

Abstract
Diverse oncogenic transformations result in the constitutive expression of tissue factor (TF) in cancer cells. The local and systemic activation of the coagulation cascade has long been a recognized hallmark for aggressive cancer, but genetic mouse models and new experimental therapeutics have only recently demonstrated crucial roles for TF initiated cell signaling in the pathogenesis of cancer. On tumor cells, the TF-VIIa binary complex mediates activation of protease activated receptor (PAR) 2 and thereby shapes the tumor microenvironment by inducing an array of pro-angiogenic and immune modulating cytokines, chemokines and growth factors. PAR2 also uniquely triggers tumor cell migration by G protein-independent pathways through β-arrestin scaffolding. Metastatic tumor cells utilize additional signaling networks of the coagulation cascade by activating PAR1 through thrombin and/or the ternary TF-VIIa-Xa signaling complex in the vascular and potentially lymphatic system. Selective antagonists of TF-VIIa-PAR2 signaling may be used as anti-angiogenic therapy without increasing the risk of bleeding, whereas coagulation and associated signaling pathways on platelets and other host cells may be targeted for therapeutic benefit in advanced cancer and metastatic disease.
Introduction
A prothrombotic state is one of the hallmarks of advanced cancer and thromboembolic disease contributes significantly to the mortality of cancer patients (reviewed in 1). Tissue factor (TF), the cellular activator of the coagulation cascade, is central to the hypercoagulable state of cancer patients and responsible for local thrombin generation and fibrin deposition in the tumor stroma. TF also triggers remote thrombotic complications involving procoagulant TF+ microparticles 2 with potential contribution from other cancer procoagulants (reviewed in 3). TF-dependent coagulation generates thrombin and induces pleiotrophic cellular effects of thrombin on platelets through G-protein coupled, protease activated receptors (PARs) 4 as well as thrombin-initiated, vascular-protective signaling of the endogenous activated protein C-EPCR-PAR1 pathway 5. Direct signaling by TF-associated proteases are mediated by the binary TF-VIIa enzyme complex that activates PAR2 or the ternary TF-VIIa-Xa coagulation initiation complex in which Xa efficiently cleaves PAR2 as well as PAR16. These signaling complexes involve distinct cellular pools of TF 7.
Oncogenic mutations of K-ras, upregulation of oncogenic epidermal growth factor receptors (EGFR) or loss of tumor suppressors p53 and PTEN result in constitutive upregulation of TF and hypoxia can amplify tumor cell TF expression in certain cancers (reviewed in 8). Furthermore, hypoxia induces the synthesis of coagulation factor VIIa in various cancer types and ectopically synthesized VII by TF-expressing cells can trigger X activation and tumor cell migration and invasion 9. Potentially, the ectopic synthesis of VIIa may turn on direct TF-VIIa signaling in cancer cells prior to alterations in vascular barrier function that typically maintains a separation of blood components and TF+ cells located in the extravascular space.
Progression of cancer from non-invasive to invasive disease is critically dependent on hypoxia-induced expression of VEGF that not only promotes vascular hyperpermeability and extravasation of coagulation factors, but also induces TF in the host compartment (reviewed in 10). In advanced cancer, tumor associated macrophages, endothelial cells and myofibroblasts contribute to the pool of TF expressed in the tumor microenvironment (TME). These host cells may further promote tumor progression through direct TF (reviewed in 11) or indirect thrombin-mediated coagulation signaling pathways (reviewed in 12). Importantly, severe reduction of TF in both host and tumor cells completely aborted the growth of teratomas 13. Because tumor cells can shed TF positive microparticles, thrombin generation in the TME may be restored, even when the host compartment is largely devoid of TF. Thus, TF and PAR signaling may make different contributions to tumor progression dependent on the tumor type and the stage-specific composition of the TME and different tumor models may be more sensitive to evaluating thrombin’s effects 14.
Several studies have documented that the levels of TF expression in primary colorectal, breast and pancreatic cancer correlate with aggressive cancer phenotypes and metastatic disease (reviewed in 8). Moreover, alternative spliced mRNA of TF was found in pancreatic, hepatocellular and leukemia cancer cell lines 15 and overexpression of alternative spliced, truncated TF promotes angiogenesis by incompletely understood mechanisms 16. Expression of full-length TF also increases tumor growth properties in several experimental models (reviewed in 17). However, TF expression in clinical and experimental tumors is not uniform. For example, cancer cell subpopulations expressing CD133, a marker for “cancer stem cells” have higher levels of procoagulant TF, indicating a possible role for TF to locally generate fibrin to establish a tumor stem cell niche 18. In contrast, propagation of tissue culture-adapted breast cancer cells in the orthotopic tumor microenvironment of the mouse mammary fat pad significantly reduced TF expression levels and essentially abolished the ternary TF-VIIa-Xa signaling response, although these cells acquired a more aggressive growth phenotype that was dependent on TF-VIIa-PAR2 signaling 19. These studies indicate that the functional state of TF, rather than expression level is a key determinant for tumor promoting activities of the TF pathway.
