

Abstract
UV B (UVB) radiation induces clones of cells mutant for the p53 tumor suppressor gene in human and murine epidermis. Here we reanalyze large datasets that report the fate of clones in mice subjected to a course of UVB radiation, to uncover how p53 mutation affects epidermal progenitor cell behavior. We show that p53 mutation leads to exponential growth of clones in UV-irradiated epidermis; this finding is also consistent with the size distribution of p53 mutant clones in human epidermis. Analysis of the tail of the size distribution further reveals that the fate of individual mutant cells is stochastic. Finally, the data suggest that ending UVB exposure results in the p53 mutant cells adopting the balanced fate of wild-type cells: the loss of mutant cells is balanced by proliferation so that the population of preneoplastic cells remains constant. We conclude that preneoplastic clones do not derive from long-lived, self-renewing mutant stem cells but rather from mutant progenitors with random cell fate. It follows that ongoing, low-intensity UVB radiation will increase the number of precancerous cells dramatically compared with sporadic, higher-intensity exposure at the same cumulative dose, which may explain why nonmelanoma skin cancer incidence depends more strongly on age than on radiation dosage. Our approach may be applied to determine cell growth rates in clonally labeled material from a wide range of tissues including human samples.
Keywords: cancer, stem cells, stochastic fate
Cancer is thought to arise from the mutation of tissue stem cells: Initial mutations generate premalignant clones, a small proportion of which undergo further oncogenic changes and develop into premalignant lesions and then into tumors (1, 2). Evidence in support of this hypothesis includes observations of human squamous cell carcinomas (SCC), which develop in skin subjected to long-term exposure to UV B (UVB; 280–320 nm) radiation. UVB induces a characteristic type of mutation (a C-to-T transition at a dipyrimidine site) (3). These signature mutations are commonly found in the p53 gene in sun-exposed (yet apparently normal) skin, sun-damaged epidermis, premalignant lesions known as actinic keratoses, and in squamous carcinomas, consistent with p53 mutation being an early event in the clonal evolution of UVB-induced SCC (4–7). Keratinocytes lacking functional p53 have a greatly increased risk of malignant transformation from UVB irradiation, suggesting that the size of the p53 mutant population in epidermis will determine cancer risk (8).
Oncogenic mutations of p53 stabilize the protein, leading mutant cells to accumulate sufficient p53 to allow them to be visualized by immunostaining (6). Clusters of p53 mutant cells, termed p53 mutant clones (PMC), are detected in sun-exposed human epidermis and also in murine epidermis treated with repeated doses of UVB (6, 9–11). In the murine model, the size and frequency of PMCs increases with the duration of UVB exposure. The clusters of stained cells range in size from just a single cell up to as many as 1,000 cells after 10 weeks of intermittent UVB irradiation (9–11). PMCs in the interfollicular epidermis are cohesive, irregular in shape, and often independent of hair follicles. Indeed, large clones are observed to flow around multiple unlabeled follicles, indicating that they do not arise from mutant hair follicle stem cells (9, 12–14). Murine PMCs resemble equivalent lesions seen in human interfollicular epidermis, in both size distribution and appearance.
An intriguing observation is that when UVB irradiation ceases, the number of PMCs in mice falls dramatically, although a proportion of PMCs may persist for many weeks. The loss of clones seems paradoxical if PMCs arise from mutant long-lived, self-renewing interfollicular epidermal stem cells. In contrast, continuing irradiation results in a relentless increase in the number and size of PMCs (9).
The progressive expansion of PMCs in UVB-exposed skin raises the question of how UVB and p53 mutation affect cell behavior. UVB not only makes mutations but also induces S-phase delay, G2 arrest, apoptosis, cytokine secretion, and epidermal hyperplasia, several of which rely on P53 (5, 15–17). If cells within a PMC acquire an increased rate of proliferation and/or a decreased rate of terminal differentiation or apoptosis, the clone will expand exponentially. However, a recent theoretical analysis of PMC growth in mice has proposed an alternative hypothesis (18). Normal cells are more sensitive than mutants to UVB-induced apoptosis because of their wild-type P53. Therefore, PMCs might expand because of the apoptotic death of adjacent normal cells outside their clonal boundary, or frontier. This “Frontier model” is an example of a general class of nonexponential growth models in which cell proliferation is “contact inhibited” so that cells are able to divide only when the local cell density falls through differentiation, or in the case of UVB irradiation, apoptosis (19). The contact inhibition model has significant clinical implications: If the death of normal cells can be prevented, PMC growth is arrested, and the number of p53 mutant cells available for further mutation may be reduced.
