

Abstract
A375 human malignant melanoma cells undergo mitotic arrest-associated apoptosis when treated with pharmacological concentrations of sodium arsenite, a chemotherapeutic for acute promyelocytic leukemia. Our previous studies indicated that decreased arsenite sensitivity correlated with reduced mitotic spindle checkpoint function and reduced expression of the checkpoint protein BUBR1. In the current study, arsenite induced securin and cyclin B stabilization, BUBR1 phosphorylation, and spindle checkpoint activation. Arsenite also increased activating cyclin dependent kinase 1 (CDK1) Thr161 phosphorylation but decreased inhibitory Tyr15 phosphorylation. Mitotic arrest resulted in apoptosis as indicated by colocalization of mitotic phospho-Histone H3 with active caspase 3. Apoptosis was associated with BCL-2 Ser70 phosphorylation. Inhibition of CDK1 with roscovitine in arsenite-treated mitotic cells inhibited spindle checkpoint maintenance as inferred from reduced BUBR1 phosphorylation, reduced cyclin B expression, and diminution of mitotic index. Roscovitine also reduced BCL-2 Ser70 phosphorylation and protected against apoptosis, suggesting mitotic arrest caused by hyperactivation of CDK1 directly or indirectly leads to BCL-2 phosphorylation and apoptosis. In addition, suppression of BUBR1 with siRNA prevented arsenite-induced mitotic arrest and apoptosis. These findings provide insight into the mechanism of arsenic’s chemotherapeutic action and indicate a functional spindle checkpoint may be required for arsenic-sensitivity.
Keywords: Arsenite, mitotic arrest, apoptosis, spindle checkpoint
Introduction
Arsenic trioxide induces complete remission in both newly diagnosed and relapsed patients with acute promyelocytic leukemia (APL) (Miller et al., 2002). However, the mechanism through which arsenic eliminates cancer cells is unresolved. Arsenic induces the degradation of the PML-RARα fusion protein that is characteristic of APL cells (Shao et al., 1998). Arsenic also causes mitochondrial transmembrane potential loss that is inhibited by BCL-2 overexpression (Larochette et al., 1999), deregulates proteins such as IκB kinase via binding of sulfhydryls (Kapahi et al., 2000) and causes oxidative stress that can affect the function of proteins like pyruvate dehydrogenase (Samikkannu et al., 2003) or lead to DNA damage (Kessel et al., 2002). Arsenic alters signal transduction by the MAP kinase (Lau et al., 2004) and NFκB (Bode and Dong, 2002) pathways. Arsenic also has a profound effect on cell cycle progression. Arsenic delays progression through all cell cycle phases (McCollum et al., 2005) and may arrest cells in G1 (Chow et al., 2004) or S-phase (Hernandez-Zavala et al., 2005), depending upon the cell line. Arsenic can also cause accumulation of cells in the G2/M compartment (States et al., 2002;Park et al., 2001) and induces mitotic arrest that is associated with apoptosis in transformed cells (States et al., 2002;Taylor et al., 2006;McCollum et al., 2005;Cai et al., 2003;Huang et al., 2000;Ling et al., 2002;Halicka et al., 2002).
Arsenite-induced mitotic arrest has been attributed to perturbations of mitotic spindle kinetics resulting from arsenic’s binding to tubulin, similar to paclitaxel and nocodazole. The binding of paclitaxel to the β-tubulin monomers that, along with α-tubulin, comprise the mitotic spindle, causes hyperpolymerization of the spindle and arrest at metaphase/anaphase (Jordan et al., 1996). Conversely, agents like nocodazole inhibit tubulin polymerization, preventing the formation of the mitotic spindle and arresting cells in prophase/prometaphase (Zieve et al., 1980). Trivalent arsenicals have been shown to bind both α-tubulin and β-tubulin (Hoffman and Lane, 1992), although the cellular consequences of such binding are unresolved (Ling et al., 2002;Huang and Lee, 1998;Kligerman et al., 2005;Li and Broome, 1999). In our studies, arsenite allows for spindle formation in cells with no noticeable alteration of spindle morphology. An alternative explanation for the induction of mitotic arrest in the absence of alteration to spindle formation is that arsenic activates the mitotic spindle checkpoint through signal transduction cascades.
The mitotic spindle checkpoint arrests cells in mitosis to prevent unequal segregation of chromosomes during cell division. Activation of the checkpoint is signaled by a lack of microtubule-kinetochore attachment or improper tension imposed on sister kinetochores (Tan et al., 2005). The mechanism of mitotic spindle checkpoint-mediated arrest involves inhibition of the anaphase promoting complex (APC), a ubiquitin ligase that is responsible for directing the ubiquitination and resulting proteolysis of securin and cyclin B. Securin inhibits separase, a protease that cleaves the cohesin complexes holding sister chromatids together. Proteolysis of securin is necessary for anaphase (Yu, 2007). Proteolysis of cyclin B, which directs the activity of the master mitotic kinase CDK1, is necessary for mitotic exit (Chang et al., 2003). Activation of the spindle checkpoint involves the formation of the mitotic checkpoint complex (MCC). The constituents of the MCC are the checkpoint proteins MAD2, BUB1 and MAD3/BUBR1 (Tan et al., 2005). Interaction of the MCC with CDC20, a subunit of the APC, inhibits APC-mediated ubiquitination of securin and cyclin B (Sironi et al., 2001).
We have used arsenite-sensitive and arsenite-resistant melanoma cell lines as a model system to uncover the mechanism of arsenite-induced mitotic arrest-associated apoptosis. Our previous studies examining arsenite cytotoxicity in melanoma lines found that cell lines sensitive to apoptosis induced by pharmacological concentrations of arsenite arrested in mitosis (McNeely et al., 2008). In contrast, arsenite-resistant cell lines failed to undergo mitotic arrest due to a weakened spindle checkpoint. The current study indicates that arsenite activates the spindle checkpoint, causing mitotic arrest. Arrest in mitosis results in sustained activity of CDK1 which likely initiates apoptosis. Our data suggest that sensitivity to arsenite depends upon a functional spindle checkpoint dependent on hyperactivation of CDK1.
