

Abstract
A proteomic method that purifies and identifies palmitoylated proteins from complex protein extracts is described below. Using the fatty acid exchange labeling chemistry (described in the preceding report), palmitoyl modifications are exchanged for biotinylated compounds, allowing the subset of palmitoyl-proteins to be affinity-purified and then identified by mass spectroscopic protein identification technologies. The advantages and pitfalls of this new technology are discussed within the context of the recent application of this method in the yeast Saccharomyces cerevisiae.
Keywords: palmitoylation, proteomics, Saccharomyces cerevisiae, protein acyl transferase, DHHC cysteine-rich domain, MudPIT
INTRODUCTION
Protein palmitoylation or protein S-acylation is a post-translational modification in which a fatty acid, usually palmitic acid, is thioesterified to the cysteine thiol. Palmitoylation is a reversible lipid modification that allows regulated membrane tethering for key proteins in cell signaling, cancer, neuronal transmission, and membrane trafficking (for reviews, see (1–3)). In addition, many membrane-spanning integral membrane proteins are palmitoylated. The function of transmembrane protein palmitoylation, where tethering seems unnecessary, may be reflective of palmitoylation roles in directing protein partitioning into "lipid-ordered" domains, e.g. lipid rafts and caveolae (4, 5).
Given the cell biological importance of protein palmitoylation, it is surprising how poorly understood its underlying mechanisms remain. The first example enzymes for mediating palmitoylation, i.e. the first protein acyl transferases (PATs), were only recently identified in yeast (6, 7). The two identified yeast PATs, both members of the DHHC protein family, has led to an exploration of this family of polytopic integral membrane proteins, as a likely family of PAT specificities (1–3). While several enzymes with demonstrable in vitro protein thioesterase activity have been identified (8, 9), the extent of their in vivo participation in regulating membrane tethering through de-palmitoylation remains unclear. The slow progress towards an understanding of the functional roles of palmitoylation reflects in large part the difficulty of its associated experimental methodology. For instance, the standard method for demonstrating protein palmitoylation uses metabolic [3H]-palmitic acid labeling. This method is notoriously tedious, typically requiring large amounts of radioactive input label with week- or month-long film exposures needed to detect palmitoylation. This weak labeling is presumably a reflection presumably, of the multiple pathways both catabolic and metabolic beyond palmitoylation that palmitate may enter. Furthermore, due to the wide variety of sequence contexts in which palmitoylation occurs, consensus motifs that would allow palmitoylation to be predicted from sequence are not available. With such difficulties in both predicting and determining palmitoylation status, many palmitoyl-proteins, perhaps a great many, likely remain to be identified and the overall scope of palmitoylation's participation in the eukaryotic cell remains unclear. Proteomic approaches, such as the one described below, should go a long way towards illuminating the role of this important modification.
Fatty Acyl Exchange Labeling
Our proteomic method for palmitoyl-protein identification is based upon the fatty acid exchange chemistry described in the preceding article (10, 11). This method, a much welcomed alternative to in vivo [3H]-palmitate labeling, relies on proven thiol chemistries to replace protein palmitoyl modifications with easily-detectable labeled compounds. The method consists of three chemical steps: 1) blockade of free thiols with N-ethylmaleimide (NEM), 2) cleavage of the palmitoyl-cysteine thioester linkage by neutral pH hydroxylamine, 3) labeling of the thiols, newly-exposed at palmitoylation sites by hydroxylamine, with a thiol-specific label. As the thiol-specific labeling compound, Drisdel et al. (10, 11) have utilized either a [3H]-NEM which allows for robust autoradiographic detection, or biotin-BMCC (essentially a biotinylated NEM) which is detected through blotting with avidin-HRP.
Overview of Proteomic Strategy
The capacity of exchanging of biotin for protein palmitoyl modifications potentiates a possible proteomic approach. The high thiol specificity of the chemistries used in this labeling (10, 11) should allow just the palmitoylated protein subset from within a complex protein extract to be selectively biotinylated. Once biotinylated, this set of proteins could be specifically purified using avidin or streptavidin affinity matrices and then identified via MS-based protein identification technologies. Such a method is described below.
As the starting material for the development and optimization of this proteomic methodology, we have relied on total cellular membranes from the yeast Saccharomyces cerevisiae. An account of our first application of this protocol in yeast has been published recently (12); thirty-five new palmitoyl-proteins were identified, tripling the known size of the yeast palmitoyl-proteome. In addition, application of this proteomic methodologies to mutant yeast strains deficient for seven members of the yeast DHHC protein family, i.e. the recently-identified family of putative PAT specificities, both confirmed the general involvement of these proteins in palmitoylation and also provided a first mapping of palmitoyl-proteins to their cognate modifying PATs (12). While these methods have worked well in yeast, these methods also should be equally applicable to cells or tissues from any organism with sequence availability for MS-based protein identification. Indeed, these methods are currently being applied with good success towards a proteomic characterization of palmitoylation in rodent brain (J. Wan, R. Kang, R. Singaraja, M. Hayden, J. Yates, N. Davis, and A. El-Husseini unpublished results). Following below is a discussion both of the rationale underlying the development of this method and of the particular advantages and pitfalls of this approach. While the global reach of this method is impressive, there remains, nonetheless, ample room for protocol improvement and it is hoped the following discussion will help spur such improvements.
