

Abstract
Macrocycles are found in important and diverse molecules such as naturally occurring peptides (e.g., cyclosporine), oligosaccharides (cyclodextrins), and polyketides (erythromycin) and synthetic compounds such as crown ethers and polyenes. The most common strategy to prepare macrocyclic lactones (macrolides) is by intramolecular C–O bond formation to provide the lactone functional group itself.[1] Though often successful and high yielding, this approach is also highly context-dependent and in some cases provides little to none of the desired macrocycle.[1] The development of methods for macrocyclization has thus received much attention.[2] Herein we report a new C–C bond forming strategy for macrocyclization, nickel-catalyzed epoxide alkyne reductive coupling,[3] and illustrate its use in the synthesis of the macrolide natural product (−)-gloeosporone (1).[4]
Keywords: macrocyclization, reductive coupling, nickel, total synthesis, C-C coupling
In the eight reported syntheses of gloeosporone, the 14-membered ring was constructed by either macrolactonization[5a–g] or ring-closing metathesis (Scheme 1).[5h–i] The nickel-catalyzed case described here represents a departure from these strategies, and for the first time uses the C5–C6 bond as the site of macrocyclization, whereby the homoallylic alcohol product (2) of an epoxide-alkyne reductive coupling reaction corresponds to the β-hydroxyketone pattern in gloeosporone. Previously we described the total synthesis of two amphidinolide T natural products using stereoselective, Ni-catalyzed aldehyde-alkyne reductive coupling reactions to form a 19-membered ring.[6] Montgomery has investigated how the size of the ring being formed affects the regioselectivity of alkyne addition in related reactions.[7] Also utilized in our amphidinolide syntheses was an intermolecular, Ni-catalyzed alkyne-epoxide reductive coupling, a process that we have also used to construct 5- and 6-membered rings.[3] Since success in smaller-ring cases is often not an accurate predictor of the outcome of cyclizations to form larger rings, it was not clear whether these Ni-catalyzed coupling reactions could be extrapolated to the case of gloeosporone.
Scheme 1.

Retrosynthetic analysis of (−)-gloeosporone (1).
Thus, we began our investigation of this question with a series of esters (3a–g, eq 1 and 2) selected to determine not only the suitability of the macrocyclization to the synthesis of gloeosporone, but also the generality of this strategy (Table 1). A preliminary survey of reaction conditions (not shown) revealed a critical interdependence of the concentrations of substrate and catalyst. To summarize, competing intermolecular reductive coupling processes were minimized under two sets of conditions: [3a]0 = 0.15 M, 20 mol% Ni(cod)2 and [3a]0 = 0.075 M, 100 mol% Ni(cod)2. Notably, the optimum initial substrate concentrations were 1–2 orders of magnitude greater than those typically used in macrocyclization reactions.
Table 1.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | substrate | reaction conditions | m | n | product | ring size | yield (%) | |
| 1 | 3a | A | 3 | 1 | 2a | 14 | 40 | |
| 2 | 3a | B | 3 | 1 | 2a | 14 | 50 | |
| 3 | 3b | A | 2 | 2 | 2b | 14 | 5 | |
| 4 | 3b | B | 2 | 2 | 2b | 14 | 18 | |
| 5 | 3c | A | 4 | 0 | 2c | 14 | <5 | |
| 6 | 3c | B | 4 | 0 | 2c | 14 | <5 | |
| 7 | 3d | A | 4 | 1 | 2d | 15 | 28 | |
| 8 | 3d | B | 4 | 1 | 2d | 15 | 53 | |
| 9 | 3e | A | 1 | 1 | 2e | 12 | 12 | |
| 10 | 3e | B | 1 | 1 | 2e | 12 | 26 | |
| 11 | 3f | A | 3 | 1 | 2f | 14 | <5 | |
| 12 | 3f | B | 3 | 1 | 2f | 14 | <5 | |
| 13 | 3g | A | 2 | 2 | 2g | 14 | <5 | |
| 14 | 3g | B | 2 | 2 | 2g | 14 | <5 | |
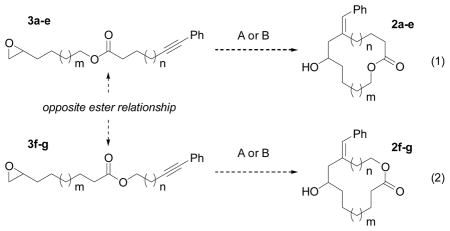
See Supporting Information and eq. (1) and (2).