Materials and Methods
Cell culture and specialty chemicals
A375 cells, a gift from Dr. Donald Miller (University of Louisville), were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were grown at 37 °C, 90% humidity, and 5% CO2.
Working solutions of sodium arsenite (Sigma, St. Louis, MO) were prepared freshly in water on the day of use and filter sterilized. Paclitaxel (Sigma) and nocodazole (Sigma) were dissolved in DMSO. Z-VAD-FMK (BIOMOL, Plymouth Meeting, PA) was dissolved in water.
Viability assay
Assays were performed as described previously (Taylor et al., 2006). Briefly, cells were seeded in 96-well plates and allowed to attach overnight. The following day media were changed and replaced with media supplemented with sodium arsenite. AlamarBlue (Biosource International, Inc, Camarillo, CA) was added directly to culture media to 10% volume 6 h prior to plate reading and plates were returned to the incubator until fluorescence was read with an excitation wavelength of 530 nm and an emission wavelength of 590 nm.
Mitotic index determination
For mitotic index determination, cells were plated and allowed to attach overnight. Cells were treated with a range of sodium arsenite concentrations for 24 h and harvested via trypsinization. Cells were swollen in 0.4% KCl solution at 37 °C for 10 min. After 2 min centrifugation at 100 × g, pellets were resuspended in fixative (3:1 methanol:acetic acid) and incubated for at least 1 h on ice. Samples were then dropped onto glass slides. Metaphase spreads and intact nuclei were scored and the mitotic index calculated by dividing the number of spreads by the total number of interphase nuclei plus mitoses. At least 500 cells were scored per sample.
Immunobotting
Cells were lysed with a solution of 10 mM Tris-HCl pH 7.4, 1 mM EDTA, 0.1% SDS, and 180 μg/ml PMSF. Lysates were sonicated and centrifuged at 4 °C for 30 min. Protein concentrations were determined with Bradford assay (BioRad, Hercules, CA). Proteins were resolved by electrophoresis in 12% polyacrylamide SDS gels. The resolved proteins were electro-blotted onto supported nitrocellulose. After staining with Ponceau S (Acros Organics, Geel, Belgium) to ensure equal loading and transfer, membranes were blocked in 5% milk in TBST (10 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween 20) at room temperature for 2 h. Blots were probed with antibodies for β-actin (clone AC-15) (Sigma), active caspase-3 (Asp175) (Cell Signaling Technology, Danver, MA), PARP (Cell Signaling Technology), BUBR1 (BD Biosciences Pharmingen, San Diego, CA), cyclin B1 (BD Biosciences Pharmingen), CDC27 (H-300) (Santa Cruz Biotechnology, Santa Cruz, CA), securin/Pds1 (Lab Vision/Neomarkers, Fremont, CA), CDK1 (Upstate Biotechnology, Lake Placid, NY), phospho-CDK1 (Thr161)(Cell Signaling Technology), phospho-CDK1 (Tyr15)(Cell Signaling Technology), BCL-2 (BD Biosciences), and phospho-BCL-2 (Ser70)(Cell Signaling Technology). Blots probed with phospho-Thr161 CDK1 and phospho-Ser70 BCL-2 were block and probed in 5% BSA in TBST rather than 5% milk. Secondary antibodies conjugated to horseradish peroxidase were purchased from Zymed Laboratories, Inc. (South San Francisco, CA). Blots were incubated with chemi-luminescent substrate (Pierce, Rockford, IL) and exposed to film. Films were photographed using a Kodak DC290 digital camera (Kodak, Rochester, NY) and Adobe Photoshop 6.0 (Adobe Systems, Inc., New York, NY). Figures were assembled using CorelDRAW9, (Corel Corporation, Eden Prairie, MN).
Immunofluorescence
Detached paclitaxel-, nocodazole-, or arsenite-treated cells were harvested and re-plated on poly-D-lysine coated coverslips (BD Biosciences). Cells were centrifuged at 100 × g for 2 min to aid in adherence. Cells were washed twice with PBS and fixed in 4% paraformaldehyde (Sigma) in PBS for 10 min at room temperature. Following two washes in PBS, cells were permeabilized by incubation in methanol (Fisher Scientific, Pittsburgh, PA) at −20 °C for 10 min. Cells were washed twice with TBST (Tris buffered saline with 0.2% Triton X-100) and blocked in 10% normal goat serum (Sigma) in PBS for 1 h. Antibodies against α-tubulin (Upstate Biotechnology), active caspase 3 (Cell Signaling Technologies), or Histone H3-Ser10P (Cell Signaling Technology), were preconjugated with either AlexaFluor 594 or 488 Zenon reagent as per manufacturer directions (Molecular Probes, Invitrogen, Eugene, Oregon). Cells were stained with conjugated antibody for 1 h in a room temperature humidity chamber. Cells were washed twice with PBS for 5 min before secondary fixation in 4% formaldehyde (Fisher Scientific) for 10 min. Cells were washed twice with PBS and mounted on glass slides with SlowFade® Gold antifade reagent containing DAPI (Molecular Probes, Invitrogen). Pictures were taken using an Olympus IX50 inverted fluorescence microscope (Olympus America Inc., Center Valley, PA) with QImaging Retiga EXi Fast 1394 12-bit cooled monochromatic camera (QImaging Corp., Burnaby, BC, Canada) and examined via Northern Eclipse Image Analysis Software (Empix Imagine, North Tonawanda, NY).