Proteomic Application of Acyl-Biotin Exchange (ABE) Chemistry: Chloroform-Methanol Precipitation & Protein Denaturation
At several points in the acyl-biotin exchange (ABE) protocol, the chemical reagents from the preceding step must be fully removed before continuing to the next step. For instance, NEM that persists from the initial blocking step into the second, hydroxylamine-mediated thioester cleavage step, will block new thiols as they become exposed, preventing biotinylation. For the fatty acid exchange labeling protocol (10, 11), the protein under analysis is first immune-precipitated; then, remaining bound to antibody resin, the protein can be easily moved between treatment steps, with prior reagents being efficiently washed free of the antigen-antibody complex before proceeding to the next treatment step. The proteomic scaling of ABE requires the movement of large protein collections between treatment steps. For this, we have relied on a precipitation approach, with proteins precipitated out of one treatment and then redissolved into the next. In some instances, for example for complete NEM removal, multiple re-precipitations are required. Following each precipitation step, the protein pellet is redissolved in a small volume of SDS-containing buffer which is then diluted into the subsequent chemical treatment condition. A consequence of these multiple protein precipitations and SDS re-dissolutions is that this protocol is denaturing. Denaturation is seen as a benefit in a couple of respects. Denaturation may help in exposing to chemical reagents, palmitoylation thioester linkages that normally are burrowed into the membrane. Denaturation also should eliminate potential co-purification and thus, identification, of non-palmitoylated proteins that happen to complex with the palmitoyl-proteins.
Chloroform-methanol (CM) precipitation (13) was found to afford several benefits. First, CM precipitation appears to be fairly quantitative, with good recovery of most proteins. Second, CM precipitation efficiently removes the chemicals and detergents introduced at prior steps. Indeed, the detergent removal afforded by CM precipitation is a particular benefit with regard to final steps of preparing the protein for MS analysis – detergent contamination being a frequent cause of failed MS analysis.
Proteomic Application of ABE: Biotinylation Reagent and Affinity Matrices
A variety of commercially-available thiol-specific biotinylation reagents as well as avidin- or streptavidin-based affinity matrices were tested. As discussed below, we settled on biotin-HPDP as the biotinylation reagent and streptavidin-agarose as the affinity matrix. With its KD of 10−14, the avidin-biotin interaction is one of biology's strongest non-covalent interactions. While this affords efficient biotinylated protein capture, the downside of this high affinity is that extremely harsh conditions typically are required for biotinylated protein elution. This can be tempered somewhat by using matrices with reduced biotin affinity, e.g. monomeric avidin or streptavidin. However, an additional negative feature of these resins relates to their substantial non-specific binding capacities for non-biotinylated proteins.
Initial efforts at purifying palmtioyl-proteins from protein extracts utilized biotin-BMCC as the biotinylation reagent and avidin-agarose as the affinity matrix. Biotin-BMCC gave strong and specific labeling, assessed by blotting labeled extracts with avidin-HRP. Furthermore, the capture of biotinylated proteins by the avidin-agarose resin also was extremely efficient (not shown). Substantial problems were encountered, however, both with elution and with a high-capacity, non-specific binding. The extremely harsh conditions required for elution – 10 mM biotin, 2% SDS, 100°C, 10 min – eluted, not only the biotinylated proteins, but also large quantities of non-biotinylated proteins which had bound, apparently non-specifically. Indeed, the complement of proteins that were specifically purified was masked by the contaminating non-specifically purified proteins that were stripped from the resin under the harsh elution conditions required. A breakthrough was achieved with a switch to biotin-HPDP as the biotinylation reagent. Biotin-HPDP, which disulfide bonds to thiols, is, like biotin-BMCC, very thiol-specific. The main advantage of biotin-HPDP is the ease of elution that it affords; quantitative elution is achieved through cleavage of the protein-biotin disulfide linkage with reducing agents (e.g. 1% β-mercaptoethanol) under otherwise mild conditions, leaving most of the non-specifically bound proteins attached to the affinity resin.
Minus-Hydroxylamine Control
An important control used by Drisdel et al. (10, 11), adopted by us both for the development and implementation of our proteomic analysis, is to process one-half of the analyzed sample through a parallel protocol that omits the key hydroxylamine cleavage step. In the absence of hydroxylamine, palmitoylation thioester linkages should remain intact, new thiols should not become exposed to the biotinylation reagent, and, in principle, no protein should be affinity-purified. Unfortunately, this is not the case. Figure 1 shows a silver-stained gel of proteins, derived from total yeast membranes that have been subjected to parallel plus- and minus-hydroxylamine ABE protocols and then purified by streptavidin-agarose. While there are many proteins that appear to be found in just the plus-hydroxylamine sample (Fig. 1 arrows), there also are a great many that appear in both the plus- and minus-hydroxylamine samples (Fig. 1, hashmarks). The proteins that are exclusive to the plus-hydroxylamine sample, we presumed to be the purified palmitoyl-proteins. Proteins present in both the plus- and minus-hydroxylamine samples presumably are purified non-specifically, due either to inappropriate, hydroxylamine- independent biotinylation, or non-specific, biotin-independent streptavidin-agarose binding. These presumptions were borne out by the subsequent MS analysis (12) – i.e. palmitoyl-proteins were identified exclusively from the plus-hydroxylamine sample, while the proteins equivalently present in both plus- and minus-hydoxylamine samples, typically corresponded to highly abundant yeast cell proteins (12) that though not palmitoylated, are purified non-specifically. The challenge is to distinguish palmitoyl-proteins from this substantial contaminant background.
FIGURE 1.