Only a small fraction of cellular TF contributes to procoagulant activity and a large pool of TF on intact cell surfaces is inert or “encrypted” 7. TF procoagulant activity is suppressed by limiting amounts of procoagulant membrane lipids 20, the chaperone protein Grp78/BIP 21, and by thiol pathways 7, 22. Extracellular protein disulfide isomerase (PDI) is associated with TF on epithelial cells and contributes to the regulation of the switch between procoagulant and signaling conformations of TF. TF retains TF-VIIa-PAR2 signaling, but not coagulant function when the allosteric Cys186-Cys209 extracellular disulfide is broken 7. Cancer cells appear to lack the PDI regulatory pathway 23, 24 and, while VIIa induced the association of TF with β1 integrins in non-cancerous cells, cancer cell TF is constitutively associated with the integrins α3β1 and α6β1 19. In this brief review, we focus on cancer cell TF-induced signaling pathways and describe distinct roles for TF-dependent protease pathways in regulating the TME versus successful metastatic implantation at distant vascular sites.
TF-VIIa-PAR2 signaling in tumor progression
PAR2 cleavage results in the activation of classical G protein-coupled intracellular signals as well as G protein-independent pathways mediated by the recruitment of the intracellular adaptor protein β-arrestin. G protein-coupled signaling is involved in diverse signaling responses by which tumor cells shape the TME (Fig. 1). TF-VIIa-PAR2 signaling of breast cancer cells induces a broad repertoire of pro-angiogenic factors such as VEGF 25, Cyr61, VEGF-C, CTGF, CXCL1 and IL8, and immune regulators such as granulocyte-macrophage colony stimulating factor (GM-CSF or CSF2) and macrophage colony stimulating factor (M-CSF or CSF1) 26. Although some of these genes were also induced by PAR1 signaling in breast cancer cells, TF-VIIa-PAR2 signaling was the major stimulus for upregulation of the immune and angiogenesis regulators CXCL1, IL8, GM-CSF and M-CSF.
Figure 1. Cancer cell TF-VIIa-PAR2 signaling.
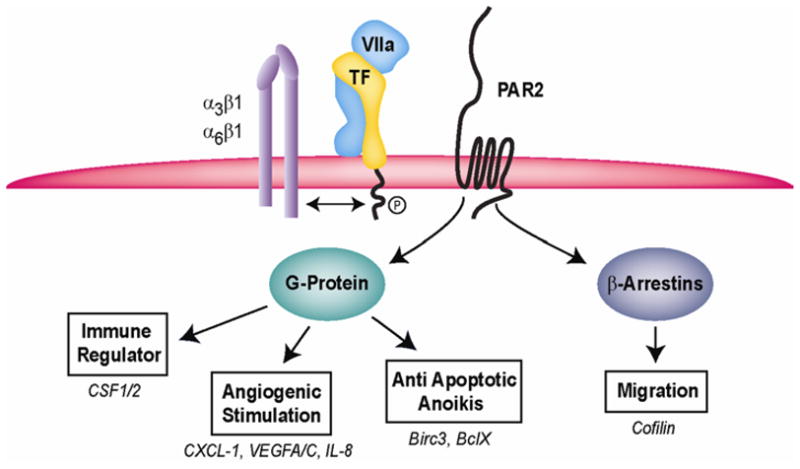
TF is constitutively associated with β1 integrins that regulate TF-VIIa-PAR2 signaling. PAR2 signaling through canonical G-protein pathways or by recruiting β-arrestins to the carboxyl-terminal tail regulates the TME and supports tumor cell migration and metastasis.
GM-CSF and M-CSF play critical roles in the recruitment and differentiation of myeloid cell populations to the TME. Typically myeloid cells initiate adaptive immune responses, but tumor cells have developed protective mechanisms to prevent CD8 T and natural killer cell-dependent tumor killing. Myeloid populations in the TME are heterogeneous and include macrophages polarized to an immune suppressive M2 phenotype as well as immature dendritic cell populations that develop under the influence of GM-CSF 27. One of the key mechanisms of myeloid suppressor cell mediated immune suppression is the release of arginase that reduces available pools of Arg required for T cell activation. In addition, myeloid suppressor cells and macrophages promote angiogenesis. The broad proangiogenic and potentially immune suppressive effects of TF-VIIa-PAR2 signaling are consistent with repeatedly documented roles of TF to act as a tumor promoter in vivo, without altering tumor cell proliferation in vitro. The emerging role of M-CSF as a regulator of lymphangiogenesis 28 also points to potential roles of TF-VIIa-PAR2 signaling in changing the TME to facilitate the exit of tumor cells for metastatic tumor dissemination throughlymphatic routes.