Roscovitine treatment
A375 cells were treated with 5 μM sodium arsenite for 6 h and mitotic, detached cells were collected via centrifugation. Cell pellets were washed once with PBS and re-incubated in fresh media containing 5 μM sodium arsenite and either vehicle (DMSO), 50 μM Z-VAD-FMK, or 20 μM roscovitine (Calbiochem, La Jolla, CA). Samples were collected for mitotic index and western blot analysis. Phase contrast photographs were taken on a Nikon TMS-F light microscope with a Kodak DC290 digital camera (Kodak) using Adobe Photoshop 6.0 (Adobe Systems incorporated, New York, NY) software. Samples re-incubated with arsenite alone were also harvested for immunofluorescent staining of phospho-Histone H3 and DNA as described above.
siRNA
Cells were transfected with either 30 nM BUBR1 SMARTpool siRNA (Dharmacon, Lafayette, CO), non-specific control (NSC) siRNA (Dharmacon) or mock-transfected with only Nucleofector solution V (amaxa, Gaithersburg, MD) using program T-20 of the amaxa Nucleofector system. Cells were allowed to recover overnight and then treated as described.
Statistical Analyses
Comparison of data was performed with independent student t-test using SlideWrite software (Advanced Graphics Software, Encinitas, CA).
Results
Our previous studies found that arsenite arrests A375 human malignant melanoma cells in mitosis (McNeely et al, 2008). Arsenite-induced mitotic arrest has been attributed to perturbations of mitotic spindle kinetics similar to paclitaxel and nocodazole. To test if arsenic affected mitotic spindle formation like these drugs, A375 cells were treated with 5 μM arsenite, 10 nM paclitaxel or 0.3 μM nocodazole for 18 hours. Detached mitotic cells were harvested and stained for the mitotic marker phospho-Histone H3 and α-tubulin. The top row of Figure 1 illustrates the appearance of a normal, bipolar spindle in an untreated anaphase cell. In nocodazole-treated cells, spindle formation was completely precluded and cells exhibited a diffuse uniform staining for tubulin along with some small punctate areas (Figure 1, bottom row). In contrast, hyperpolymerization due to paclitaxel treatment caused bright-staining aggregates of tubulin (Figure 1, row 3). Arsenite treatment had neither paclitaxel- nor nocodazole-like effects and appeared to allow normal spindle formation (Figure 1, row 2). However, it should be noted that cells with multi-polar spindles were observed at a low frequency, consistent with findings in our laboratory (Taylor et al., 2008) and those of others (Yih et al., 2006) indicating that arsenite causes centrosome amplification. However, the study by Yih et al reported that centrosome amplification was a consequence and not a cause of arsenite-induced mitotic arrest.
Figure 1.
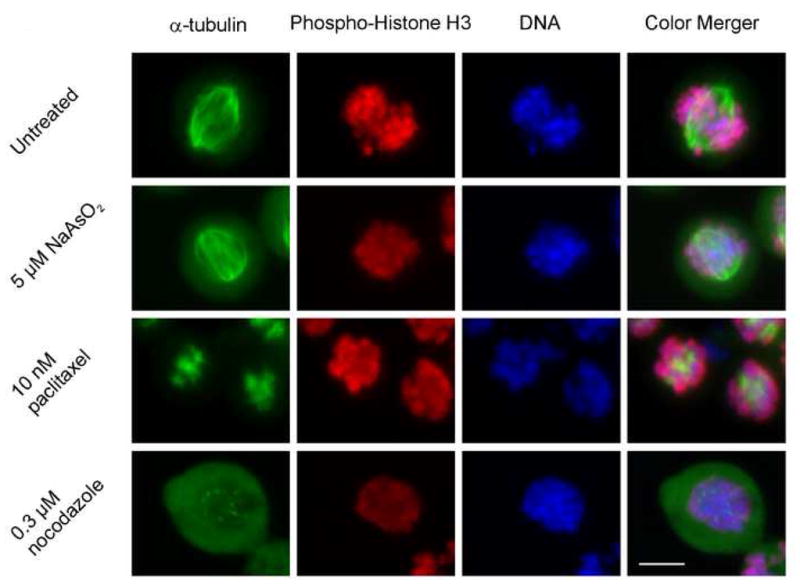
Arsenite does not alter spindle formation like either paclitaxel or nocodazole. A375 cells were treated as indicated for 18 h. Detached cells were collected, centrifuged onto glass coverslips and stained for α-tubulin (green), phospho-Histone H3 (red) and DNA (blue). Bar, 10 μm
In the absence of any observable effect on spindle formation, we examined the expression of proteins involved in mitotic progression to determine if the spindle checkpoint was activated by arsenite. Arsenite treatment resulted in the appearance of a doublet on blots probed for BUBR1, indicating retardation of electrophoretic mobility due to phosphorylation (Figure 2). Phosphorylation of BUBR1 is associated with spindle checkpoint activation by microtubule-inhibiting agents (Rancati et al., 2005). Additionally, CDC27, a subunit of the APC, was phosphorylated in arsenite-treated cells (Figure 2). CDC27 phosphorylation occurs during mitosis and has been observed in response to spindle checkpoint activation by treatment with nocodazole (Wu et al., 2000;Wassmann and Benezra, 1998). The levels of the APC-specificity subunit CDC20 were unaffected by arsenite (Figure 2). APC substrates securin and cyclin B were stabilized by arsenite (Figure 2), indicating inhibition of their APC-mediated ubiquitination and degradation necessary for mitotic exit.
Figure 2.

Analysis of arsenite-induced alterations of mitotic proteins in A375 cells. A375 cells were treated with indicated concentrations of NaAsO2 for 24 h and examined via western blot.
In addition to undergoing mitotic arrest, A375 cells treated with arsenite also become apoptotic. To determine whether apoptosis occurred in mitotically arrested cells, A375 cells were treated with arsenite and the detached fraction of the culture was stained for immunofluorescent examination of active caspase 3, a marker of apoptosis, and phosphorylated Histone H3, a marker of mitosis (Crosio et al., 2002). Active caspase 3 (Figure 3A1, green) and phosphorylated Histone H3 (Figure 3A2, red) colocalized, indicating mitotic cells that were apoptotic (Figure 3A4, merged image). Additionally, the DNA (Figure 3A3, blue) in cells demonstrating colocalization had apoptotic morphology. We examined the kinetics involved with the appearance of apoptotic DNA by harvesting detached, 5 μM arsenite-treated A375 cells from cultures and re-incubating cells in arsenite-supplemented media for 3, 6, and 12 h at which points the cells were stained for both phospho-Histone H3 and DNA (Figure 3B). At 6 h of arsenite treatment, 75% of the harvested cells were positive for phospho-Histone H3 while only 0.5% exhibited DNA of apoptotic morphology. After 12 h of re-incubation in arsenite-supplemented media, the phospho-Histone H3-positive cells declined to 13% while cells with DNA of apoptotic morphology increased to 11%.