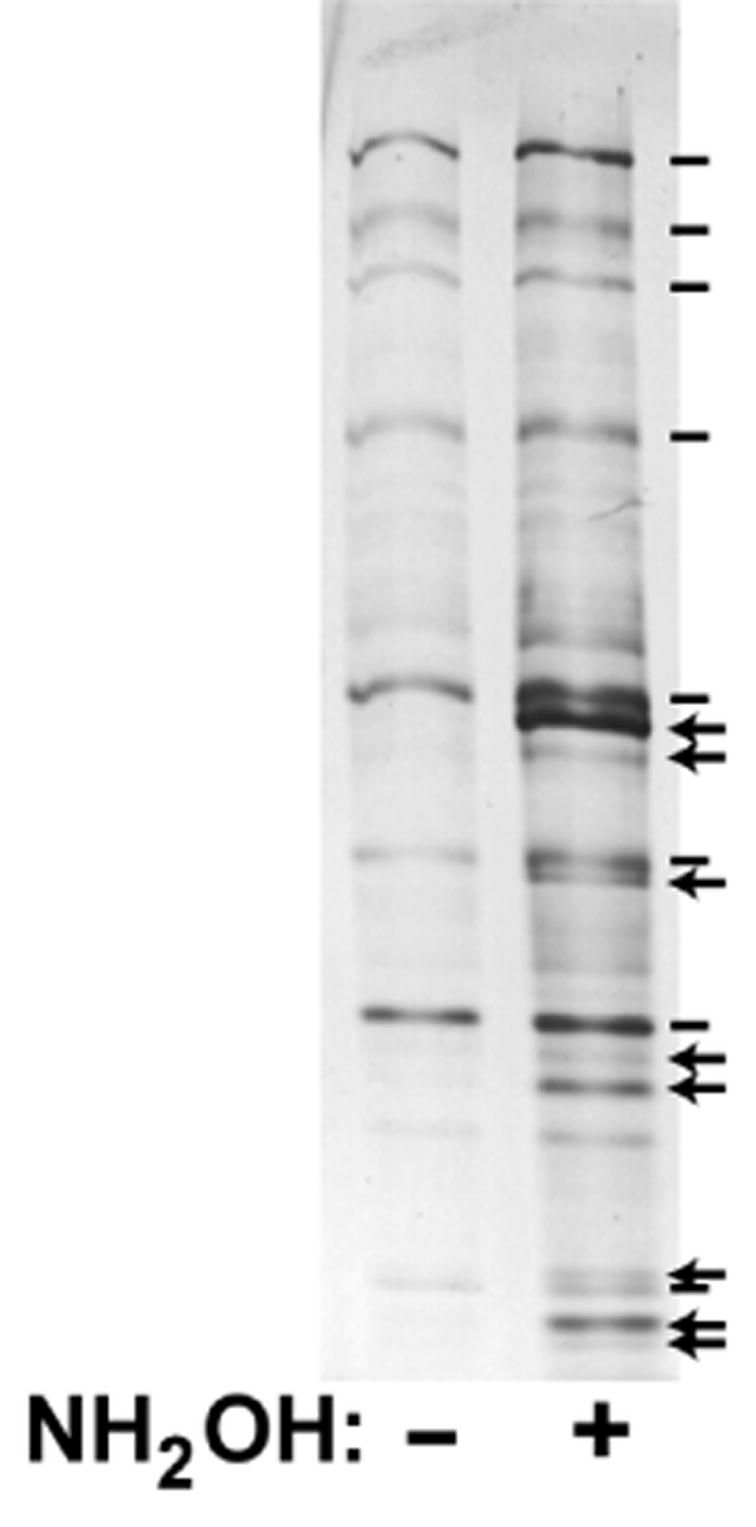
ABE-purified proteins from yeast membranes. Proteins, detergent-extracted from total yeast membranes, were subjected to the parallel plus- and minus-hydroxylamine ABE protocols. The resulting biotinylated proteins, affinity purified by streptavidin-agarose and eluted through cleavage of protein-biotin links with 1% β-mercaptoethanol, were subjected to SDS-PAGE and silver-staining. Protein bands present exclusively in just the plus-hydroxylamine sample, i.e. the candidate palmitoyl-proteins, are indicated with arrows, while protein species that are present in both samples, i.e. non-specifically purified are indicated by hash marks.
MS-Based Protein Identification Strategy
Proteins are identified made by tandem MS analysis of their component peptides. Proteins are proteolyzed and the resulting peptides are sequenced by the tandem MS and then searched against the relevant protein sequence database for matches. An important decision for our proteomic analysis was whether to digest proteins before or after the streptavidin-agarose purification step. Reserving the proteolysis until after the affinity purification, purifies the entire palmitoyl-protein. Proteolyzing prior to purification purifies just the acylated peptide. This second option, i.e. prior proteolysis, has two obvious benefits – a concentration on just the acylation site peptides both simplifies sample complexity and also leads, potentially, to immediate palmitoylation site identification (a substantial benefit considering the wide variety of sequence contexts in which palmitoylation occurs). However, a major downside to this approach is that, by discarding most of the peptides, one also discards most of the protein sequence information. We were concerned that by relying on just the palmitoylation site peptides for the protein identifications, that many palmitoyl-proteins would be missed (if the palmitoylation site peptide happens to be poorly identified by the MS analysis). For the alternative, post-purification proteolysis strategy, multiple peptides would contribute to each protein identification; this redundancy serves both to reinforce the identifications and, more importantly, as further discussed below, allows a quantitative analysis that has proved invaluable in distinguishing bona fide palmitoyl-proteins from co-purifying contaminants. Thus, the post-purification proteolysis approach was used to achieve robust protein identification.
MudPIT Analysis
We have relied on MudPIT (Multi-Dimensional Protein Identification Technology) as the protein identification technology. MudPIT is a large-scale, tandem MS-based methodology, which has proved to be particularly powerful in the analysis of highly complex protein samples (14, 15). For MudPIT, the protein sample, which may contain thousands of different proteins, is proteolyzed and the resulting peptides are fractionated by a two-dimensional chromatographic separation with direct elution into the tandem MS over an 8- to 12-hr timecourse. Protein identifications are made by correlating tandem mass spectra to theoretical spectra derived from virtual digestion of database protein sequences.