However, TF-PAR2 signaling also influences other aspects of tumor cell behavior and some of these may play important roles in metastasis. Breast cancer cell TF-VIIa-PAR2 signaling induces anti-apoptotic proteins, such as Birc3 26, and prevents apoptosis and death following the loss of cell adhesion (anoikis) 29, 30. Gα12/13 proteins promote breast cancer migration through activation of the rho pathway 31 and PAR2 recruits the intracellular adaptor β-arrestin that acts as a scaffolding molecule for activated ERK 32. Although β-arrestins regulate receptor internalization 33, activated PAR2 recruits β-arrestin and ERKto pseudopodia of migrating cells 34. PAR2-dependent recruitment of β-arrestins promotes breast cancer migration 35 and leads to dephosphorylation of cofilin by activating a phosphatase (chronophin) and inhibiting LIM kinase 36. The activation of the cofilin pathway severs actin filaments required for cytoskeleton reorganization and is crucial for breast cancer invasion and metastasis 37.
TF-VIIa-PAR2-dependent breast cancer migration is also in part dependent on the secretion of chemokines, such as IL8 38, and conversely the induction of chemokines is dependent on the adhesion of tumor cells to specific extracellular matrices 19. In non-cancerous epithelial cells, TF regulates integrin α3β1-dependent migration on laminin 5 39 and binding of VIIa promotes the association of TF with integrins α3β1 and α6β1. The association of TF with these integrins is constitutive in cancer cells and crucial for TF-VIIa-PAR2 signaling 19. The constitutive association of TF with integrins may facilitate the escape of tumor cells from controlling cues of the extracellular environment. PAR1 signaling is also deregulated in tumor cells and deregulated trafficking of PAR1 leads to increased transactivation of the EGF and ErbB2 receptors 40, 41 and tumor cell invasion, in particular when breast cancer cell PAR1 is activated by matrix metalloprotease (MMP) 1 42. In melanoma cells, the prometastatic activities of PAR1 are dependent on PAR2 43, pointing to important cooperative effects of PAR signaling in promoting tumor progression.
Mouse models support a crucial role of TF-VIIa-PAR2 signaling in regulating the TME
Transgenic animals provide excellent tools to evaluate the contributions of host and tumor cell factors to tumor progression. Hematogenous metastasis models in syngeneic or immune deficient mice have been instrumental to demonstrate that the hemostatic system and platelets in particular play key roles in successful homing and tumor cell survival at distant sites (reviewed in 44). Loss of the platelet thrombin receptors PAR4 and GPIbα results in significantly reduced tumor cell metastasis to the lungs 45, 46. In contrast to these pronounced contributions of thrombin pathways, syngeneic tumor growth and metastasis were normal in mice deficient in PAR1 that, unlike in humans, is not expressed on mouse platelets, but only on other host cells 45, 47. However, transplanted tumor models poorly measure tumor cell contributions as well as complex and compensatory roles of other cell types in the TME during early stages of tumor development.
Genetic mouse models of spontaneous tumor development are powerful tools to simultaneously evaluate the role of receptors in host and tumor cells and can be used as versatile tools to study tumor progression in immune competent hosts. The mouse mammary tumor virus (MMTV) promoter-driven expression of the Polyoma Middle T antigen (PyMT) results in spontaneous development of breast cancer that typically appears with complete penetrance in each mammary gland. Tumors of the PyMT model mimic important aspects of human breast cancer. In the hyperplasia and early adenoma stage, tumors are estrogen (ER) and progesterone receptor (PR) positive and do not break through interstitial barriers of mammary gland acini. Loss of ER and PR expression and upregulation of the epidermal growth factor receptor ErbB2 and cyclin D1 48 mark the transition from adenoma to invasive carcinoma that spontaneously metastasize to the lungs.
The transition from adenoma to invasive carcinoma in this model is dependent on the angiogenic switch, characterized by an infiltration of the adenomas with neovasculature. In mice lacking M-CSF (CSF1), macrophages are inefficiently recruited to developing adenomas, resulting in delayed tumor progression 49. Macrophages infiltrate the TME during the angiogenic switch and, remarkably, are in close contact with tumor cells that escape into the blood stream 50. Mammary epithelial cell-specific deletion of the hypoxia induced factor (HIF) 1α that is crucial for VEGF induction also delays the angiogenic switch in this breast cancer model 51. Thus, the early stages of tumor progression in the PyMT model are highly dependent on tumor cell-derived angiogenic regulators.
We used the PyMT model as an unbiased approach to study contributions of PARs to spontaneous breast cancer development. PAR1-deficienty did not impair tumor progression in this model 52 which was unexpected, because PAR1 has previously been shown to be upregulated in human breast cancer samples 53. Tumor cell isolated from PAR1−/− mice lost all thrombin signaling, excluding the compensatory upregulation of other thrombin receptors. A limited role for PAR1 in breast cancer progression is also suggested from data demonstrating that overexpression of PAR1 in the mammary gland is insufficient to promote breast cancer development 54. However, PAR1 may make very specific contributions to tumor progression e.g. in the context of upregulation of the EGF receptor family member ErbB2. Excellent tumor models 55 are available for further studies of potential tumor promoting roles of PAR1 that is also widely expressed in other cancer types.