Reaction conditions: (A) 20 mol% Ni(cod)2, 40 mol% Bu3P, 1000 mol% Et3B, THF, [SM]0 = 0.15 M; (B) 100 mol% Ni(cod)2, 200 mol% Bu3P, 1000 mol% Et3B, THF, [SM]0 = 0.075 M.
As shown in Table 1, the number of atoms in the ring targeted is not as important as two other factors. Holding the ring size constant but varying the number of CH2 groups between the ester and alkyne clearly shows the superiority of 3 CH2 (n = 1) vs. 2 or 4 such units (n = 0 or 2, respectively; entries 1–6). With this requirement satisfied, 12 and 15-membered rings can also be prepared in this fashion, albeit with reduced efficiency (entries 7–10). Also significant is the orientation of the ester relative to the alkyne. With the ester oxygen in the tether (entries 11–14), no product was detected, even in cases with the same number of heavy atoms in the tether (3) as in best cases (3 CH2).
These results strongly suggest that a temporary interaction between Ni and the ester[8, 9] is necessary for effective promotion of the macrocyclization, in contrast to Fürstner’s observations that certain arrangements of ester functional groups inhibited macrocyclization via ring-closing metathesis.[5h, 9c–d]
Since the necessary ester-alkyne relationship corresponded to that in gloeosporone, we hypothesized that this strategy would be well suited for preparing the natural product. Epoxy alcohol 4 was prepared in 4 steps from 7-octen-1-ol, with Jacobsen’s hydrolytic kinetic resolution[10] establishing the absolute configuration of the epoxide (Scheme 2). As five methylene groups separate the epoxide and aldehyde, carbonyl addition reactions of 6 proceeded with no detectable diastereoselection (not shown). However, a reagent-controlled addition of diamylzinc afforded alcohol 4 in >95:5 dr and 80% yield.[11,12]
Scheme 2.

Preparation of epoxyalcohol 3. a) mCPBA, CH2Cl2; b) ((R,R)-salen)Co–OAc, H2O, 35% over 2 steps, 99% ee; c) nPr4NRuO4, NMO, 3 Å MS, CH2Cl2, 95%; d) 7, (nC5H11)2Zn, Ti(OiPr)4, −20 °C, PhMe, 80%, >95 : 5 dr; e) DCC, DMAP, CH2Cl2, 85%. DCC = 1,3-dicyclohexylcarbodiimide, DMAP = 4–dimethylaminopyridine, mCPBA = meta-chloroperbenzoic acid, MS = molecular sieves, NMO = N-methylmorpholine N-oxide
Hexynoic acid 8 was prepared from commercially available 5-hexyn-1-ol in two steps in 80% overall yield. Fragment coupling via ester formation (DCC/DMAP) provided the substrate (3) for the catalytic epoxide-alkyne reductive macrocyclization (Scheme 2). At 20% Ni loading 2 is obtained in 46% yield (eq 3). Use of stoichiometric amounts of nickel provided 2 with improved efficiency (67% yield). When compared to the studies in Table 1 (entries 1 and 2, respectively), these results indicate that the amyl group plays a small yet beneficial role in the macrocyclization.