To identify yeast palmitoyl-proteins, plus- and minus-hydroxylamine ABE samples, purified from total yeast membranes (Fig. 1), were subjected to MudPIT analysis. Typically, 500–800 different proteins were identified per sample run. Our initial strategy for identifying the candidate palmitoyl-proteins involved a simple subtraction of the list of proteins identified from the minus-hydroxylamine sample from the list of plus-hydroxylamine sample proteins. While such an approach does identify both known yeast palmitoyl-proteins and new candidate palmitoyl-proteins, it also was found to miss many palmitoyl proteins: many known palmitoyl-proteins were subtracted out, due to low-level co-detection from minus-hydroxylamine samples. Such a non-quantitative approach fails to account for a protein's relative abundance within the two samples – a protein weakly detected from the minus-hydroxylamine sample is subtracted even though it may be detected much more strongly from the plus-hydroxylamine sample.
Quantitative MudPIT
Much effort in the proteomics field has been devoted towards the development of methods that add a quantitative capacity to MS. Such methods typically rely on a differential labeling of the two samples under comparison – one sample is labeled with heavy isotopes, the other with the corresponding light isotope (16). Mixing of the two samples prior to MS analysis allows the same peptide from the two samples to be identified (by their predicted mass shift) and directly compared for peak intensity. Such methods add power, but also considerable complexity both with regards to initial sample preparation and to subsequent MS data analysis. To introduce a quantitative component into our analyses, we have made use of a semi-quantitative parameter that is provided as a standard component of the MudPIT data set, this being the spectral count number associated with each protein identification (17). Abundant proteins typically are identified multiple times from a single MudPIT run, both through identification of a protein's different component peptides and through the redundant re-identification of the same peptide, that may elute into the tandem MS in multiple fractions due to "peak spreading" within the prior in-line chromatography. The spectral count number is the total number of peptide identifications (redundant plus non-redundant) associated with each protein identification. Liu and Yates have found that the spectral count number provides a good quantitative metric useful in assessing relative abundance (17).
Yeast Proteomic Analysis
For our characterization of the yeast palmitoyl-proteome, proteins enriched from total yeast membranes were processed through the parallel plus- and minus-hydroxylamine ABE protocols and the final streptavidin-agarose-purified proteins (e.g. Fig. 1) were subjected to MudPIT. A total of 1558 different yeast proteins were identified in MudPIT analyses of four paired plus- and minus-hydroxylamine samples. The spectral count data for the ten proteins most prominently detected from the four plus-hydroxylamine samples is reported in Table I. A ratio of averaged plus-hydroxylamine sample spectral counts to averaged minus-hydroxylamine sample spectral counts was used as measure of a protein's relative representation in the two samples. Four of the top ten, namely Tef1/2, Cdc19, Tdh3, and Yeh3, were prominently detected from both plus- and minus-hydroxylamine samples and show plus:minus ratios near or below one. These four are proteins of known high abundance (18) with "housekeeping" functions, and are presumably purified non-specifically, due to either inappropriate biotinylation or non-specific streptavidin-agarose binding. The other six show the skewed plus:minus spectral count ratios expected for palmitoyl-proteins. Indeed, two of the six, Ras2 and Vac8, were known to be palmitoylated, while another two, Ycp4 and Sso2, were demonstrated to be palmitoylated by subsequent analysis (see below, "Confirmatory Tests of Palmitoylation"). The two remaining proteins, Lat1 and Pdx1, were found to represent a class of non-palmitoylated, false-positive proteins that are strongly labeled by ABE (see below, "ABE Detection of Non-Palmitoylated Proteins").
A graphical representation of the application of this quantitative analysis to all 1558 identified proteins is shown in Figure 2: each identified protein is plotted by averaged plus-hydroxylamine spectral count number (x-coordinate) versus averaged minus-hydroxylamine spectral count number (y-coordinate). The bulk of the identified proteins show significant representations in both plus- and minus-hydroxylamine analyses and map near the x,y-diagonal. Again, these proteins tend to be abundant yeast cell proteins (18), that purify non-specifically. Separating from this bulk of the proteins, a set proteins is found to cluster near the x-axis (Fig. 2, circled region) as would be expected for palmitoyl-proteins. Indeed, this cluster contains thirteen of the fifteen proteins known at the inception of this work to be palmitoylated (Fig. 2, black dots). The proteins that co-cluster with the known palmitoyl-proteins, constitute the new candidate palmitoyl-proteins.
FIGURE 2.
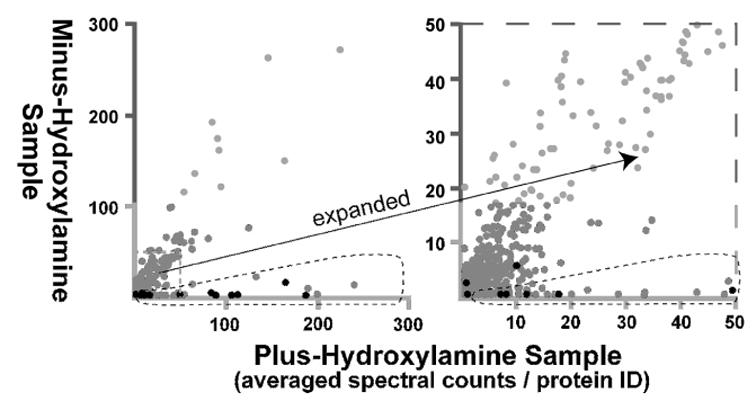
Relative plus- and minus-hydroxylamine sample representations for all of the identified proteins. Each of the 1558 proteins identified from the MudPIT analyses of four paired plus- and minus-hydroxylamine samples is plotted by averaged, normalized spectral counts from the plus-hydroxylamine samples (x-coordinate) and from the minus-hydroxylamine samples (y-coordinate). At right, the indicated portion of the plot is expanded. Known palmitoyl-proteins are indicated in black. The encircled area includes the new candidate palmitoyl-proteins co-clustering with the known palmitoyl-proteins.