Figure 3.

(A) Mitotic marker phosphorylated Histone H3 and apoptotic marker active caspase 3 colocalize in arsenite-treated A375 cells. (A1–4) A375 cells were treated for 24 h with 5 μM NaAsO2. Detached cells were harvested and centrifuged onto glass coverslips. Cells were stained for (A1) active caspase 3 (green), (A2) phosphorylated Histone H3 (red), and (A3) DNA (blue); (A4) merged color images. (B) Loss of Histone H3 phosphorylation precedes apoptotic DNA fragmentation. A375 cells were treated for 6 h with 5 μM NaAsO2. Detached cells were harvested and centrifuged onto glass coverslips and re-incubated in 5 μM NaAsO2 for indicated times. Cells were stained for phosphorylated Histone H3 and DNA. Data are means ± standard deviation of three independent experiments. (C) Caspase inhibition augments arsenite-induced mitotic arrest. Mitotic indices were determined after A375 cells were treated with 0 or 5 μM NaAsO2 for 24 h in the absence or presence of 50 μM Z-VAD-FMK. Significance of difference between cells treated with NaAsO2 or NaAsO2 plus Z-VAD-FMK determined by student t-test denoted by *; p < 0.05. (D) Western blot showing loss of cleaved caspase 3 in cells treated with Z-VAD-FMK as described for (C).
The effect of caspase inhibition on mitotic index was then assessed. Cells were treated with arsenite in the absence and presence of Z-VAD-FMK, a pan-caspase inhibitor. Inhibition of caspase activity increased the mitotic index of arsenite-treated cultures from 42 to 59% (Figure 3C). Inhibition of caspase activation was confirmed via western blot (Figure 3D).
Ectopic, concurrent overexpression of cyclin B and CDK1 in HeLa cells activates caspase 3, resulting in the initiation of apoptosis (Gu et al., 2003). Consequently, we considered whether overactive CDK1 was a possible initiator of arsenite-induced apoptosis. CDK1 activity requires association with cyclin B, which was stabilized by arsenite (Figure 2). Arsenite did not alter CDK1 expression in A375 cells (Figure 4) but did affect its regulatory phosphorylation. Arsenite increased activating phosphorylation at Thr161 and reduced inhibitory phosphorylation at Tyr15 (Figure 4). Arsenite induces BCL-2 phosphorylation prior to apoptosis (Park et al., 2001). Furthermore, CDK1 can phosphorylate BCL-2 in vitro (Furukawa et al., 2000). Arsenite increased phosphorylation at serine 70 of BCL-2 (Figure 4). Phosphorylation of BCL-2 was also demonstrated by the presence of a BCL-2 band with slower mobility in the blot for total BCL-2 (Figure 4).
Figure 4.

Arsenite activates CDK1 and induces BCL-2 phosphorylation. A375 cells were treated with indicated concentrations of NaAsO2 for 24 h and analyzed by western blot for the indicated proteins.
To test if CDK1 inhibition allows for mitotic exit and protects against apoptotic cell death, the detached fraction of cells (mitotic plus apoptotic) from cultures of A375 cells treated with arsenite for 6 h was harvested and re-incubated in either media containing arsenite alone or arsenite plus roscovitine. Roscovitine is an inhibitor of CDK1 (De Azevedo et al., 1997). In lieu of the proteosomal degradation of cyclin B that is required for mitotic exit, inhibition of the cyclin B/CDK1 complex with roscovitine allowed for arsenite-treated cells to escape mitosis. As shown in Figure 5A, roscovitine significantly lowered the mitotic index within 0.5 h. Figure 5B panels 1–3 show photographs of cultures that were re-incubated either without arsenite (5B1), with arsenite (5B2) or with arsenite plus roscovitine (5B3) for 18 h. Arsenite-treated cells (5B2) had round, highly refractile morphology, indicating they were either mitotic or apoptotic. In contrast, cells re-incubated with arsenite and roscovitine (5B3) had the flattened morphology of untreated cells (5B1). Markers of apoptosis were examined to determine the impact of roscovitine on cell death (Figure 5C). Cells re-incubated in arsenite in the presence of Z-VAD-FMK were included as a negative control for apoptosis. At both 6 and 18 h of re-incubation, cyclin B expression was reduced by roscovitine (Figure 5C, lanes 2 and 5 compared to lanes 1, 3, 4, and 6). Roscovitine also reduced phosphorylation of BUBR1 (Figure 5C, lanes 2 and 5 compared to lanes 1, 3, 4, and 6). By 18 h of re-incubation, apoptotic cleavage of both PARP and caspase 3 was observed in cells treated with arsenite alone (Figure 5C, 4th lane) but not in samples re-incubated with arsenite plus either roscovitine or Z-VAD-FMK (Figure 5C, lanes 5 and 6, respectively). Arsenite also induced BCL-2 phosphorylation at both 6 and 18 h of re-incubation (Figure 5C, 1st and 4th lanes). Z-VAD-FMK did not affect BCL-2 phosphorylation (Figure 5C, lanes 3 and 6). In contrast, roscovitine reduced BCL-2 phosphorylation (Figure 5C, lanes 2 and 5).
Figure 5.