In contrast to PAR1-deficiency, there was a significant delay in the transition from adenomas to invasive carcinoma in PAR2−/− mice 52. Highly vascularized tumors appeared later in PAR2−/− mice relative to wild-type, consistent with a role for PAR2 signaling in the angiogenic switch. We focused on the TF-VIIa-induced ELR CXC-type chemokine CXCL1 that binds CXCR2 56 and has proangiogenic activities on endothelial cells 57. Levels of CXCL1 were significantly reduced in early tumors of PAR2−/− relative to wild-type mice, implicating this axis as an important component of the pathway by which TF-VIIa-PAR2 signaling regulates the angiogenic switch. Macrophages were also less abundant in early tumors of PAR2−/− mice, providing initial evidence that the recruitment of myeloid cells is another pathway that is dependent on PAR2 signaling. We succeeded to establish PAR2−/− tumor cell lines that grew slower relative to a similar wild-type line when transplanted into either wild-type or PAR2-deficient mice 52. This indicated that tumor cell, rather than host PAR2 signaling makes the major contribution to breast cancer progression. This notion that is further supported by data demonstrating improved growth properties of PAR2−/− PyMT cells upon transduction of PAR2 (Schaffner et al., unpublished 2009). The PyMT model should also provide additional insight into the role of the TF cytoplasmic domain in tumor progression and metastasis, a topic that remains controversial in the literature 58–62.
Thrombin signaling dominates in metastatic tumor dissemination
For metastatic homing at distant sites, tumor cells are dependent on invasion and migration to exit the TME and on mechanisms to survive in a rapidly changing environment that they encounter in the lymphatic compartment and blood stream. A role for TF in the extravasation from the TME was recently uncovered in a model of spontaneous metastasis from the chicken chorioallantoic membrane (CAM) 63. Tumor cells selected for more efficient metastasis in this assay showed increased TF expression, detected by cell surface proteomic profiling. Blocking the TF substrate interaction and coagulation with a selective antibody decreased spontaneous metastasis of these highly metastatic cells from the CAM to the chicken embryo. While TF-VIIa binary complex signaling predominated in shaping the TME for optimal tumor growth, these data indicated that TF ternary complex regulated cell migration 64 and/or thrombin pathways become the determining mechanisms, as tumors progress to metastatic disease. Consistently, inhibiting thrombin has a profound effect of spontaneous tumor cell metastasis 14.
This notion is further supported by experiments in the hematogenous metastasis assay in which blocking TF signaling has no effect on initial homing and survival of tumor cells. In contrast, inhibition of the ternary complex leads to rapid loss of viable tumor cells at metastatic sites and profoundly attenuates late stage metastatic disease 19, 65. In this setting, thrombin is crucial not only for the generation of a protective platelet and fibrin rich envelope that protects the metastatic tumor cell from rapid clearance by natural killer cells, but also for the exposure of subendothelial matrix and tumor cell adhesion66, 67. Thrombin is known to have endothelial barrier disruptive effects, but these are counterbalanced by signaling of the endogenous protein C (PC) pathway. Endothelial cell PC receptor (EPCR)/aPC-PAR1 signaling is barrier protective in vivo by crossactivating sphingosine 1 phosphate receptor 1 5. Intriguingly, overexpression of EPCR attenuates hematogenous metastasis 68, pointing to novel control mechanisms at the vascular interface that can prevent metastasis.
Therapeutic opportunities in TF initiated signaling pathways
Thus, tumor cells rely on TF to accomplish environment specific tasks, i.e. utilize TF signaling to induce the angiogenic switch and TF coagulation to accomplish successful metastatic homing. These data suggest that appropriate targeting of the TF pathway can prevent tumor progression both in early and late stages of disease. Blocking TF-VIIa-PAR2 signaling on tumor cells by a selective antibody is sufficient to attenuate angiogenesis and tumor growth 19. Profound reductions in tumor growth 69 and spontaneous colorectal cancer development 70 were also observed with NAPc2, a nematode-derived inhibitor that blocks TF-VIIa. NAPc2 has a complex, TFPI-like inhibitory mechanism 71 that blocks coagulation, but holds nascent product Xa in an active conformation to cleave PAR1 or PAR2 6, making the interpretation of its anti-tumor effects less straightforward. The tumor growth suppressive activities of NAPc2 may result from tumor cell effects and indicate that binary TF-VIIa, rather than TF-VIIa-Xa ternary complex signaling promotes tumor progression. Alternatively, inhibition of local thrombin generation may in complex ways synergize with sustained ternary complex signaling and/or loss of binary signaling to promote tumor cell apoptosis. Consistent with efficient thrombin blockade, NAPc2 blocks metastasis and NAPc2 may be thus effective in both the early and late stages of tumor progression. NAPc2 has anti-angiogenic effects in matrigel plug assays and retinal neovascularization 47, 69, indicating additional targets on the host compartment. Despite these impressive effects, potential concerns of bleeding remain if the coagulant limb of the TF pathway is blocked efficiently 72.