Selection of Candidate Palmitoyl-Proteins
Candidate yeast palmitoyl-protein selection was based on two quantitative parameters: (1) plus-hydroxylamine sample abundance, estimated from averaged plus-hydroxylamine spectral count numbers, and (2) relative plus- versus minus-hydroxylamine representation, i.e. the plus/minus spectral count ratio. Thus, from the 432 most abundant plus-hydroxylamine sample proteins, the 70 proteins having plus/minus spectral count ratios of 5.5 or greater were selected. The top group of 70 proteins included 12 of the 15 yeast known palmitoyl-proteins, plus 58 new candidate palmitoyl-proteins. Further testing (see below, "Confirmatory Tests of Palmitoylation") confirmed palmitoylation for 35 of these 58 candidates (12).
False-Positives
For proteins detected by high spectral count numbers (Table 1), our quantitative approach easily distinguishes candidate palmitoyl-proteins from non-specifically purified proteins. At smaller spectral counts numbers, however, this discrimination of candidates from statistical noise becomes more problematic. Pooling data from multiple repeats of MudPIT analysis serves to increase confidence. Our yeast analysis relied on data pooled from MudPIT analyses of four paired plus- and minus-hydroxylamine samples (12). Nonetheless, given the large total number of proteins being sampled, many proteins may present as palmitoyl-proteins, i.e. with high plus/minus ratios, for purely statistical reasons, e.g. due to chance minus-hydroxylamine sample under-detection. Table 2 shows data for a cluster of ten candidate palmitoyl-proteins ranking from 52 through 61 within the top 70 grouping; these ten proteins were exclusively detected only from the plus-hydroxylamine samples, but with relatively low spectral count numbers. Follow-up tests confirmed palmitoylation for six of these proteins, for Ybr016w, Psr2, Ygl108c, Ypl236c, Sam3 and Rho2 (12). However, palmitoylation could be detected for the remaining four, Rps10A/B, Ybr014c, Sec63, and Adr1, suggesting that these proteins may be false-positives. This is certainly the case for Rps10, a small ribosomal subunit protein, whose sequence lacks cysteines for possible palmitoyl acceptance. As one proceeds even further down to proteins detected with even lower spectral count numbers, one obviously expects the list to be increasingly dominated by such false-positives. Nonetheless, the 70 protein cut-off used for our yeast analysis is arbitrary and there certainly are additional palmitoyl-proteins that fall below this threshold.
Table 1.
Spectral count data for the ten most-abundant plus-hydroxylamine sample proteins.1
|
Individual MudPIT Runs3 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MudPIT #1 | MudPIT #2 | MudPIT #3 | MudPIT #4 | Combined Data5 | ||||||||
| Protein | PS2 | plus | minus | plus | minus | plus | minus | plus | minus | plus | minus | ratio6 |
| LAT1 | FP | 78 | 2 | 535 | 2 | 285 | 14 | 460 | 12 | 239.7 | 12.7 | 18.8 |
| TEF1/TEF2 | FP | 21 | 134 | 642 | 65 | 391 | 205 | 429 | 266 | 224.2 | 271.4 | 0.8 |
| PDX1 | FP | 41 | 0 | 995 | 0 | 107 | 3 | 309 | 6 | 199.7 | 2.9 | 68.2 |
| YCP4 | N | 75 | 8 | 343 | 4 | 220 | 3 | 355 | 7 | 189.5 | 9.1 | 20.8 |
| RAS2 | K | 70 | 0 | 726 | 0 | 102 | 0 | 258 | 5 | 187.0 | 1.0 | 193.7 |
| VAC8 | K | 26 | 8 | 204 | 7 | 204 | 4 | 598 | 27 | 165.1 | 15.5 | 10.6 |
| CDC19 | FP | 73 | 192 | 597 | 64 | 146 | 14 | 92 | 97 | 163.4 | 150.1 | 1.1 |
| TDH3 | FP | 50 | 212 | 343 | 233 | 146 | 63 | 312 | 364 | 148.1 | 349.2 | 0.4 |
| YEF3 | FP | 33 | 184 | 269 | 75 | 199 | 87 | 362 | 441 | 144.9 | 263.3 | 0.6 |
| SSO2
|
N
|
45
|
28
|
613
|
15
|
51
|
0
|
172
|
6
|
133.1 | 22.8 | 5.8 |
| Total Spec Counts4 | 3148 | 5897 | 24132 | 3836 | 11645 | 4240 | 18803 | 12945 | ||||
Data is presented for the ten most abundant plus-hydroxylamine sample proteins judged by normalized and averaged plus-hydroxylamine sample spectral counts.
Palmitoylation Status (PS) for each protein is indicated: known palmitoyl-protein (K), newly-identified palmitoyl-proteins (N), and non-palmitoylated false-positive protein (FP).
For each protein, raw spectral counts from the MudPIT runs of the four paired plus- and minus-hydroxylamine samples are shown.
Total spectral counts from each MudPIT run.
Raw spectral counts from each MudPIT were normalized (individual spectral count values were divided by the total spectral count number from that run and then multiplied by 10,000). Then, the four normalized values were averaged.
The ratio of averaged, normalized plus-hydroxylamine sample spectral counts to averaged, normalized minus-hydroxylamine sample spectral counts.
Table 2.
Proteins of lower abundance that have high plus-minus ratios1.