Here, we analyze a range of high-quality published datasets recording the evolution of the PMC size distribution in chronically UVB-irradiated mouse epidermis. From simple statistical considerations, we argue that the variation of the PMC size distribution provides an unambiguous fingerprint of the clone growth characteristics that discriminates between exponential and “contact-inhibited” PMC growth (9, 10). The data provide compelling evidence for exponential PMC growth. Further analysis reveals that p53-mutated cells have a stochastic fate similar to that of the normal progenitor cells that self-renew to maintain the interfollicular epidermis during homeostasis (20). However, instead of the rates of production of cycling and postmitotic cells being perfectly balanced as they are in normal epidermis, UVB exposure results in a small excess of proliferating cells over those lost through differentiation and apoptosis in PMCs. Clone size distributions in sun-exposed human epidermis are also consistent with p53 mutation conferring this imbalanced stochastic fate. Following cessation of UVB exposure, p53 mutant cells appear to revert to the same homeostatic behavior as wild-type progenitors in the absence of irradiation. The result is that the number of PMCs falls through stochastic differentiation whereas the remaining clones expand through proliferation, so the proportion of preneoplastic cells is maintained at a constant level. These findings have important implications for cancer prevention strategies. Moreover, the statistical approach that we use may have broad applicability, allowing cell fate to be inferred in tissues containing mutant clones.
Results
p53 Mutant Cells Proliferate Exponentially During Ongoing UVB Treatment.
Prolonged exposure of mouse epidermis to UVB leads to the ongoing induction of PMCs through mutation. These clones have different sizes, a reflection of their different ages as well as potentially different patterns of cell fate (Fig. 1A). The ensemble of surviving p53-mutated clones at some time, t, following UVB exposure can therefore be represented by a size distribution, Pn(t), which describes the probability of finding a clone of total size n > 0 cells. We analyzed this distribution to provide a direct signature of the mechanism of cell turnover associated with the p53-mutated clones.
Fig. 1.

Illustration of the relationship between p53 mutant clone (PMC) size distributions and the average clone growth rate during ongoing UVB radiation. (A) Schematic showing age-related variability in PMC size following a period texp of ongoing radiation. Clones induced earlier in the experiment are expected to have a larger average size than younger clones. (B) Hypothetical growth curve, N(t), showing clone size as a function of age t at the time of analysis. Clones (1)–(3) from A are indicated; the oldest clones induced by the first pulse of UVB radiation are of age texp. As illustrated in A, at texp the growth curve consists of large clones induced early in the experiment and small clones induced a short time before the experiment is terminated. (C) Histogram of clone sizes at time texp. Dotted lines in B and C indicate the histogram binning boundaries, with the height of each bar corresponding to the number of data points (clones) demarcated in B. Therefore, the shape of the histogram depends only on the shape of the growth curve N(t) in B, as formally expressed in Eq. (1). Black curve shows the clone size distribution directly inferred from B using Eq. (1), with a sharp cutoff corresponding to the maximum size seen in the experiment. (D) Same-aged clones in reality acquire a broad distribution of sizes about the average size N(t) (Inset cartoon and colored curves; purple indicates younger clones; green, older clones). For a wide range of heterogeneous fates, the combined clone size distribution (black curve) is the same as that predicted by Eq. (1) (SI Text); however, the cutoff is now smooth as a result of heterogeneity in the fate of the oldest clones.
To develop the analysis, we defined a time-specific clone size distribution, gn(t), as the probability of finding a clone of size n > 0 following a single pulse of UVB radiation at some earlier time, t. The average size of these clones is given simply by the sum, 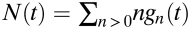 . As all surviving clones start with just one mutant cell, we have the initial condition, gn(0) = δn,1 (where δ is the Kronecker-delta symbol), from which it follows that N(0) = 1. Clones induced at some time t1 will have, at a later time t2, an average number of cells N(t2−t1).