While selective inhibition of TF-VIIa signaling appears to be feasible either by TF-directed antibody 19 or potentially by antagonists of PAR2 73, it is currently unclear whether targeting the signaling of thrombin can prevent tumor progression, metastasis and ultimately prolong survival. Antagonists of PAR1 may not only block the important contributions of platelets to metastatic disease, but also attenuate directly tumor cell and host proangiogenic signaling in tumor progression 12. However, PAR2 may support alternative pro-metastatic pathways that can results in an escape from PAR1 antagonistic therapy 43.
Our experiments in the PyMT model found not major contributions of PAR2 to late stage tumor growth. These data and similar findings with HIF1α-deficient tumor cells indicate that the genetic instability and dynamic adaptations of tumor cells results in compensatory mechanisms to escape rate limiting early proangiogenic pathways. The increased production of thrombin in the hyperpermeable TME may represent one such pathway. It is worth exploring whether inhibition thrombin provides additive or synergistic effects with TF-VIIa-PAR2 blockade to optimally suppress the multiple contributions of the coagulation cascade to cancer progression. Another important area for further research is to understand whether blockade of direct TF signaling provides additional benefits with anti-angiogenic therapy. The established animal models and prototypic inhibitors of the TF pathway will be instrumental in addressing these important questions. The continuing basic research and general interest in this area will identify potential innovate avenues to interrupt these pathway for improved cancer therapy.
Acknowledgments
Sources of Funding: The work reviewed here was funded by CBCRP 13FB-0125 (F.S.) and NIH grant HL-60742 (WR).
We would like to thank Cheryl Johnson for preparation of the manuscript and illustration.
Footnotes
Disclosure: WRhas pending patent applications on the use of antibodies described in this review.
References
- 1.Ten Cate H, Falanga A. Overview of the postulated mechanisms linking cancer and thrombosis. Pathophysiol Haemost Thromb. 2007;36:122–30. doi: 10.1159/000175150. [DOI] [PubMed] [Google Scholar]
- 2.Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S, Kyrle PA, Weltermann A. Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost. 2007;97:119–23. [PubMed] [Google Scholar]
- 3.Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood. 2007;110:1723–9. doi: 10.1182/blood-2006-10-053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–64. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 5.Niessen F, Furlan-Freguia C, Fernandez JA, Mosnier LO, Castellino FJ, Weiler H, Griffin JH, Ruf W. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood. 2009;113:2859–66. doi: 10.1182/blood-2008-12-192385. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Riewald M, Ruf W. Mechanistic coupling of protease signaling and initiation of coagulation by tissue factor. Proc Natl Acad Sci USA. 2001;98:7742–7. doi: 10.1073/pnas.141126698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006;103:13932–7. doi: 10.1073/pnas.0606411103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milsom C, Rak J. Tissue factor and cancer. Pathophysiol Haemost Thromb. 2007;36:160–76. doi: 10.1159/000175154. [DOI] [PubMed] [Google Scholar]
- 9.Koizume S, Jin M-S, Miyagi E, Hirahara F, Nakamura Y, Piao J-H, Asai A, Yoshida A, Tsuchiya E, Ruf W, Miyagi Y. Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res. 2006;66:9453–60. doi: 10.1158/0008-5472.CAN-06-1803. [DOI] [PubMed] [Google Scholar]
- 10.Ruf W. Hemostasis and angiogenesis. In: Khorana AA, Francis CW, editors. Cancer-Associated Thrombosis: New Findings in Translational Science, Prevention and Treatment. New York: Informa Healthcare USA; 2008. pp. 17–34. [Google Scholar]
- 11.Belting M, Ahamed J, Ruf W. Signaling of the Tissue Factor Coagulation Pathway in Angiogenesis and Cancer. Arterioscler Thromb Vasc Biol. 2005;25:1545–50. doi: 10.1161/01.ATV.0000171155.05809.bf. [DOI] [PubMed] [Google Scholar]
- 12.Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell. 2006;10:355–62. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Yu J, May L, Milsom C, Anderson GM, Weitz JI, Luyendyk JP, Broze G, Mackman N, Rak J. Contribution of host-derived tissue factor to tumor neovascularization. Arterioscler Thromb Vasc Biol. 2008;28:1975–81. doi: 10.1161/ATVBAHA.108.175083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu L, Lee M, Campbell W, Perez-Soler R, Karpatkin S. Role of endogenous thrombin in tumor implantation, seeding, and spontaneous metastasis. Blood. 2004;104:2746–51. doi: 10.1182/blood-2004-03-1047. [DOI] [PubMed] [Google Scholar]