|
Individual MudPIT Runs3 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MudPIT #1 | MudPIT #2 | MudPIT #3 | MudPIT #4 | Combined Data5 | ||||||||
| Protein | PS2 | plus | minus | plus | minus | plus | minus | plus | minus | plus | minus | ratio6 |
| YBR016W | N | 0 | 0 | 5 | 0 | 18 | 0 | 4 | 0 | 4.9 | 0 | 24.6 |
| PSR2 | N | 0 | 0 | 8 | 0 | 9 | 0 | 16 | 0 | 4.9 | 0 | 24.4 |
| RPS10A/B | FP | 0 | 0 | 9 | 0 | 12 | 0 | 10 | 0 | 4.8 | 0 | 24.2 |
| YGL108C | FP | 2 | 0 | 27 | 0 | 0 | 0 | 0 | 0 | 4.4 | 0 | 21.9 |
| YPL236C | N | 3 | 0 | 6 | 0 | 4 | 0 | 4 | 0 | 4.4 | 0 | 22.0 |
| YBR014C | N | 3 | 0 | 10 | 0 | 0 | 0 | 5 | 0 | 4.1 | 0 | 20.4 |
| SEC63 | FP | 3 | 0 | 6 | 0 | 0 | 0 | 8 | 0 | 4.1 | 0 | 20.3 |
| SAM3 | N | 2 | 0 | 4 | 0 | 3 | 0 | 9 | 0 | 3.8 | 0 | 19.2 |
| ADR1 | FP | 0 | 0 | 20 | 0 | 5 | 0 | 5 | 0 | 3.8 | 0 | 19.1 |
| RHO2
|
N
|
0
|
0
|
5
|
0
|
9
|
0
|
9
|
0
|
3.6 | 0 | 18.2 |
| Total Spec Counts4 | 3148 | 5897 | 24132 | 3836 | 11645 | 4240 | 18803 | 12945 | ||||
Spectral count data for the ten proteins ranking between 52 and 61 from the 70 top-ranking proteins (see "Selection of Candidate Palmitoyl-Protein").
Palmitoylation Status (PS) is indicated: new palmitoyl-proteins (N) and non-palmitoylated false-positive proteins (FP).
For each protein, raw spectral counts from the MudPIT runs of the four paired plus- and minus-hydroxylamine samples are shown.
Total spectral counts from each MudPIT run.
Raw spectral counts from each MudPIT were normalized (individual spectral count values were divided by the total spectral count number from that run and then multiplied by 10,000). Then, the four normalized values were averaged. To avoid division by zero, a value of 0.2 was substituted for those proteins having an averaged, normalized minus-hydroxylamine sample spectral count value of zero.
The ratio of averaged, normalized plus-hydroxylamine sample spectral counts to averaged, normalized minus-hydroxylamine sample spectral counts.
ABE Detection of Non-Palmitoylated Proteins
Protein palmitoylation is detected by ABE through detection of the thioester linkage. Thus, another class of false-positives that were specifically and sometimes strongly detected in our yeast analysis, are proteins that use thioesters for biochemistries other than acylation. Examples of this class include the two proteins most prominently detected in our analysis, Lat1 and Pdx1, which are subunits of the mitochondrial pyruvate dehydrogenase complex (Table 1). In the decarboxylation of pyruvate, the Lat1 and Pdx1 subunits transiently accept acetyl moieties in thioester linkage to their lipoic acid prosthetic groups. Though strongly ABE labeled, Pdx1 is not detected as palmitoylated in [3H]-palmitic acid labeling experiments (12). Other thioester-using proteins that were detected in our yeast proteomic analysis include Gcv3 which also uses the lipoic acid prosthetic group (glycine decarboxylation), the E2 ubiquitin conjugase Ubc1 which transiently accepts ubiquitin moieties in thioester linkage, and the acyl-carrier protein Acp1 which carries growing fatty acyl chains in thioester linkage to a phosphopantetheinyl prosthetic group. Preliminary application of these proteomic methods to mammalian samples detects the orthologous mammalian false-positive class (R. Kang, J. Wan, R. Singaraja, M. Hayden, J. Yates, N. Davis, and A. El-Husseini, unpublished results).
Confirmatory Tests of Palmitoylation
Palmitoylation must be independently confirmed for each of the new candidate proteins. Our yeast analysis utilized two regimens to test for palmitoylation, either a scaled-down ABE labeling protocol or standard [3H]-palmitate incorporation analysis (12). While tests of ABE labeling may eliminate statistical false positives, [3H]-palmitic acid labeling provides a more definitive test given the prominence of the thioester-utilizing false-positives. Both strategies require that the candidate protein under analysis be immunoprecipitated. For this, we have relied on an epitope-tagging strategy and have constructed for each protein, a plasmid driving GAL1 promoter overexpression of either the N- or C-terminally epitope-tagged protein. The choice of N- or C-terminal tagging was based on predictions of the likely palmitoylation site. Lipidation often occurs near polypeptide chain ends and thus tags were engineered to the end opposite the site harboring the presumed palmitoylation site. A dual HA/FLAG epitope tag was used - the FLAG epitope being used to immunoprecipitate the candidate protein from the labeled extract and the HA tag for Western blot monitioring of protein recovery.
While positive [3H]-palmitate labeling confirms palmitoylation, the negative result, i.e. the absence of labeling, does not fully eliminate possible palmitoylation, as the test conditions, i.e. either the epitope-tagging or the GAL1-driven over-expression, may interfere, in some instances, with proper acylation. Indeed, a couple examples of this were encountered in our yeast analysis (12). For instance, Rho3, a Rho GTPase not previously known to be palmitoylated, was strongly detected as palmitoylated in our proteomic analysis. Like other yeast and mammalian Rho proteins, Rho3 has a C-terminal CaaX consensus prenylation motif; however unlike all other proteins that are known to be dually prenylated-palmitoylated, Rho3 lacks cysteines in sequence proximity to its prenylation motif for service as potential palmitoyl acceptors. Surprisingly, an N-terminal cysteine, Cys5, was found to function as the Rho3 palmitoyl acceptor (12). Given this, it is not surprising that individual tests, utilizing either N- or C-terminally-tagged constructs, failed to yield evidence of Rho3 palmitoylation; N-terminal tags may directly interfere with the N-terminal palmitoylation, while C-terminal tags disrupt C-terminal prenylation and thus likely interfere indirectly; for dually prenylated-palmitoylated proteins, prior prenylation typically is a prerequisite for subsequent palmitoylation. Confirmation of Rho3 palmitoylation required that the HA/FLAG tag be engineered internally within the RHO3 ORF.