. As all surviving clones start with just one mutant cell, we have the initial condition, gn(0) = δn,1 (where δ is the Kronecker-delta symbol), from which it follows that N(0) = 1. Clones induced at some time t1 will have, at a later time t2, an average number of cells N(t2−t1).
If we assume that the induction rate is constant during the period of UVB exposure, one obtains the following expression for the normalized size distribution (SI Text),
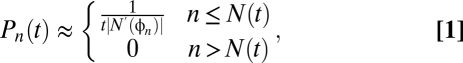 |
where φn is the inverse function to N(t), i.e., N(φn) = n, and N′(φn) denotes the time-derivative of N(t) evaluated at time t = φn. An intuitive illustration of how this result emerges is shown in Fig. 1 B and C. This result is strictly correct when all clones have precisely the average number of cells for their age, N(t); however Eq. (1) still holds, with minor modification, when clone sizes depart from their average value (Fig. 1D and SI Text). As N′(φn) is independent of the time t, Eq. (1) implies that the form of the clone size distribution is time-independent, with the time entering only through the position of the large clone size cutoff, set by the size of the oldest mutant clones, N(t) (Fig. 1), and the (1/t) prefactor arising from the normalization. In particular, if PMCs expand exponentially, N(t) ≈ exp[νt], the resulting distribution is a power-law of the form
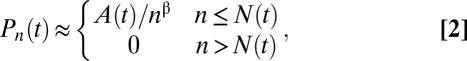 |
where β = 1 and A(t) is a time-varying normalization constant (exact expression given in SI Text). In contrast, polynomial growth, with N(t)~tα, leads to a power-law distribution with β = (α−1)/α. Note that, should early PMC expansion be conditional on other mutations in addition to the one in p53, then Eqs. (1) and (2) change in a manner that does not alter the conclusions of this study (SI Text).
In interpreting the PMC size distribution following prolonged UVB exposure, a recent study has placed emphasis on the Frontier model, in which clones expand in response to the loss of adjacent normal cells (18). In this model, the restriction of growth to the boundary of the clone leads to an expansion in the average PMC size that is quadratic with time (i.e., α = 2), leading in turn to an inverse square root dependence of the probability distribution, Pn(t), on the clone size, n, i.e., β = 1/2 in Eq. (2). Therefore, Eq. (2) provides a clear prediction for the clone size distributions following quadratic and exponential growth (Fig. 2A).
Fig. 2.

Empirical lesion size distributions follow a 1/n power law, indicative of exponential growth. (A–C) PMC size distributions in mouse back skin following 11, 9, and 7 weeks of daily UVB radiation (data from ref. 10). Data points show raw size distributions; black curves show smoothed distributions (Methods). When plotted on a log–log scale, exponential growth predicts a size distribution with slope of −1 (corresponding to a 1/n power law; solid straight curves), whereas quadratic growth predicts a slope of −1/2 (corresponding to a 1/n1/2 power law; dashed curve in A). Regression analysis of clones of size n < 500 cells (chosen to avoid the cutoff size after which the distribution decays rapidly; main text) reveals that the data have slopes of β = −0.99 ± 0.04, −1.08 ± 0.04, and −1.1 ± 0.05 at 11, 9, and 7, weeks respectively, indicating that the data follow a 1/n power-law before dropping off at large clone sizes. This deviation occurs at larger clone sizes for later time points, as expected for an age-dependent cutoff, as illustrated in Fig.1C and D and Eq. (1). (D) Size distribution of p53 mutant clones in chronically exposed human epidermis (data from ref. 6). The distribution shows the same power law decay as in the mouse, consistent with exponential growth of PMCs while exposed to UVB.
With these predictions, we analyzed the detailed p53-mutated clone size distributions measured after a relatively low level of UVB exposure (750 J/m2 administered five times per week, for 7–11 weeks) in a study comparing wild-type and immunocompromised (Rag−/−) mice (10). (In the main text, we focus on wild-type mice; similar results in Rag−/− animals are discussed in the SI Text). Figure 2 A–C shows the clone size distribution for three time points following chronic UVB exposure (for evaluation of distributions, see Methods). In each case, the raw experimental data provide a strikingly good fit to a power law decay, Pn(t) = A/nβ, with β lying in the range 0.99–1.11. The power-law behavior appears to terminate at increasing sizes at progressive time points, with the most persistent power law manifest at 11 weeks (Fig. 2A), consistent with an age-dependent truncation of the clone size distribution described in Eq. (2). The same power law behavior is found for Rag−/− animals (Fig. S1). Although these results are incompatible with the Frontier model, they provide powerful evidence for exponential growth.
