara A, Bergersen L, Usami S, Wenthold RJ, Ottersen OP. Differential localization of delta glutamate receptors in the rat cerebellum: coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J Neurosci. 1997;17:834–842. doi: 10.1523/JNEUROSCI.17-02-00834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli H, Sprengel R, Laurie DJ, Köhr G, Herb A, Seeburg PH, Wisden W. The rat delta-1 and delta-2 subunits extend the excitatory amino acid receptor family. FEBS Lett. 1993;315:318–322. doi: 10.1016/0014-5793(93)81186-4. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Yuzaki M. Mutation in hotfoot-4J mice results in retention of delta2 glutamate receptors in ER. Eur J Neurosci. 2002;16:1507–1516. doi: 10.1046/j.1460-9568.2002.02219.x. [DOI] [PubMed] [Google Scholar]
- Mayat E, Petralia RS, Wang YX, Wenthold RJ. Immunoprecipitation, immunoblotting, and immunocytochemistry studies suggest that glutamate receptor delta subunits form novel postsynaptic receptor complexes. J Neurosci. 1995;15:2533–2546. doi: 10.1523/JNEUROSCI.15-03-02533.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML. Glutamate receptors at atomic resolution. Nature. 2006;440:456–462. doi: 10.1038/nature04709. [DOI] [PubMed] [Google Scholar]
- Naur P, Hansen KB, Kristensen AS, Dravid SM, Pickering DS, Olsen L, Vestergaard B, Egebjerg J, Gajhede M, Traynelis SF, Kastrup JS. Ionotropic glutamate-like receptor delta2 binds d-serine and glycine. Proc Natl Acad Sci U S A. 2007;104:14116–14121. doi: 10.1073/pnas.0703718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Bruggencate GT, Steinberg R, Stöckle H. Calcium modulation in brain extracellular microenvironment demonstrated with ion-selective micropipette. Proc Natl Acad Sci U S A. 1977;74:1287–1290. doi: 10.1073/pnas.74.3.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, ten Bruggencate G, Stöckle H, Steinberg R. Calcium and potassium changes in extracellular microenvironment of cat cerebellar cortex. J Neurophysiol. 1978;41:1026–1039. doi: 10.1152/jn.1978.41.4.1026. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- Oh BH, Pandit J, Kang CH, Nikaido K, Gokcen S, Ames GF, Kim SH. Three-dimensional structures of the periplasmic lysine/arginine/ornithine-binding protein with and without a ligand. J Biol Chem. 1993;268:11348–11355. [PubMed] [Google Scholar]
- Plested AJ, Mayer ML. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Plested AJ, Vijayan R, Biggin PC, Mayer ML. Molecular basis of kainate receptor modulation by sodium. Neuron. 2008;58:720–735. doi: 10.1016/j.neuron.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Rusakov DA. The role of perisynaptic glial sheaths in glutamate spillover and extracellular Ca(2+) depletion. Biophys J. 2001;81:1947–1959. doi: 10.1016/S0006-3495(01)75846-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid SM, Hollmann M. To gate or not to gate: are the delta subunits in the glutamate receptor family functional ion channels? Mol Neurobiol. 2008;37:126–141. doi: 10.1007/s12035-008-8025-0. [DOI] [PubMed] [Google Scholar]
- Shannon RD. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides. Acta Crystallogr A. 1976;32:751–767. [Google Scholar]
- Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron. 1994;13:929–936. doi: 10.1016/0896-6273(94)90258-5. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino S, Sakimura K, Nagahari K, Mishina M. Mutations in a putative agonist binding region of the AMPA-selective glutamate receptor channel. FEBS Lett. 1992;308:253–257. doi: 10.1016/0014-5793(92)81286-u. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Mitchel J, Vassilev M, Kanazirska M, Brown EM. Assessment of frequency-dependent alterations in the level of extracellular Ca2+ in the synaptic cleft. Biophys J. 1997;72:2103–2116. doi: 10.1016/S0006-3495(97)78853-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissel B, Krupp JJ, Heinemann SF, Westbrook GL. A use-dependent tyrosine dephosphorylation of NMDA receptors is independent of ion flux. Nat Neurosci. 2001;4:587–596. doi: 10.1038/88404. [DOI] [PubMed] [Google Scholar]
- Vogel MW, Caston J, Yuzaki M, Mariani J. The Lurcher mouse: fresh insights from an old mutant. Brain Res. 2007;1140:4–18. doi: 10.1016/j.brainres.2005.11.086. [DOI] [PubMed] [Google Scholar]
- Watase K, Sekiguchi M, Matsui TA, Tagawa Y, Wada K. Dominant negative mutant of ionotropic glutamate receptor subunit GluR3: implications for the role of a cysteine residue for its channel activity and pharmacological properties. Biochem J. 1997;322:385–391. doi: 10.1042/bj3220385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MC, Schuck P, Ghosal A, Rosenmund C, Mayer ML. Conformational restriction blocks glutamate receptor desensitization. Nat Struct Mol Biol. 2006;13:1120–1127. doi: 10.1038/nsmb1178. [DOI] [PubMed] [Google Scholar]
- Williams K, Chao J, Kashiwagi K, Masuko T, Igarashi K. Activation of N-methyl-d-aspartate receptors by glycine: role of an aspartate residue in the M3–M4 loop of the NR1 subunit. Mol Pharmacol. 1996;50:701–708. [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Jatzke C, Seeburg PH, Heintz N, Zuo J. The Lurcher mutation identifies δ2 as an AMPA/kainate receptor-like channel that is potentiated by Ca2+ J Neurosci. 2000;20:5973–5980. doi: 10.1523/JNEUROSCI.20-16-05973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, Fay AM, Bowie D. External ions are coactivators of kainate receptors. J Neurosci. 2006;26:5750–5755. doi: 10.1523/JNEUROSCI.0301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, MacLean DM, Bowie D. Na+/Cl− dipole couples agonist binding to kainate receptor activation. J Neurosci. 2007;27:6800–6809. doi: 10.1523/JNEUROSCI.0284-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HM, Wenthold RJ, Petralia RS. Glutamate receptor targeting to synaptic populations on Purkinje cells is developmentally regulated. J Neurosci. 1998;18:5517–5528. doi: 10.1523/JNEUROSCI.18-14-05517.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]