The Yeast Palmitoyl-Proteome
Forty-nine of the 58 candidates from the 70 top-ranking proteins from our proteomic analysis were individually tested for palmitoylation (12). The new and known proteins, comprising the yeast palmitoyl-proteome are listed and grouped in Figure 3. Like the known yeast and mammalian palmitoyl-proteins, many of the new yeast palmitoyl-proteins also participate in aspects of signaling transduction and/or membrane trafficking – a variety of G proteins, phosphatases, and SNAREs are identified. In addition, many of the new palmitoyl-proteins have cysteines positioned in typical palmitoylation sequence contexts – for instance, having cysteines that map proximal to either N-terminal myristoylation or C-terminal prenylation consensuses. Others proteins that lack myristoylation or prenylation consensuses, do show clusters of cysteines intriguingly mapping near either N or C termini. Furthermore, many new transmembrane proteins also were identified as palmitoylated including subsets of the yeast SNARE proteins and amino acid permeases (AAPs).
FIGURE 3.
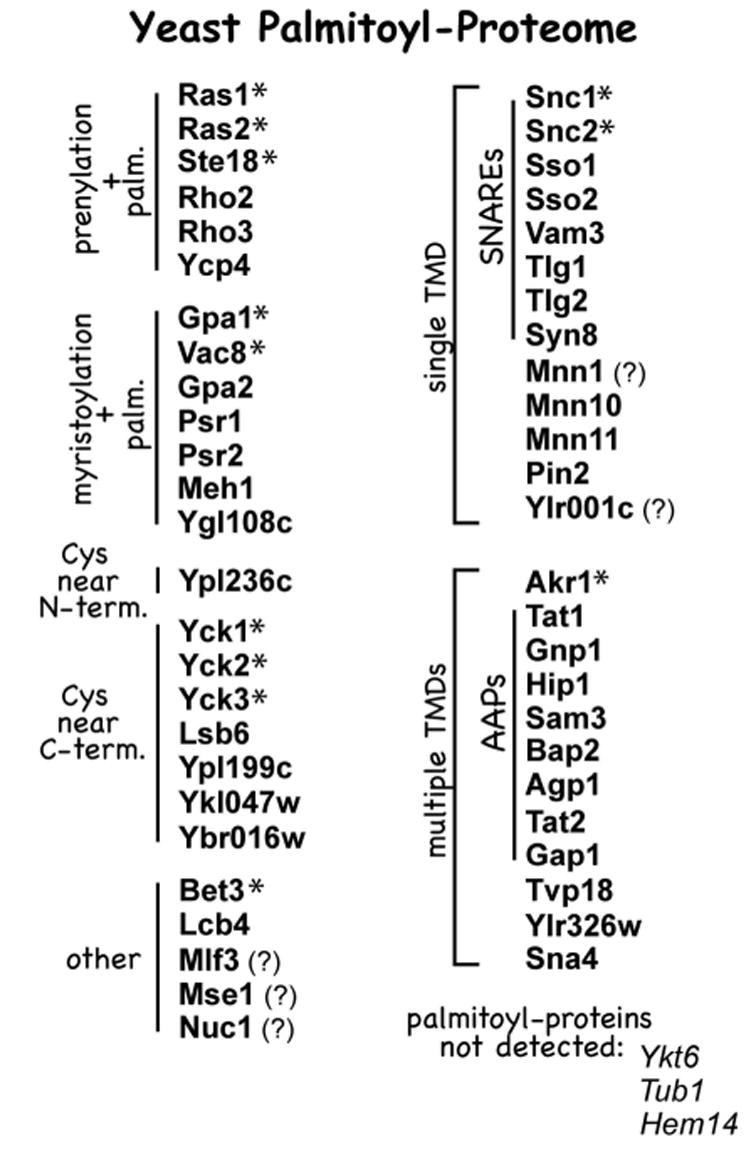
Yeast proteins, from the 70 top-ranking identified by the proteomic analysis, with confirmed palmitoylation. Proteins are grouped by likely palmitoylation sequence features. The 12 detected proteins known at the outset of this work to be palmitoylated, are indicated by asterisks, while the three known palmitoyl-proteins that were not detected from within the top 70 grouping, are listed at the bottom. Our analysis detected palmitoylation for many yeast SNARE proteins and for many amino acid permeases (AAPs). Proteins lacking strong independent confirmation of palmitoylation are indicated by question marks. Four of the palmitoyl-proteins, newly-identified by this analysis, have also been demonstrated to be palmitoylated in recent published reports, these are Gpa2, Lcb4, Tlg1, and Syn8 (20–22).
Comprehensiveness of the Analysis
Though somewhat arbitrary, we believe that the 70-protein cut-off used for our yeast analysis does include the bulk of the palmitoyl-proteins that are likely to be present in vegetatively-growing yeast cells. In addition to including 80% (12 of 15) of the known yeast palmitoyl-proteins, this top 70 grouping also apparently includes the complete collection of palmitoylated yeast SNAREs (12). Eight SNARE proteins were identified as palmitoylated from the top 70: two, Snc1 and Snc2, were previously known to be palmitoylated, while the other six, Sso1, Sso2, Tlg1, Tlg2, Syn8, and Vam3, are new (Fig. 4). For Snc1 and Snc2, as is often true for palmitoylated transmembrane proteins, the palmitoyl-accepting cysteines map to the cytoplasmic transmembrane domain (TMD) boundary (19). Interestingly, the six SNAREs newly-identified as palmitoylated by our analysis and by a recent study on SNARE protein palmitoylation (12, 20), all have similarly positioned cysteines (Fig. 4). Strikingly, the nine other yeast SNAREs from this class of TMD anchored SNAREs that were not detected by our proteomic analysis, all lack such juxta-TMD cysteines (Fig. 4). Thus, at least with regards to palmitoylated SNAREs, our analysis appears to be saturating.
FIGURE 4.