UVB-induced p53 mutant clones have also been described in human epidermis. In humans, such clones arise over an unknown time period following variable levels of UVB exposure at variable intervals (6). Remarkably, analysis of PMC size distributions in human skin chronically exposed to UVB reveals the same 1/n power law behavior (Fig. 2D) indicating the same pattern of exponential clone growth.
Stochastic Exponential Lesion Growth.
An important question is the mechanism by which individual p53 mutant cells depart from homeostatic behavior during UVB irradiation to give rise to exponential growth. Recent studies of epidermal homeostasis in mouse tail have revealed that, during homeostasis, interfollicular epidermis is maintained by a population of committed progenitor (CP) cells that acquire one of three stochastic fates upon division (Fig. 3A) (20). Qualitative evidence consistent with CP cell behavior in the back skin of the mouse was also reported (20). In homeostasis, loss of CP cells through differentiation is perfectly balanced by progenitor cell self-duplication (i.e., CP→2CP) to ensure self-renewal (21). In this case, CP cell fate is fully specified by three parameters: the cell division rate, λ; the ratio, r, relating the relative fraction of symmetric and asymmetric divisions (Fig. 3A); and the fraction of basal layer cells that are cycling, ρ (19). Therefore, we further analyzed the PMC size distributions to determine whether stochastic fate could play an important role in the exponential growth of p53 mutant lesions.
Fig. 3.

Stochastic fate in normal and mutant clones. (A) Model of stochastic population self-renewal in homeostasis, characterized by the probability of symmetric division r. Each CP cell (red) may divide into two cycling CP daughter cells with probability r; or it may divide asymmetrically into one cycling and one postmitotic cell (blue, probability 1–2r); or it may divide symmetrically into two postmitotic cells, with probability r. In homeostasis the creation of new CP cells is balanced by differentiation into postmitotic cells, which are ultimately shed from the tissue (Fig. S2 for tissue organization). (B) p53 Mutant CP cells follow the same fate choices as normal cells (Left) but with an imbalance between the probabilities of CP cell multiplication and differentiation (i.e., the parameter Δ). The result is exponential clone growth at a rate of 2rΔ per cell division. Halting UVB exposure (Right) restores this balance (i.e., Δ = 0), so that the average number of mutant cells is constant. (C and D) Tests of stochastic fate in murine and human epidermis, showing the first incomplete moment of the lesion size distributions. Stochastic cell fate is predicted to give an exponential decay of the form 1− μ1(n,t)/μ1(∞,t) = e-n/N(t) [after Eq. (4); for details, see main text]. Data points are calculated from the PMC size distributions in Fig. 2 A–C for murine (C) and from Fig. 2D for human (D). Lines show fits to an exponential decay. A log-plot of the average clone size after a single UVB pulse, N(t), obtained from the fits, is shown in the Inset.
During ongoing UVB exposure, the wild-type progenitor cell population compensates for increased cell loss to maintain a new steady state. By contrast, mutant cells may become imbalanced toward exponential growth if they multiply faster than they are lost. This imbalance can be captured by a relative increase, Δ, in the number of cells self-duplicating compared with those that are lost (Fig. 3B), leading to an exponential growth of the average size of the surviving clone population with
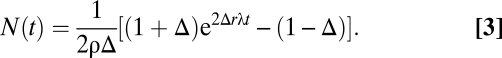 |
With Δ = 0, this description recovers the conventional CP cell dynamics in which the loss of one cell because of differentiation is compensated for by the creation of a new CP cell through self-duplication. With Δ = 1, mutant CP cells continue to divide indefinitely without loss. When 0 < Δ < 1, cell fate remains stochastic but biased toward self-duplication. In all cases, note that asymmetric divisions do not alter the number of CP cells (Fig. 3B). Interestingly, this description is analogous to the well-known Luria-Delbrück model, which was initially proposed to investigate the spread of mutations in bacterial populations (refs. 22–25 and SI Text).