- 15.Chand HS, Ness SA, Kisiel W. Identification of a novel human tissue factor splice variant that is upregulated in tumor cells. Int J Cancer. 2006;118:1713–20. doi: 10.1002/ijc.21550. [DOI] [PubMed] [Google Scholar]
- 16.Signaevsky M, Hobbs J, Doll J, Liu N, Soff GA. Role of alternatively spliced tissue factor in pancreatic cancer growth and angiogenesis. Semin Thromb Hemost. 2008;34:161–9. doi: 10.1055/s-2008-1079256. [DOI] [PubMed] [Google Scholar]
- 17.Ruf W. Tissue factor and PAR signaling in tumor progression. Thromb Res. 2007;120(Suppl 2):S7–S12. doi: 10.1016/S0049-3848(07)70125-1. [DOI] [PubMed] [Google Scholar]
- 18.Milsom C, Anderson GM, Weitz JI, Rak J. Elevated tissue factor procoagulant activity in CD133-positive cancer cells. J Thromb Haemost. 2007;5:2550–2. doi: 10.1111/j.1538-7836.2007.02766.x. [DOI] [PubMed] [Google Scholar]
- 19.Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008;111:190–9. doi: 10.1182/blood-2007-07-101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bach RR. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–61. doi: 10.1161/01.ATV.0000202656.53964.04. [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharjee G, Ahamed J, Pedersen B, El Sheikh A, Mackman N, Ruf W, Liu C, Edgington TS. Regulation of Tissue Factor-Mediated Initiation of the Coagulation Cascade by Cell Surface Grp78. Arterioscler Thromb Vasc Biol. 2005;25:1737–43. doi: 10.1161/01.ATV.0000173419.31242.56. [DOI] [PubMed] [Google Scholar]
- 22.Chen VM, Ahamed J, Versteeg HH, Berndt MC, Ruf W, Hogg PJ. Evidence for activation of tissue factor by an allosteric disulfide bond. Biochemistry. 2006;45:12020–8. doi: 10.1021/bi061271a. [DOI] [PubMed] [Google Scholar]
- 23.Pendurthi UR, Ghosh S, Mandal SK, Rao LV. Tissue factor activation: is disulfide bond switching a regulatory mechanism? Blood. 2007;110:3900–8. doi: 10.1182/blood-2007-07-101469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang HP, Hogg PJ. Critical importance of the cell system when studying tissue factor de-encryption. Blood. 2008;112:912–3. doi: 10.1182/blood-2008-05-158766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Mueller BM. Protease-activated receptor-2 regulates vascular endothelial growth factor expression in MDA-MB-231 cells via MAPK pathways. Biochem Biophys Res Commun. 2006;344:1263–70. doi: 10.1016/j.bbrc.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 26.Albrektsen T, Sorensen BB, Hjortoe GM, Fleckner J, Rao LVM, Petersen LC. Transcriptional program induced by factor VIIa-tissue factor, PAR1 and PAR2 in MDA-MB-231 cells. J Thromb Haemost. 2007;5:1588–1597. doi: 10.1111/j.1538-7836.2007.02603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–66. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, Saya H, Suda T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 2009;206:1089–1102. doi: 10.1084/jem.20081605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Versteeg HH, Spek CA, Richel DJ, Peppelenbosch MP. Coagulation factors VIIa and Xa inhibit apoptosis and anoikis. Oncogene. 2004;23:410–7. doi: 10.1038/sj.onc.1207066. [DOI] [PubMed] [Google Scholar]
- 30.Sorensen BB, Rao LVM, Tornehave D, Gammeltoft S, Petersen LC. Anti-apoptotic effect of coagulation factor VIIa. Blood. 2003;102:1708–15. doi: 10.1182/blood-2003-01-0157. [DOI] [PubMed] [Google Scholar]
- 31.Kelly P, Casey PJ, Meigs TE. Biologic functions of the G12 subfamily of heterotrimeric g proteins: growth, migration, and metastasis. Biochemistry. 2007;46:6677–87. doi: 10.1021/bi700235f. [DOI] [PubMed] [Google Scholar]
- 32.DeFea KA, Zalevsky J, Thoma MS, Déry O, Mullins RD, Bunnett N. β-Arrestin-dependent endocyrosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–81. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar P, Lau CS, Mathur M, Wang P, DeFea KA. Differential effects of beta-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am J Physiol Cell Physiol. 2007;293:C346–C357. doi: 10.1152/ajpcell.00010.2007. [DOI] [PubMed] [Google Scholar]
- 34.Ge L, Ly Y, Hollenberg M, DeFea K. A β-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2 induced chemotaxis. J Biol Chem. 2003;278:34418–26. doi: 10.1074/jbc.M300573200. [DOI] [PubMed] [Google Scholar]