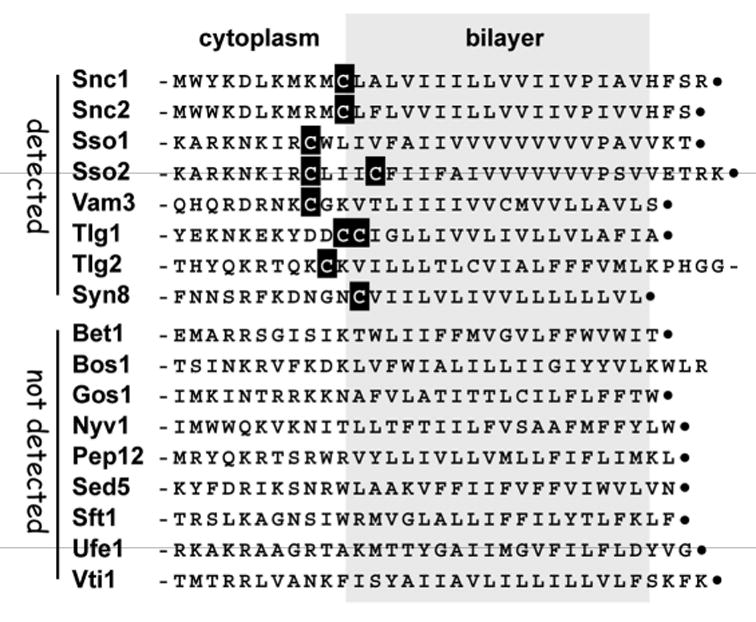
Yeast SNARE proteins detected and not detected by the proteomic analysis. For the 17 yeast SNARE proteins having typical single-TMD architecture, the sequence surrounding the TMD is shown. Cysteines predicted to map near the membrane-cytoplasm interface are indicated. C termini are indicated with dots.
Nonetheless, this sort of analysis can never be fully comprehensive. Palmitoyl-proteins that may be poorly expressed under the growth conditions used for this analysis or that have low fractional palmitoylation may be passed over. Indeed, the "glass-half-empty" view of our 12 of 15 detection is that 3 of 15 known palmitoyl-proteins were missed, suggesting that additional palmitoyl-proteins remain for mining from below the 70-protein cut-off.
Additional ABE Proteomic Uses
The proteomic methods described above should be applicable to cells or tissues from any other organism with the available sequence information to facilitate protein identification. In addition to its utility in surveying palmitoylation, this technology also should prove useful in mechanistic studies of palmitoylation as well. Indeed, in addition to characterizing palmitoylation in the wild-type yeast cell, we have also applied the same proteomic approach to mutant yeast strains defective for seven members of the DHHC protein family, an emerging family of putative PAT specificities (12). Identifying the proteins that drop out of the palmitoyl-proteome from different DHHC mutant strains has allowed many palmitoyl-proteins to be tentatively mapped to their cognate modifying DHHC PAT, providing valuable insights regarding the features driving PAT substrate recognition. Again, such an approach should translate well to mammalian systems with either DHHC gene knockout mice or RNAi-suppressed cells. More generally, this analysis should allow changes to the palmitoyl-proteome induced by any number of different drugs, perturbants, or environmental conditions, to be monitored globally.
Acknowledgments
This work was supported by NIH grants GM65525 (N.G.D.) and P41 RR11823 (J.R.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smotrys JE, Linder ME. Annu Rev Biochem. 2004;73:559–87. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- 2.Huang K, El-Husseini A. Curr Opin Neurobiol. 2005 doi: 10.1016/j.conb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. J Lipid Res 2006 [Google Scholar]
- 4.Brown DA, London E. Annu Rev Cell Dev Biol. 1998;14:111–36. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 5.Zacharias DA, Violin JD, Newton AC, Tsien RY. Science. 2002;296:913–6. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 6.Lobo S, Greentree WK, Linder ME, Deschenes RJ. J Biol Chem. 2002;277:41268–73. doi: 10.1074/jbc.M206573200. [DOI] [PubMed] [Google Scholar]
- 7.Roth AF, Feng Y, Chen L, Davis NG. J Cell Biol. 2002;159:23–8. doi: 10.1083/jcb.200206120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camp LA, Hofmann SL. J Biol Chem. 1993;268:22566–74. [PubMed] [Google Scholar]
- 9.Duncan JA, Gilman AG. J Biol Chem. 1998;273:15830–7. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 10.Drisdel RC, Green WN. Biotechniques. 2004;36:276–85. doi: 10.2144/04362RR02. [DOI] [PubMed] [Google Scholar]
- 11.Drisdel RC, Alexander JK, Sayeed A, Green WN. Methods. 2006 doi: 10.1016/j.ymeth.2006.04.015. this issue. [DOI] [PubMed] [Google Scholar]
- 12.Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR, 3rd, Davis NG. Cell. 2006 doi: 10.1016/j.cell.2006.03.042. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wessel D, Flugge UI. Anal Biochem. 1984;138:141–3. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 14.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Nat Biotechnol. 1999;17:676–82. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 15.Washburn MP, Wolters D, Yates JR., 3rd Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 16.Aebersold R, Mann M. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 17.Liu H, Sadygov RG, Yates JR., 3rd Anal Chem. 2004;76:4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 18.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Nature. 2003;425:737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 19.Couve A, Protopopov V, Gerst JE. Proc Natl Acad Sci U S A. 1995;92:5987–91. doi: 10.1073/pnas.92.13.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Valdez-Taubas J, Pelham H. EMBO J. 2005;24:2524–32. doi: 10.1038/sj.emboj.7600724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harashima T, Heitman J. Mol Biol Cell. 2005;16:4557–71. doi: 10.1091/mbc.E05-05-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kihara A, Kurotsu F, Sano T, Iwaki S, Igarashi Y. Mol Cell Biol. 2005;25:9189–97. doi: 10.1128/MCB.25.21.9189-9197.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]