From Eq. (2), we anticipated that mutant CP cells would generate a size distribution that decays as 1/n. A more careful analysis, which includes the stochastic fate of individual cells, shows that the lesion size distribution acquires the form (SI Text),
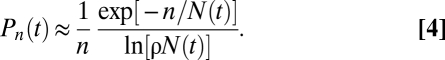 |
This result allows us to experimentally test the imbalanced CP cell growth model by focusing on the departure of large PMCs from the 1/n power-law size distribution. In particular, by analyzing the product, nPn(t), we can explore whether the distributions conform to the predicted exponential tail seen in Eq. (4). To further reduce the effect of fluctuations resulting from counting statistics, in Fig. 3C we plotted the cumulative sum of nPn(t), known as the first incomplete moment, μ1(n,t), of the experimental data, which is predicted to follow the same exponential decay (SI Text and Fig. 3) (26).
Figure 3C shows that Eq. (4) provides an excellent fit to the data at all three time points (with r2 = 0.996, 0.997, and 0.98 for the 7-, 9-, and 11-week data points, respectively; Fig. S1 shows similar results in Rag−/− mice). Further analysis of the naturally occurring p53 mutant clone fate data from human epidermis (6) shows an equally favorable fit (Fig. 3D). These striking observations confirm that the experimental data has all of the signatures of stochastic exponential growth of a mutant CP cell that may be lost by chance. The observations therefore rule out long-lived stem cells as supporting lesion growth. A more detailed analysis of the data reveals the parameter values rλ = 1.34 ± 0.42/week, Δ = 0.11 ± 0.03, and 1/ρ = 8.9 ± 2.0 (SI Text).
Fate of Mutant Clones After UVB Exposure.
When PMC size distributions are scored at 2–4 weeks following the cessation of UVB, a striking observation is a decline of 60–70% in the number of PMCs per square millimeter of epidermis, which is surprising in light of the exponential growth of the same clones during UVB exposure (ref. 10 and Fig. S3). Following the initial decline, the number and average clone size plateau, indicating that the proportion of mutant cells in the epidermis remains approximately constant (9, 10, 27). Therefore, we considered whether the stochastic fate of epidermal progenitor cells could account for these observations.
After a prolonged period of UVB exposure, Texp, the clone size distribution acquires the form given by Eq. (4) with Nexp = N(Texp). Then, if cells continued to follow the same unbalanced pattern of fate but without new clones being created, stochastic loss (e.g., through progenitor cell differentiation; Fig. 3A), would result in only a fraction,
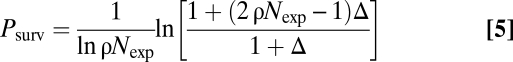 |
of PMCs surviving post UVB exposure (SI Text). If instead the p53 mutant cells revert to the balanced fate of normal CP cells after cessation of UVB (i.e., Δ = 0), then the number of PMCs would inexorably decline through stochastic differentiation (20). In this case, the remaining PMCs would expand so that the proportion of mutant cells in the epidermis remains constant. Because the observed 60–70% depletion in PMC density corresponds to a survival value of Psurv <30–40%, Eq. (5) can be used to estimate the degree of stochastic imbalance between proliferation and loss, Δ, in p53 mutant cells in the absence of UVB. Interestingly, using the values of ρ, Nexp obtained in the previous section (Fig. 3C, inset), the observed depletion in clone density is consistent with p53-mutated cells returning to balanced (or near-balanced) behavior in the absence of UVB, with Δ < 0.02. Therefore, the drop in the PMC density is consistent with a cessation of exponential growth in the absence of UVB (Fig. 3B).
Discussion
Implications of Stochastic Exponential Growth.
An immediate implication of exponential PMC growth is that ongoing, low-intensity UV radiation may carry significantly more cancer risk than sporadic higher-intensity exposure to the same cumulative UV dosage. In both cases, the number of cells acquiring de novo p53 mutations is likely to be similar; however, PMCs will expand further during ongoing exposure than during sporadic exposure, leading to many more p53 mutant cells (Fig. 4A). This time–dose effect is consistent with the observation that cancer incidence rates depend more strongly on age than on cumulative radiation dosage (28).
Fig. 4.