- 35.Ge L, Shenoy SK, Lefkowitz RJ, DeFea K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J Biol Chem. 2004;279:55419–24. doi: 10.1074/jbc.M410312200. [DOI] [PubMed] [Google Scholar]
- 36.Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem. 2007;282:20634–46. doi: 10.1074/jbc.M701391200. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Wyckoff JB, Goswami S, Wang Y, Sidani M, Segall JE, Condeelis JS. Coordinated regulation of pathways for enhanced cell motility and chemotaxis is conserved in rat and mouse mammary tumors. Cancer Res. 2007;67:3505–11. doi: 10.1158/0008-5472.CAN-06-3714. [DOI] [PubMed] [Google Scholar]
- 38.Hjortoe GM, Petersen LC, Albrektsen T, Sorensen BB, Norby PL, Mandal SK, Pendurthi UR, Rao LV. Tissue factor-factor VIIa specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated via PAR-2 and results in increased cell migration. Blood. 2004;103:3029–37. doi: 10.1182/blood-2003-10-3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorfleutner A, Hintermann E, Tarui T, Takada Y, Ruf W. Crosstalk of integrin α3β1 and tissue factor in cell migration. Mol Biol Cell. 2004;15(10):4416–25. doi: 10.1091/mbc.E03-09-0640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arora P, Cuevas BD, Russo A, Johnson GL, Trejo J. Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene. 2008;27:4434–45. doi: 10.1038/onc.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Booden MA, Eckert LB, Der CJ, Trejo J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol. 2004;24:1990–9. doi: 10.1128/MCB.24.5.1990-1999.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 Is a Matrix Metalloprotease-1 Receptor that Promotes Invasion and Tumorigenesis of Breast Cancer Cells. Cell. 2005;120:303–13. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 43.Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM. Protease-activated receptor 1 (PAR1) and PAR2 contribute to tumor cell motility and metastasis. Mol Cancer Res. 2004;2:395–402. [PubMed] [Google Scholar]
- 44.Ruf W, Mueller BM. Thrombin generation and the pathogenesis of cancer. Semin Thromb Hemost. 2006;32(Suppl 1):61–8. doi: 10.1055/s-2006-939555. [DOI] [PubMed] [Google Scholar]
- 45.Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104:397–401. doi: 10.1182/blood-2004-02-0434. [DOI] [PubMed] [Google Scholar]
- 46.Jain S, Zuka M, Liu J, Russell S, Dent J, Guerrero JA, Forsyth J, Maruszak B, Gartner TK, Felding-Habermann B, Ware J. Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proc Natl Acad Sci U S A. 2007;104:9024–8. doi: 10.1073/pnas.0700625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uusitalo-Jarvinen H, Kurokawa T, Mueller BM, Andrade-Gordon P, Friedlander M, Ruf W. Role of Protease Activated Receptor 1 and 2 Signaling in Hypoxia-Induced Angiogenesis. Arterioscler Thromb Vasc Biol. 2007;27:1456–62. doi: 10.1161/ATVBAHA.107.142539. [DOI] [PubMed] [Google Scholar]
- 48.Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–26. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–46. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 50.Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, Segall JE, Pollard JW, Condeelis J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 51.Liao D, Corle C, Seagroves TN, Johnson RS. Hypoxia-inducible factor-1alpha is a key regulator of metastasis in a transgenic model of cancer initiation and progression. Cancer Res. 2007;67:563–72. doi: 10.1158/0008-5472.CAN-06-2701. [DOI] [PubMed] [Google Scholar]
- 52.Versteeg HH, Schaffner F, Kerver M, Ellies LG, Andrade-Gordon P, Mueller BM, Ruf W. Protease activated receptor (PAR)2, but not PAR1 signaling promotes the development of mammary adenocarcinoma in PyMT mice. Cancer Res. 2008;68:7219–27. doi: 10.1158/0008-5472.CAN-08-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Even-Ram S, Uziely B, Cohen P, Grisaru-Granovsky S, Maoz M, Ginzburg Y, Reich R, Vlodavsky I, Bar-Shavit R. Thrombin receptor overexpression in malignant and physiological invasion processes. Nature Med. 1998;4:909–14. doi: 10.1038/nm0898-909. [DOI] [PubMed] [Google Scholar]
- 54.Yin YJ, Katz V, Salah Z, Maoz M, Cohen I, Uziely B, Turm H, Grisaru-Granovsky S, Suzuki H, Bar-Shavit R. Mammary gland tissue targeted overexpression of human protease-activated receptor 1 reveals a novel link to beta-catenin stabilization. Cancer Res. 2006;66:5224–33. doi: 10.1158/0008-5472.CAN-05-4234. [DOI] [PubMed] [Google Scholar]