CDK1 inhibitor roscovitine promotes mitotic exit and inhibits apoptosis. (A) A375 cells were treated with 5 μM NaAsO2 for 6 h. Detached cells were collected and re-incubated in media containing 5 μM NaAsO2 with and without 20 μM roscovitine. Mitotic index was determined at 0–3 hour of re-incubation. Data are means ± standard deviation of three independent experiments. * p < 0.05 (B1–3) Phase contrast microscopy of A375 cells after 18 h re-incubation either untreated (1) or treated as in (A) in NaAsO2 alone (2) or NaAsO2 plus roscovitine (3) Bar, 50 μm. (C) Cells treated as described in (A) were re-incubated for 6 and 18 h. Cells re-incubated with NaAsO2 and Z-VAD-FMK were also included as negative controls for apoptosis. Samples were collected and analyzed by western blot for indicated proteins.
Our previous work indicated arsenite-resistant SK-Mel-3 and SK-Mel-28 cells failed to arrest in mitosis like arsenite-sensitive A375 and SK-Mel-2 cells, suggesting arsenite sensitivity is related to susceptibility to arsenite-induced mitotic arrest. SK-Mel-3 and SK-Mel-28 cells were found to have reduced expression of BUBR1 (McNeely et al, 2008). To test the impact of spindle checkpoint loss on sensitivity to arsenite, expression of BUBR1 was suppressed using siRNA. A375 cells were transfected with either BUBR1-specific siRNA, non-specific control siRNA (NSC), or mock transfected. BUBR1 expression was suppressed at 0 and 24 h but began to return at 48 h. The impact of BUBR1 suppression on the induction of mitotic arrest by arsenite was then examined (Figure 6B). The mitotic index of A375 cells transfected with NSC siRNA or mock transfected increased to as much as 28% after arsenite exposure, while BUBR1 siRNA-transfected cells had only a modest increase in mitotic index with a peak of 7%. The impact of BUBR1 suppression on cellular sensitivity to arsenite was then tested (Figure 6C). BUBR1 siRNA-transfected A375 cells were significantly less sensitive than NSC- or mock-transfected cultures at concentrations of arsenite ≥ 5 μM. The impact of BUBR1 on the initiation of apoptosis was examined with western blotting (Figure 6D). Apoptotic cleavage of both PARP and caspase 3 was observed in mock and NSC-transfected cells treated with 5 μM arsenite. Suppression of BUBR1 eliminated PARP and caspase 3 cleavage indicating prevention of arsenite-induced apoptosis.
Figure 6.

Suppression of BUBR1 prevents arsenite-induced mitotic arrest in A375 cells. (A) A375 cells were transfected with BUBR1 siRNA, non-specific control siRNA (NSC) or mock-transfected with buffer alone and allowed to recover for 24 h. Suppression of BUBR1 was confirmed via western blot of untreated samples at indicated times after recovery period. (B) Cells treated as in (A) were exposed to indicated concentrations of NaAsO2 for 24 h and harvested for determination of mitotic indices. Data are means ± standard deviation of three independent experiments. * p < 0.05, ** p < 0.01 (C) Cells transfected as in (A) were exposed to indicated concentrations of NaAsO2 for 48 h and assayed for viability. Data are means ± standard deviation of three independent experiments. ** pval < 0.01. (D) Cells transfected as in (A) were treated with 0 or 5 μM NaAsO2 for 24 h and harvested for western blot analysis of the indicated proteins.
Discussion
Our previous studies investigating the mechanism of arsenic-induced cell death in melanoma cell lines indicated that sensitivity of melanoma cell lines to arsenic at clinically relevant concentrations was dependent upon susceptibility to mitotic arrest (McNeely et al, 2008). In the current study, we show that mitotic arrest induced in A375 malignant melanoma cells by arsenite is associated with the phosphorylation of the mitotic checkpoint kinase BUBR1/MAD3. Phosphorylation of BUBR1 occurs during spindle checkpoint activation by mitotic disrupting agents (Rancati et al., 2005). Activation of the spindle checkpoint by arsenite resulted in stabilization of cyclin B and securin, thereby inhibiting mitotic exit. In addition to cyclin B stabilization, arsenite also promoted activating Thr161 phosphorylation of CDK1 and reduced inhibitory phosphorylation at Tyr15. These alterations in CDK1 phosphorylation likely cause increased CDK1 activity that maintains spindle checkpoint activation and further inhibits exit from mitosis (D’Angiolella et al., 2003). To examine if mitotic arrest directly resulted in apoptosis, we tested detached arsenite-treated cells for colocalization of active caspase 3 and the mitotic marker phosphorylated Histone H3. Colocalization was observed, indicating mitotic cells were undergoing apoptosis, consistent with our previous observations of mitotic SV40-transformed human fibroblasts undergoing apoptosis in EM micrographs (States et al., 2002). It should be noted that intense staining for either active caspase 3 or phosphorylated Histone H3 only occurred in detached, arsenite-treated A375 cells without colocalization, whereas cells demonstrating colocalization had a lower intensity staining for both markers. These data suggest colocalization occurs at a transitional point where the phosphorylation of Histone H3 is lost as the apoptotic machinery is activated. Consistent with these data, the percentage of cells exhibiting Histone-H3 phosphorylation in detached, arsenite-treated cells declined prior to the appearance of cells with disintegrated DNA, a late marker of apoptosis. Loss of Histone H3 phosphorylation may have been due to caspase-mediated mitotic exit into a polyploid state, also termed mitotic slippage. BUBR1 as well as BUB1, another checkpoint protein, are targets of caspases (Kim et al., 2005;Baek et al., 2005) and it has been proposed that apoptotic pathways may feed back to the spindle checkpoint, affecting the maintenance of the mitotic checkpoint (Yamada and Gorbsky, 2006). Consequently, there may be indistinct boundaries between mitotic and post-mitotic death as activation of the apoptotic machinery may allow for mitotic exit in dying cells. Inhibition of caspase activity with Z-VAD-FMK may have prevented mitotic slippage which resulted in the observed increase in arsenite-induced mitotic arrest.