Implications of exponential clone growth for skin cancer risk. (A) Brief exposure to UV radiation leads to fewer p53 mutant cells than ongoing, low-intensity exposure to the same cumulative UV dose. The number of de novo p53 mutants is expected to depend mainly on UV dose, giving similar clone counts in both cases. Ongoing exposure drives exponential growth of mutant clones, resulting in a higher number of mutant cells. (B) Skin cancer risk increases through exponential growth of p53 mutant cells. Growth is initially driven by direct UVB-induced mutations (black curve). Ongoing cell division following UVB damage and sunburn leads to exponential growth of existing mutant cell clones (gray curve), which significantly amplifies the effects of UVB exposure. AU, arbitrary units.
Stochastic cell behavior has important implications for the acquisition of the additional oncogenic mutations. In normal homeostatic epidermis, the acquisition of multiple mutations in a progenitor cell is very unlikely, as an individual mutant clone has a high probability of loss through differentiation before a second mutation occurs, unless the mutation is sufficient to alter cell fate (20) (Fig. 3A). In contrast, in irradiated epidermis, p53 mutation alters cell fate in favor of self-duplication, so the probability of a cell within a mutant clone acquiring an additional oncogenic mutation and then persisting is increased. Accordingly, acquiring a large p53 mutant cell population is thus predicted to increase oncogenic mutation rate and cancer risk in later life (29).
Restricting Growth of Large Clones.
We also analyzed PMC fate data from an earlier study by Zhang et al. (9), which again reveal that clone growth is exponential, refuting a recent claim that these data support the Frontier model by which clones expand quadratically in response to the death of normal cells beyond the clone perimeter (Fig. S4) (18). Our analysis explains the full range of clone fate data, unlike the Frontier model, which only provides a good fit to the size distribution of large clones.
Yet, although the exponential growth of small PMCs will not affect the tissue, if left unchecked, such behavior would result in many PMCs generating tumors without acquiring additional mutations (30). How is the expansion of PMCs slowed? A possible explanation is through the regulation of cell division, e.g., by contact-inhibition (19). In this case, the evolution of the largest clones may indeed cross over to Frontier model–type dynamics. A narrowing of the large-size tail of the PMC distribution, as seen in the first incomplete moment of the distribution (Fig. S4), is consistent with such a change in behavior (18). Alternatively, the packing density of cells in PMCs may increase, with more cells occupying the same area (9).
EPUs vs. Stochastic Progenitors.
Earlier studies of p53 mutant clones identified oscillations in PMC size distributions with maxima occurring roughly every 12–16 cells per clone (5, 9, 31). It was proposed that these oscillations arise as a result of the epidermis being organized into epidermal proliferative units or EPUs, each supported by a single stem cell (32, 33). In contrast, we found that the pattern of oscillations may arise from the timing of UV radiation: the peaks in the PMC size distribution are a consequence of pulsed induction of cohorts PMC (SI Text and Fig. S5).
Mechanism of PMC Expansion.
How do PMC expand? It is well established that chronic UVB exposure leads to radiation-induced apoptosis and an increase in the proportion of cycling cells in normal epidermis (5, 31). However, despite this, tissue homeostasis is maintained (10). This indicates that tissue-wide signals to wild-type keratinocytes increase the rate of production of proliferating cells to compensate for cell loss through UVB-induced apoptosis. Previous studies provide strong evidence for the resistance of p53-mutated cells to apoptosis, growth arrest, or terminal differentiation induced by UVB (5, 34). If both normal and p53 mutant progenitor cells respond equally to UVB by generating more proliferating cells, the reduced loss of p53-mutated cells will result in exponential growth, as there is an effective imbalance between the rate of mutant cell loss and self-duplication (Fig. 3B), giving the mutant cells a competitive advantage over wild-type cells (35). It is important to note that this advantage emerges only during UV irradiation.
In summary, these findings show that most p53 mutant cells in irradiated skin are created by proliferation of existing mutant clones and not by induction of new mutations by UVB (Fig. 4). Once formed the pool of preneoplastic cells is self-maintaining, until further UVB exposure occurs, when exponential growth resumes. Such behavior may be a key contributor to the observed supralinear increase in the incidence of nonmelanoma skin cancer with age in humans (28). These results suggest that individuals with the highest lifetime dose of UVB have the most to gain from avoiding further sun exposure: a summer of sun or a week in a suntan parlor has a far greater effect on skin that already contains a significant population of p53 mutant cells.