- 55.Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–97. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 56.Ben-Baruch A. The multifaceted roles of chemokines in malignancy. Cancer Metastasis Rev. 2006;25:357–71. doi: 10.1007/s10555-006-9003-5. [DOI] [PubMed] [Google Scholar]
- 57.Caunt M, Hu L, Tang T, Brooks PC, Ibrahim S, Karpatkin S. Growth-regulated oncogene is pivotal in thrombin-induced angiogenesis. Cancer Res. 2006;66:4125–32. doi: 10.1158/0008-5472.CAN-05-2570. [DOI] [PubMed] [Google Scholar]
- 58.Mueller BM, Ruf W. Requirement for binding of catalytically active factor VIIa in tissue factor dependent experimental metastasis. J Clin Invest. 1998;101:1372–8. doi: 10.1172/JCI930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Hu Z, Barney KA, Degen JL. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and -independent mechanisms. Blood. 2007;110:133–41. doi: 10.1182/blood-2007-01-065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abe K, Shoji M, Chen J, Bierhaus A, Danave I, Micko C, Casper K, Dillehay DL, Nawroth PP, Rickles FR. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc Natl Acad Sci USA. 1999;96:8663–8. doi: 10.1073/pnas.96.15.8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bromberg ME, Sundaram R, Homer RJ, Garen A, Konigsberg WH. Role of tissue factor in metastasis: functions of the cytoplasmic and extracellular domains of the molecule. Thromb Haemost. 1999;82:88–92. [PubMed] [Google Scholar]
- 62.Bromberg ME, Konigsberg WH, Madison JF, Pawashe A, Garen A. Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc Natl Acad Sci USA. 1995;92:8205–9. doi: 10.1073/pnas.92.18.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conn EM, Madsen MA, Cravatt BF, Ruf W, Deryugina EI, Quigley JP. Cell surface proteomics identifies molecules functionally linked to tumor cell intravasation. J Biol Chem. 2008;283:26518–27. doi: 10.1074/jbc.M803337200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang X, Bailly MA, Panetti TS, Cappello M, Konigsberg WH, Bromberg ME. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J Thromb Haemost. 2004;2:93–101. doi: 10.1111/j.1538-7836.2004.00545.x. [DOI] [PubMed] [Google Scholar]
- 65.Mueller BM, Reisfeld RA, Edgington TS, Ruf W. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc Natl Acad Sci USA. 1992;89:11832–6. doi: 10.1073/pnas.89.24.11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Im JH, Fu W, Wang H, Bhatia SK, Hammer DA, Kowalska MA, Muschel RJ. Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res. 2004;64:8613–9. doi: 10.1158/0008-5472.CAN-04-2078. [DOI] [PubMed] [Google Scholar]
- 67.Wang H, Fu W, Im JH, Zhou Z, Santoro SA, Iyer V, DiPersio CM, Yu QC, Quaranta V, Al Mehdi A, Muschel RJ. Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J Cell Biol. 2004;164:935–41. doi: 10.1083/jcb.200309112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bezuhly M, Cullen R, Esmon CT, Morris SF, West KA, Johnston B, Liwski RS. Role of activated protein C and its receptor in inhibition of tumor metastasis. Blood. 2009;113:3371–4. doi: 10.1182/blood-2008-05-159434. [DOI] [PubMed] [Google Scholar]
- 69.Hembrough TA, Swartz GM, Papathanassiu A, Vlasuk GP, Rote WE, Green SJ, Pribluda VS. Tissue factor/factor VIIa inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism. Cancer Res. 2003;63:2997–3000. [PubMed] [Google Scholar]
- 70.Zhao J, Aguilar G, Palencia S, Newton E, Abo A. rNAPc2 inhibits colorectal cancer in mice through tissue factor. Clin Cancer Res. 2009;15:208–16. doi: 10.1158/1078-0432.CCR-08-0407. [DOI] [PubMed] [Google Scholar]
- 71.Bergum PW, Cruikshank A, Maki S, Kelly CR, Ruf W, Vlasuk G. Role of zymogen and activated factor X as scaffolds for the inhibition of the blood coagulation factor VIIa-tissue factor complex by recombinant nematode anticoagulant protein c2. J Biol Chem. 2001;276:10063–71. doi: 10.1074/jbc.M009116200. [DOI] [PubMed] [Google Scholar]
- 72.Snyder LA, Rudnick KA, Tawadros R, Volk A, Tam SH, Anderson GM, Bugelski PJ, Yang J. Expression of human tissue factor under the control of the mouse tissue factor promoter mediates normal hemostasis in knock-in mice. J Thromb Haemost. 2008;6:306–14. doi: 10.1111/j.1538-7836.2008.02833.x. [DOI] [PubMed] [Google Scholar]
- 73.Kelso EB, Lockhart JC, Hembrough T, Dunning L, Plevin R, Hollenberg MD, Sommerhoff CP, McLean JS, Ferrell WR. Therapeutic promise of proteinase-activated receptor-2 antagonism in joint inflammation. J Pharmacol Exp Ther. 2006;316:1017–24. doi: 10.1124/jpet.105.093807. [DOI] [PubMed] [Google Scholar]