Roscovitine-mediated inhibition of CDK1 promoted exit from mitosis and prevented the onset of apoptosis in arsenite-treated A375 cells. This result indicates that continued kinase activity was required for the mitotic arrest, and that the mitotic arrest is required for the onset of apoptosis. Similarly, the kinase inhibitor 2-aminopurine protected mitotic arsenite-treated HeLa S3 cells from apoptosis (Huang et al., 2000). Inhibition of CDK1 also reduced BUBR1 phosphorylation. Reduced BUBR1 phosphorylation may have been due to roscovitine-induced mitotic exit or slippage. Alternatively, a reduction in BUBR1 phosphorylation by CDK1 inhibition with roscovitine suggests that CDK1 may directly or indirectly mediate spindle checkpoint activation through BUBR1-phosphorylation-dependent checkpoint signaling. A link between CDK1 activity and the spindle checkpoint may provide insight into a potential mechanism for the induction of mitotic arrest by arsenite. In the absence of any arsenite-induced changes in mitotic spindle formation, arsenic might induce mitotic arrest by causing the accumulation of cyclin B and sustained activation of CDK1. CDK1 may then impinge upon the spindle checkpoint to maintain arrest in mitosis as observed in another study using nocodazole (D’Angiolella et al., 2003). Consistent with this idea, cyclin B expression is regulated through ubiquitin-dependent proteolytic pathways (Koepp et al., 1999), which are a target of inhibition by arsenite (Klemperer and Pickart, 1989;Berleth et al., 1992).
Increased CDK1 activity has been observed under a variety of apoptotic conditions (Castedo and Kroemer, 2004). Furthermore, inhibition of CDK1 blocks paclitaxel-induced mitotic death (Shen et al., 1998). It is possible that increased activity of CDK1 due to arsenite-induced stabilization of cyclin B and increased CDK1 Tyr15 phosphorylation coupled with decreased Thr161 phosphorylation leads to the initiation of apoptosis. Consistent with this hypothesis, the inhibition of CDK1 with roscovitine prevented apoptosis in the current study. CDK1 may phosphorylate BCL-2 on Ser70 and Ser87, suppressing its anti-apoptotic function (Pathan et al., 2001). Roscovitine reduced BCL-2 phosphorylation in the current study suggesting that BCL-2 is a possible substrate of CDK1. However, results of in vitro tests of direct BCL-2 phosphorylation by CDK1 are equivocal (Furukawa et al., 2000;Vantieghem et al., 2002). It is possible that loss of BCL-2 phosphorylation is due to inhibition of an unidentified BCL-2 kinase by roscovitine directly or indirectly through CDK1 or other mitotic kinases. Deletion of the loop containing Ser70 on BCL-2 prevents paclitaxel-induced apoptosis (Srivastava et al., 1999). Our results suggest the possibility that inappropriate arsenite-induced hyperactivity of CDK1 resulted in apoptosis due to Ser70 phosphorylation of BCL-2. BCL-2 Ser70 phosphorylation may result in apoptosis through the intrinsic pathway (Haldar et al., 1995).
Because BUBR1 expression was a possible determinant of cellular sensitivity to arsenite in our previous study, we determined if BUBR1 suppression in spindle checkpoint-competent A375 cells would impact their sensitivity to arsenite. Consistent with arsenite-induced mitotic arrest-associated apoptosis being dependent on BUBR1, siRNA-mediated suppression of BUBR1 dramatically inhibited the induction of mitotic arrest by arsenite. Furthermore, suppression of BUBR1 also reduced the sensitivity of A375 cells to arsenite and abrogated 5 μM arsenite-induced apoptosis. Suppression of either BUBR1 or MAD2 also inhibited arsenite-induced mitotic death in CGL-2 cells (Yih et al., 2006).
In summary, the current study demonstrates that the mechanism of apoptosis initiated by pharmacological concentrations of arsenite involves induction of spindle checkpoint-dependent mitotic arrest. Consequently, a weakened spindle checkpoint likely confers resistance to arsenic and other antimitotic drugs. The development of methods for assessing spindle checkpoint function in cancer cells will aid in predicting which cancers will respond to arsenic monotherapy. Future studies aimed as elucidating the pathway through which arsenic activates the spindle checkpoint may identify new molecular targets and provide valuable information in the design of new antimitotic drugs.