Methods
Evaluation of p53 Mutant Clone Size Distributions in Murine and Human Epidermis.
Experimental data used for generating size distributions was drawn from previously published studies (9, 10) that scored the number of cells per clone {n1, n2, …, nN} for clones 1, 2,…N at each time point. From these data, the fraction of lesions, Pn, with exactly n cells was calculated in two ways. First, Pn was evaluated as a coarse-grained (or kernel-smoothed) probability density from the data (curves in Fig. 2 A–D). Second, for a direct evaluation of Pn based on the raw data, we calculated the experimental fraction P*n as the total number of lesions found to have n cells, and then divided the result by the separation between consecutive nonzero values of P*n. Thus, the corrected lesion size distribution is
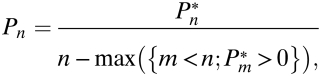 |
giving the data points in Fig. 2A–D. Coarse graining was performed using the MATLAB (v7.7.0) ksdensity function; power laws were fitted by regression analysis using XMGRACE (v5.1.22).
Supplementary Material
Acknowledgments
We thank a reviewer for pointing out the connection to the Luria-Delbruck model. AMK thanks Pedro Bordalo, Michael Springer, and Yifat Merbl for stimulating discussions, and the EPSRC for support.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0909738107/DCSupplemental.
References
- 1.Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 2.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 3.Brash DE, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci USA. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakazawa H, et al. UV and skin cancer: Specific p53 gene mutation in normal skin as a biologically relevant exposure measurement. Proc Natl Acad Sci USA. 1994;91:360–364. doi: 10.1073/pnas.91.1.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziegler A, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372:773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 6.Jonason AS, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci USA. 1996;93:14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling G, et al. Persistent p53 mutations in single cells from normal human skin. Am J Pathol. 2001;159:1247–1253. doi: 10.1016/S0002-9440(10)62511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang W, Ananthaswamy HN, Muller HK, Kripke ML. p53 Protects against skin cancer induction by UV-B radiation. Oncogene. 1999;18:4247–4253. doi: 10.1038/sj.onc.1202789. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Remenyik E, Zelterman D, Brash DE, Wikonkal NM. Escaping the stem cell compartment: Sustained UVB exposure allows p53-mutant keratinocytes to colonize adjacent epidermal proliferating units without incurring additional mutations. Proc Natl Acad Sci USA. 2001;98:13948–13953. doi: 10.1073/pnas.241353198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Remenyik E, Wikonkál NM, Zhang W, Paliwal V, Brash DE. Antigen-specific immunity does not mediate acute regression of UVB-induced p53-mutant clones. Oncogene. 2003;22:6369–6376. doi: 10.1038/sj.onc.1206657. [DOI] [PubMed] [Google Scholar]
- 11.Berg RJ, et al. Early p53 alterations in mouse skin carcinogenesis by UVB radiation: Immunohistochemical detection of mutant p53 protein in clusters of preneoplastic epidermal cells. Proc Natl Acad Sci USA. 1996;93:274–278. doi: 10.1073/pnas.93.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rebel H, et al. Early p53-positive foci as indicators of tumor risk in ultraviolet-exposed hairless mice: Kinetics of induction, effects of DNA repair deficiency, and p53 heterozygosity. Cancer Res. 2001;61:977–983. [PubMed] [Google Scholar]
- 13.Levy V, Lindon C, Harfe BD, Morgan BA. Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev Cell. 2005;9:855–861. doi: 10.1016/j.devcel.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Ito M, et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med. 2005;11:1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 15.Berg RJ, et al. Impact of global genome repair versus transcription-coupled repair on ultraviolet carcinogenesis in hairless mice. Cancer Res. 2000;60:2858–2863. [PubMed] [Google Scholar]