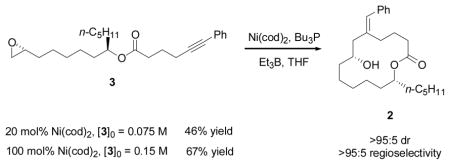 |
(3) |
With the macrocycle in hand, the major remaining challenge in the synthesis of (−)–gloeosporone was site-selective oxidation of the C4 methylene group, rather than that at C6. Ozonolytic cleavage of the alkene in 2 and protection of the secondary alcohol afforded 9 in 94% yield over the two steps (Scheme 3).[13] Heating 9 in t-butoxy-bis(dimethylamino)methane (Bredereck’s reagent[14]) generated enamine 10 with excellent regiocontrol (>95:5 site-selectivity[15]) that was immediately exposed to singlet oxygen.[16] The ensuing [2 + 2] and retro-[2 + 2] cycloaddition reactions afforded the desired 1,2-diketone (11) in 70% overall yield (two steps) by way of a dioxetane. Removal of the Et3Si group provided (−)-gloeosporone (1) in 90% yield.
Scheme 3.

Completion of the synthesis of (−)-gloeosporone (1). a) O3, then Ph3P b) Et3SiOTf, 2,6–lutidine, 94% yield over 2 steps; c) t-butoxybis(dimethylamino)methane, 60 °C, >95 :5 site selectivity; d) O2, hν, Rose Bengal, CH2Cl2, 70% yield over 2 steps; e) HF•pyridine, THF, 90% yield.
In summary, Ni-catalyzed epoxide-alkyne reductive coupling represents a novel strategy for the preparation of large rings and proceeds with high regioselectivity for both alkyne and epoxide components. This approach enabled the synthesis of (−)-gloeosporone in 10 steps (longest linear sequence) and 6% overall yield at 20% catalyst loading in the macrocyclization (9% overall yield at 100 mol% loading). A critical component of the end game was a notably site-selective oxidation using Bredereck’s reagent. Other applications of macrocyclization via catalytic epoxide-alkyne reductive coupling are under investigation.
Supplementary Material
Footnotes
Support for this work was provided by the National Institute of General Medical Sciences (GM-72566). We are grateful to Li Li for obtaining mass spectrometric data for all compounds (MIT Department of Chemistry Instrumentation Facility, which is supported in part by the NSF (CHE-9809061 and DBI-9729592) and the NIH (1S10RR13886-01)).
Contributor Information
Dr. James D. Trenkle, Gilead Sciences, 333 Lakeside Dr., Foster City, CA 94404
Prof. Dr. Timothy F. Jamison, Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139 (USA).
References
- 1.Parenty A, Moreau X, Campagne JM. Chem Rev. 2006;106:911. doi: 10.1021/cr0301402. [DOI] [PubMed] [Google Scholar]
- 2.RCM: Gradillas A, Perez-Castells J. Angew Chem. 2006;118:6232. doi: 10.1002/anie.200600641.Angew Chem Int Ed. 2006;45:6086.C–H oxidation: Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC. J Am Chem Soc. 2006;128:9032. doi: 10.1021/ja063096r.Ni-mediated: Namba K, Kishi Y. J Am Chem Soc. 2005;127:15382. doi: 10.1021/ja055966v.