Supplementary Material
Acknowledgments
This work was supported in part by USPHS grants R01ES011314, P30ES014443, T32ES011564 and F30ES013372. Portions of this work constituted partial fulfillment for the Ph.D. in Pharmacology and Toxicology awarded to Samuel C. McNeely from the University of Louisville.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Baek KH, Shin HJ, Jeong SJ, Park JW, McKeon F, Lee CW, Kim CM. Caspases-dependent cleavage of mitotic checkpoint proteins in response to microtubule inhibitor. Oncol Res. 2005;15:161–168. doi: 10.3727/096504005776367906. [DOI] [PubMed] [Google Scholar]
- Berleth ES, Kasperek EM, Grill SP, Braunscheidel JA, Graziani LA, Pickart CM. Inhibition of ubiquitin-protein ligase (E3) by mono- and bifunctional phenylarsenoxides. Evidence for essential vicinal thiols and a proximal nucleophile. J Biol Chem. 1992;267:16403–16411. [PubMed] [Google Scholar]
- Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit Rev Oncol Hematol. 2002;42:5–24. doi: 10.1016/s1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- Cai X, Yu Y, Huang Y, Zhang L, Jia PM, Zhao Q, Chen Z, Tong JH, Dai W, Chen GQ. Arsenic trioxide-induced mitotic arrest and apoptosis in acute promyelocytic leukemia cells. Leukemia. 2003;17:1333–1337. doi: 10.1038/sj.leu.2402983. [DOI] [PubMed] [Google Scholar]
- Castedo M, Kroemer G. Mitotic catastrophe: a special case of apoptosis. J Soc Biol. 2004;198:97–103. [PubMed] [Google Scholar]
- Chang DC, Xu N, Luo KQ. Degradation of cyclin B is required for the onset of anaphase in Mammalian cells. J Biol Chem. 2003;278:37865–37873. doi: 10.1074/jbc.M306376200. [DOI] [PubMed] [Google Scholar]
- Chow SK, Chan JY, Fung KP. Inhibition of cell proliferation and the action mechanisms of arsenic trioxide (As2O3) on human breast cancer cells. J Cell Biochem. 2004;93:173–187. doi: 10.1002/jcb.20102. [DOI] [PubMed] [Google Scholar]
- Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–885. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Mari C, Nocera D, Rametti L, Grieco D. The spindle checkpoint requires cyclin-dependent kinase activity. Genes Dev. 2003;17:2520–2525. doi: 10.1101/gad.267603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M, Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem. 1997;243:518–526. doi: 10.1111/j.1432-1033.1997.0518a.x. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Iwase S, Kikuchi J, Terui Y, Nakamura M, Yamada H, Kano Y, Matsuda M. Phosphorylation of Bcl-2 protein by CDC2 kinase during G2/M phases and its role in cell cycle regulation. J Biol Chem. 2000;275:21661–21667. doi: 10.1074/jbc.M906893199. [DOI] [PubMed] [Google Scholar]
- Gu L, Zheng H, Murray SA, Ying H, Jim Xiao ZX. Deregulation of Cdc2 kinase induces caspase-3 activation and apoptosis. Biochem Biophys Res Commun. 2003;302:384–391. doi: 10.1016/s0006-291x(03)00189-x. [DOI] [PubMed] [Google Scholar]
- Haldar S, Jena N, Croce CM. Inactivation of Bcl-2 by phosphorylation. Proc Natl Acad Sci USA. 1995;92:4507–4511. doi: 10.1073/pnas.92.10.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halicka HD, Smolewski P, Darzynkiewicz Z, Dai W, Traganos F. Arsenic trioxide arrests cells early in mitosis leading to apoptosis. Cell Cycle. 2002;1:201–209. [PubMed] [Google Scholar]
- Hernandez-Zavala A, Cordova E, Del Razo LM, Cebrian ME, Garrido E. Effects of arsenite on cell cycle progression in a human bladder cancer cell line. Toxicology. 2005;207:49–57. doi: 10.1016/j.tox.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Hoffman RD, Lane MD. Iodophenylarsine oxide and arsenical affinity chromatography: new probes for dithiol proteins. Application to tubulins and to components of the insulin receptor-glucose transporter signal transduction pathway. J Biol Chem. 1992;267:14005–14011. [PubMed] [Google Scholar]
- Huang S, Huang CF, Lee T. Induction of mitosis-mediated apoptosis by sodium arsenite in HeLa S3 cells. Biochem Pharmacol. 2000;60:771–780. doi: 10.1016/s0006-2952(00)00397-x. [DOI] [PubMed] [Google Scholar]
- Huang SC, Lee TC. Arsenite inhibits mitotic division and perturbs spindle dynamics in HeLa S3 cells. Carcinogenesis. 1998;19:889–896. doi: 10.1093/carcin/19.5.889. [DOI] [PubMed] [Google Scholar]
- Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–825. [PubMed] [Google Scholar]
- Kapahi P, Takahashi T, Natoli G, Adams SR, Chen Y, Tsien RY, Karin M. Inhibition of NF-kappa B activation by arsenite through reaction with a critical cysteine in the activation loop of Ikappa B kinase. J Biol Chem. 2000;275:36062–36066. doi: 10.1074/jbc.M007204200. [DOI] [PubMed] [Google Scholar]
- Kessel M, Liu SX, Xu A, Santella R, Hei TK. Arsenic induces oxidative DNA damage in mammalian cells. Mol Cell Biochem. 2002:234–235. 301–308. [PubMed] [Google Scholar]
- Kim M, Murphy K, Liu F, Parker SE, Dowling ML, Baff W, Kao GD. Caspase-mediated specific cleavage of BubR1 is a determinant of mitotic progression. Mol Cell Biol. 2005;25:9232–9248. doi: 10.1128/MCB.25.21.9232-9248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemperer NS, Pickart CM. Arsenite inhibits two steps in the ubiquitin-dependent proteolytic pathway. J Biol Chem. 1989;264:19245–19252. [PubMed] [Google Scholar]
- Kligerman AD, Doerr CL, Tennant AH. Oxidation and methylation status determine the effects of arsenic on the mitotic apparatus. Mol Cell Biochem. 2005;279:113–121. doi: 10.1007/s11010-005-8283-3. [DOI] [PubMed] [Google Scholar]
- Koepp DM, Harper JW, Elledge SJ. How the cyclin became a cyclin: regulated proteolysis in the cell cycle. Cell. 1999;97:431–434. doi: 10.1016/s0092-8674(00)80753-9. [DOI] [PubMed] [Google Scholar]
- Larochette N, Decaudin D, Jacotot E, Brenner C, Marzo I, Susin SA, Zamzami N, Xie Z, Reed J, Kroemer G. Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp Cell Res. 1999;249:413–421. doi: 10.1006/excr.1999.4519. [DOI] [PubMed] [Google Scholar]