- 16.van Oosten M, et al. Differential role of transcription-coupled repair in UVB-induced G2 arrest and apoptosis in mouse epidermis. Proc Natl Acad Sci USA. 2000;97:11268–11273. doi: 10.1073/pnas.200226697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu YP, Lou YR, Peng QY, Xie JG, Conney AH. Stimulatory effect of topical application of caffeine on UVB-induced apoptosis in the epidermis of p53 and Bax knockout mice. Cancer Res. 2004;64:5020–5027. doi: 10.1158/0008-5472.CAN-04-0760. [DOI] [PubMed] [Google Scholar]
- 18.Chao DL, Eck JT, Brash DE, Maley CC, Luebeck EG. Preneoplastic lesion growth driven by the death of adjacent normal stem cells. Proc Natl Acad Sci USA. 2008;105:15034–15039. doi: 10.1073/pnas.0802211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein AM, Doupé DP, Jones PH, Simons BD. Kinetics of cell division in epidermal maintenance. Phys Rev E Stat Nonlin Soft Matter Phys. 2007;76:021910. doi: 10.1103/PhysRevE.76.021910. [DOI] [PubMed] [Google Scholar]
- 20.Clayton E, et al. A single type of progenitor cell maintains normal epidermis. Nature. 2007;446:185–189. doi: 10.1038/nature05574. [DOI] [PubMed] [Google Scholar]
- 21.Potten CS, Loeffler M. Stem cells: Attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development. 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 22.Dewanji A, Luebeck EG, Moolgavkar SH. A generalized Luria-Delbrück model. Math Biosci. 2005;197:140–152. doi: 10.1016/j.mbs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Q. Progress of a half century in the study of the Luria-Delbrück distribution. Math Biosci. 1999;162:1–32. doi: 10.1016/s0025-5564(99)00045-0. [DOI] [PubMed] [Google Scholar]
- 25.Zhu AJ, Haase I, Watt FM. Signaling via beta1 integrins and mitogen-activated protein kinase determines human epidermal stem cell fate in vitro. Proc Natl Acad Sci USA. 1999;96:6728–6733. doi: 10.1073/pnas.96.12.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dodge Y, Marriott FHC, Cox D. The Oxford Dictionary of Statistical Terms. Oxford: Oxford University Press; 2003. [Google Scholar]
- 27.Lu YP, et al. Administration of green tea or caffeine enhances the disappearance of UVB-induced patches of mutant p53 positive epidermal cells in SKH-1 mice. Carcinogenesis. 2005;26:1465–1472. doi: 10.1093/carcin/bgi086. [DOI] [PubMed] [Google Scholar]
- 28.Fears TR, Scotto J, Schneiderman MA. Mathematical models of age and ultraviolet effects on the incidence of skin cancer among whites in the United States. Am J Epidemiol. 1977;105:420–427. doi: 10.1093/oxfordjournals.aje.a112400. [DOI] [PubMed] [Google Scholar]
- 29.Kricker A, Armstrong BK, English DR, Heenan PJ. Pigmentary and cutaneous risk factors for non-melanocytic skin cancer—a case-control study. Int J Cancer. 1991;48:650–662. doi: 10.1002/ijc.2910480504. [DOI] [PubMed] [Google Scholar]
- 30.Rebel H, et al. Relationship between UV-induced mutant p53 patches and skin tumours, analysed by mutation spectra and by induction kinetics in various DNA-repair-deficient mice. Carcinogenesis. 2005;26:2123–2130. doi: 10.1093/carcin/bgi198. [DOI] [PubMed] [Google Scholar]
- 31.Lu YP, et al. Time course for early adaptive responses to ultraviolet B light in the epidermis of SKH-1 mice. Cancer Res. 1999;59:4591–4602. [PubMed] [Google Scholar]
- 32.Potten CS. The epidermal proliferative unit: The possible role of the central basal cell. Cell Tissue Kinet. 1974;7:77–88. doi: 10.1111/j.1365-2184.1974.tb00401.x. [DOI] [PubMed] [Google Scholar]
- 33.Mackenzie IC. Relationship between mitosis and the ordered structure of the stratum corneum in mouse epidermis. Nature. 1970;226:653–655. doi: 10.1038/226653a0. [DOI] [PubMed] [Google Scholar]
- 34.Stout GJ, et al. Epidermal transit of replication-arrested, undifferentiated keratinocytes in UV-exposed XPC mice: An alternative to in situ apoptosis. Proc Natl Acad Sci USA. 2005;102:18980–18985. doi: 10.1073/pnas.0505505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W, et al. UVB-induced apoptosis drives clonal expansion during skin tumor development. Carcinogenesis. 2005;26:249–257. doi: 10.1093/carcin/bgh300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.