- 3.a) Molinaro C, Jamison TF. J Am Chem Soc. 2003;125:8076. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]; b) Woodin KS, Jamison TF. J Org Chem. 2007;72:7451. doi: 10.1021/jo071132e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Lax AR, Templeton GE, Meyer WL. Phytopathology. 1985;75:386. [Google Scholar]; b) Meyer WL, Lax AR, Templeton GE, Brannon MJ. Tetrahedron Lett. 1983;24:5059. [Google Scholar]; c) Carling RW, Holmes AB. Tetrahedron Lett. 1986;27:6133. [Google Scholar]; d) Meyer WL, Seebach D, Schreiber SL, Schweizer WB, Beck AK, Scheifele W, Kelly SE. Helv Chim Acta. 1987;70:281. [Google Scholar]
- 5.a) Adam G, Zibuck R, Seebach D. J Am Chem Soc. 1987;109:6176. [Google Scholar]; b) Schreiber SL, Kelly SE, Porco JA, Sammakia T, Suh EM. J Am Chem Soc. 1988;110:6210. doi: 10.1021/ja00226a041. [DOI] [PubMed] [Google Scholar]; c) Takano S, Shimazaki Y, Takahashi M, Ogasawara K. J Chem Soc Chem Commun. 1988:1004. [Google Scholar]; d) Seebach D, Adam G, Zibuck R, Simon W, Rouilly M, Meyer WL, Hinton JF, Privett TA, Templeton GE, Heiny DK, Gisi U, Binder H. Liebigs Ann Chem. 1989:1233. [Google Scholar]; e) Curtis NR, Holmes AB, Looney MG, Pearson ND, Slim GC. Tetrahedron Lett. 1991;32:537. [Google Scholar]; f) Matsushita M, Yoshida M, Zhang Y, Miyashita M, Irie H, Ueno T, Tsurushima T. Chem Pharm Bull. 1992;40:524. [Google Scholar]; g) Sharma A, Gamre S, Chattopadhyay S. Lett Org Chem. 2005;2:547. [Google Scholar]; h) Fürstner A, Langemann K. J Am Chem Soc. 1997;119:9130. [Google Scholar]; i) Ley SV, Cleator E, Harter J, Hollowood CJ. Org Biomol Chem. 2003;1:3263. doi: 10.1039/b308793j. [DOI] [PubMed] [Google Scholar]
- 6.a) Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2004;126:998. doi: 10.1021/ja039716v. [DOI] [PubMed] [Google Scholar]; b) Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2005;127:4297. doi: 10.1021/ja042733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Knapp-Reed B, Mahandru GM, Montgomery J. J Am Chem Soc. 2005;127:13156. doi: 10.1021/ja054590i. [DOI] [PubMed] [Google Scholar]; b) Chaulagain MR, Sormunen GJ, Montgomery J. J Am Chem Soc. 2007;129:9568. doi: 10.1021/ja072992f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.We have observed the directing of regioselectivity in Ni-catalyzed coupling reactions of alkynes by a similarly positioned alkene: Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342. doi: 10.1021/ja0446799.Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J Am Chem Soc. 2004;126:4130. doi: 10.1021/ja0491735.
- 9.a) Feldman J, Murdzek JS, Davis WM, Schrock RR. Organometallics. 1989;8:2260. [Google Scholar]; b) Fu GC, Grubbs RH. J Am Chem Soc. 1992;114:7324. [Google Scholar]; c) Fürstner A, Langemann K. J Org Chem. 1996;61:3942. doi: 10.1021/jo960733v. [DOI] [PubMed] [Google Scholar]; d) Fürstner A, Thiel OR, Lehmann CW. Organometallics. 2002;21:331. [Google Scholar]
- 10.a) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]; b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AR, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi H, Kawakita T, Yoshioka M, Kobayashi S, Ohno M. Tetrahedron Lett. 1989;30:7095. [Google Scholar]
- 12.Generation of the dialkylzinc using Knochel’s method was critical for high stereoselectivity (3.5 : 1 dr was observed using n-C5H11MgX/ZnCl2): Rozema M, Sidduri A, Knochel P. J Org Chem. 1992;57:1956.
- 13.This ketone also undergoes high yielding and highly site-selective soft enolization at C4-C5 (Et3SiOTf, Et3N, 90% yield). (Elaboration of this enolsilane to gloeosporone via Rubottom oxidation was unsuccessful.)
- 14.Bredereck H, Simchen G, Rebsdat S, Kantlehner W, Horn P, Wahl R, Hoffman H, Grieshaber P. Chem Ber. 1968;101:41.for a recent use of Bredereck’s reagent in target-oriented synthesis, see: Peng J, Clive DLJ. Org Lett. 2007;9:2939. doi: 10.1021/ol071147z.
- 15.The regioselectivity was >95:5 as determined by crude 1H NMR spectral analysis of 11 (NMR yield of 11 was >95%).
- 16.a) Wasserman HH, Ives JL. J Am Chem Soc. 1976;98:7868. [Google Scholar]; b) Wasserman HH, Ives JL. J Org Chem. 1985;50:3573. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.