- Lau AT, Li M, Xie R, He QY, Chiu JF. Opposed arsenite-induced signaling pathways promote cell proliferation or apoptosis in cultured lung cells. Carcinogenesis. 2004;25:21–28. doi: 10.1093/carcin/bgg179. [DOI] [PubMed] [Google Scholar]
- Li YM, Broome JD. Arsenic targets tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res. 1999;59:776–780. [PubMed] [Google Scholar]
- Ling YH, Jiang JD, Holland JF, Perez-Soler R. Arsenic trioxide produces polymerization of microtubules and mitotic arrest before apoptosis in human tumor cell lines. Mol Pharmacol. 2002;62:529–538. doi: 10.1124/mol.62.3.529. [DOI] [PubMed] [Google Scholar]
- McCollum G, Keng PC, States JC, McCabe MJ. Arsenite delays progression through each cell cycle phase and induces apoptosis following G2/M arrest in U937 myeloid leukemia cells. J Pharmacol Exp Ther. 2005;313:877–887. doi: 10.1124/jpet.104.080713. [DOI] [PubMed] [Google Scholar]
- McNeely SC, Belshoff AC, Taylor BF, Fan TW, McCabe MJ, Pinhas AR, States JC. Sensitivity to sodium arsenite in human melanoma cells depends upon susceptibility to arsenite-induced mitotic arrest. Toxicol Appl Pharmacol. 2008 doi: 10.1016/j.taap.2008.01.020. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- Park JW, Choi YJ, Jang MA, Baek SH, Lim JH, Passaniti T, Kwon TK. Arsenic trioxide induces G2/M growth arrest and apoptosis after caspase-3 activation and bcl-2 phosphorylation in promonocytic U937 cells. Biochem Biophys Res Commun. 2001;286:726–734. doi: 10.1006/bbrc.2001.5416. [DOI] [PubMed] [Google Scholar]
- Pathan N, ime-Sempe C, Kitada S, Haldar S, Reed JC. Microtubule-targeting drugs induce Bcl-2 phosphorylation and association with Pin1. Neoplasia. 2001;3:70–79. doi: 10.1038/sj.neo.7900131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancati G, Crispo V, Lucchini G, Piatti S. Mad3/BubR1 phosphorylation during spindle checkpoint activation depends on both Polo and Aurora kinases in budding yeast. Cell Cycle. 2005;4:972–980. doi: 10.4161/cc.4.7.1829. [DOI] [PubMed] [Google Scholar]
- Samikkannu T, Chen CH, Yih LH, Wang AS, Lin SY, Chen TC, Jan KY. Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chem Res Toxicol. 2003;16:409–414. doi: 10.1021/tx025615j. [DOI] [PubMed] [Google Scholar]
- Shao W, Fanelli M, Ferrara FF, Riccioni R, Rosenauer A, Davison K, Lamph WW, Waxman S, Pelicci PG, Lo CF, Avvisati G, Testa U, Peschle C, Gambacorti-Passerini C, Nervi C, Miller WH., Jr Arsenic trioxide as an inducer of apoptosis and loss of PML/RAR alpha protein in acute promyelocytic leukemia cells. J Natl Cancer Inst. 1998;90:124–133. doi: 10.1093/jnci/90.2.124. [DOI] [PubMed] [Google Scholar]
- Shen SC, Huang TS, Jee SH, Kuo ML. Taxol-induced p34cdc2 kinase activation and apoptosis inhibited by 12-O-tetradecanoylphorbol-13-acetate in human breast MCF-7 carcinoma cells. Cell Growth Differ. 1998;9:23–29. [PubMed] [Google Scholar]
- Sironi L, Melixetian M, Faretta M, Prosperini E, Helin K, Musacchio A. Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J. 2001;20:6371–6382. doi: 10.1093/emboj/20.22.6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci USA. 1999;96:3775–3780. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- States JC, Reiners JJ, Jr, Pounds JG, Kaplan DJ, Beauerle BD, McNeely SC, Mathieu P, McCabe MJ., Jr Arsenite disrupts mitosis and induces apoptosis in SV40-transformed human skin fibroblasts. Toxicol Appl Pharmacol. 2002;180:83–91. doi: 10.1006/taap.2002.9376. [DOI] [PubMed] [Google Scholar]
- Tan AL, Rida PC, Surana U. Essential tension and constructive destruction: the spindle checkpoint and its regulatory links with mitotic exit. Biochem J. 2005;386:1–13. doi: 10.1042/BJ20041415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BF, McNeely SC, Miller HL, Lehmann GM, McCabe MJ, Jr, States JC. p53 suppression of arsenite-induced mitotic catastrophe is mediated by p21CIP1/WAF1. J Pharmacol Exp Ther. 2006;318:142–151. doi: 10.1124/jpet.106.103077. [DOI] [PubMed] [Google Scholar]
- Taylor BF, McNeely SC, Miller HL, States JC. Arsenite-induced mitotic death involves stress response and is independent of tubulin polymerization. Toxicol Appl Pharmacol. 2008 doi: 10.1016/j.taap.2008.02.030. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantieghem A, Xu Y, Assefa Z, Piette J, Vandenheede JR, Merlevede W, De Witte PA, Agostinis P. Phosphorylation of Bcl-2 in G2/M phase-arrested cells following photodynamic therapy with hypericin involves a CDK1-mediated signal and delays the onset of apoptosis. J Biol Chem. 2002;277:37718–37731. doi: 10.1074/jbc.M204348200. [DOI] [PubMed] [Google Scholar]
- Wassmann K, Benezra R. Mad2 transiently associates with an APC/p55Cdc complex during mitosis. Proc Natl Acad Sci USA. 1998;95:11193–11198. doi: 10.1073/pnas.95.19.11193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Lan Z, Li W, Wu S, Weinstein J, Sakamoto KM, Dai W. p55CDC/hCDC20 is associated with BUBR1 and may be a downstream target of the spindle checkpoint kinase. Oncogene. 2000;19:4557–4562. doi: 10.1038/sj.onc.1203803. [DOI] [PubMed] [Google Scholar]
- Yamada HY, Gorbsky GJ. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol Cancer Ther. 2006;5:2963–2969. doi: 10.1158/1535-7163.MCT-06-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yih LH, Tseng YY, Wu YC, Lee TC. Induction of centrosome amplification during arsenite-induced mitotic arrest in CGL-2 cells. Cancer Res. 2006;66:2098–2106. doi: 10.1158/0008-5472.CAN-05-2308. [DOI] [PubMed] [Google Scholar]
- Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell. 2007;27:3–16. doi: 10.1016/j.molcel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Zieve GW, Turnbull D, Mullins JM, McIntosh JR. Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor nocodazole. Nocodazole accumulated mitotic cells. Exp Cell Res. 1980;126:397–405. doi: 10.1016/0014-4827(80)90279-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.