

SUMMARY
Li-Fraumeni syndrome (LFS) is a highly penetrant, autosomal dominant, human familial cancer predisposition. Although a key role for the tumor suppressor p53 has been implicated in LFS, the genetic and cellular mechanisms underpinning this disease remain unknown. Therefore, modeling LFS in a vertebrate system that is accessible to both large-scale genetic screens and in vivo cell biological studies will facilitate the in vivo dissection of disease mechanisms, help identify candidate genes, and spur the discovery of therapeutic compounds. Here, we describe a forward genetic screen in zebrafish embryos that was used to identify LFS candidate genes, which yielded a p53 mutant (p53I166T) that as an adult develops tumors, predominantly sarcomas, with 100% penetrance. As in humans with LFS, tumors arise in heterozygotes and display loss of heterozygosity (LOH). This report of LOH indicates that Knudson’s two-hit hypothesis, a hallmark of human autosomal dominant cancer syndromes, can be modeled in zebrafish. Furthermore, as with some LFS mutations, the zebrafish p53I166T allele is a loss-of-function allele with dominant-negative activity in vivo. Additionally, we demonstrate that the p53 regulatory pathway, including Mdm2 regulation, is evolutionarily conserved in zebrafish, providing a bona fide biological context in which to systematically uncover novel modifier genes and therapeutic agents for human LFS.
INTRODUCTION
Familial cancers, although rare in the general population, represent important starting points to understanding the genetic components and mechanisms of cancer. Much of our knowledge of key cancer-causing genes has been gained by studying familial cancers, such as RB1 in retinoblastoma, APC in familial adenomous polyposis (FAP), WT1 in Wilms tumor, VHL in von Hippel-Lindau disease, and others (Field et al., 2007; Ganjavi and Malkin, 2002). These familial syndromes often model sporadic cancer in the general population and help in devising strategies for cancer treatment.
Li-Fraumeni syndrome (LFS) is an autosomal dominant, highly penetrant cancer predisposition that presents with a wide variety of tumor types at an early age, with sarcomas being the hallmarks of the disease (Kleihues et al., 1997; Varley, 2003). The criteria for diagnosis of LFS are that the presenting individual has a sarcoma before the age of 45 and has two first-degree relatives who either developed cancer before the age of 45 or who developed a sarcoma at any age. Li-Fraumeni-like syndrome (LFL) and incomplete LFS (LFI) are similar to LFS, but with slightly different diagnostic criteria. Germline mutations in p53 have been identified in 50–70% of LFS families, 40% of LFL families and 6% of LFI families (Birch et al., 1994; Chompret et al., 2000; Frebourg et al., 1995; Li and Fraumeni, 1969; MacGeoch et al., 1995). Checkpoint kinase 2 (Chk2, also known as Chek2) had been implicated in 5% of LFS families (Bell et al., 1999); however, subsequent patient analysis has determined that Chk2 is not the cause of LFS (Evans et al., 2008). In addition, Chk2−/− mice do not develop tumors (Takai et al., 2002). Nonetheless, the genes responsible for the remaining 25–45% of LFS families, 60% of LFL families and 94% of LFI families are unknown. In four families in which a p53 mutation was excluded, linkage mapping implicated chromosome 1q23 and at least one additional locus (Bachinski et al., 2005), indicating that multiple loci contribute to LFS. However, owing to the small size of the families, mapping to a single gene on 1q23 and other loci has been difficult.
Among LFS families there appears to be a genotype-phenotype correlation, with a subset of missense p53 alleles conferring more severe cancer predisposition (Birch et al., 1998). The idea that specific mutant p53 proteins have increased tumorigenic potential that is not found in null alleles is supported by the observation that a disproportionate number of missense, as compared with nonsense, mutations are found in Li-Fraumeni families. In vitro studies have demonstrated that mutant p53 proteins have loss-of-function (LOF) activity (Sigal and Rotter, 2000), dominant-negative (DN) activity (Milner and Medcalf, 1991) and/or gain-of-function (GOF) activity (Dittmer et al., 1993), perhaps conferring advantages for tumor progression (Cadwell and Zambetti, 2001; Varley, 2003).
In addition to LFS, p53 is mutated in 50–70% of sporadic cancers, making it one of the most widely implicated genes in cancer biology (Cadwell and Zambetti, 2001). The tumor suppressor p53 is a transcription factor that is known to induce many targets following DNA damage. The outcome of p53 activation is predominantly apoptosis (through Puma, Noxa and Bax) and cell cycle arrest (through p21 and cyclin G). Both of these functions have been shown to be important in cancer prevention.
Whereas p53 null mice display some of the dominant phenotypes seen in LFS, such as autosomal dominance and loss of heterozygosity (LOH) (Clarke et al., 1993; Donehower et al., 1992; Jacks et al., 1994), mice with missense mutations that are analogous to those found in LFS (LFS mice) are a better model, in that they display DN and GOF activity (Lang et al., 2004; Olive et al., 2004). However, the difficulty of forward genetic screens in mice and the challenges of genetic mapping in humans emphasize the importance of designing genetic approaches in other organisms to unravel the p53 pathway and identify new Li-Fraumeni cancer genes.
p53-related genes have been identified in a variety of genetically useful invertebrates, including D. melanogaster (Brodsky et al., 2000; Ollmann et al., 2000), and C. elegans (Derry et al., 2001; Schumacher et al., 2001). However, these model systems lack the p53-like genes p63 and p73, as well as the p53 regulators Mdm2 and Mdm4 (Lu and Abrams, 2006). In Drosophila, the p53 pathway appears to be simplified; DNA damage activates Chk2, which phosphorylates and activates p53 to transcribe apoptotic targets genes such as reaper and hid (also known as Wrinkled), thereby resulting in cell death. Unlike the mammalian system, Drosophila p53 does not induce growth arrest, and phosphorylation does not stabilize the p53 protein (Lu and Abrams, 2006). This lack of modulated p53 protein stability concurs with the lack of the negative regulators Mdm2 or Mdm4 in Drosophila. Therefore, although genetically powerful, these invertebrate systems do not recapitulate the complexity of the p53 regulatory pathways in mammalian systems.
Zebrafish are becoming a powerful tool for cancer research (Feitsma and Cuppen, 2008; Goessling et al., 2007), in part owing to the ease of in vivo manipulation and the ability to perform both genetic and in vivo drug screens. Zebrafish have p53, p63 and p73 genes, as well as the regulatory mdm2 and mdm4 genes (Lu and Abrams, 2006). A zebrafish p53M214K mutant was identified in a reverse genetics screen (Berghmans et al., 2005); this mutant develops tumors, suggesting that zebrafish might be a good model for cancer studies. However, these tumors were seen with low penetrance and no tumors were found in heterozygous fish, indicating that this mutant line does not recapitulate the LFS phenotype.
In this study, we characterized a p53-dependent, ionizing (gamma) irradiation (IR) sensitivity phenotype in zebrafish embryos and used this embryonic phenotype to genetically screen for novel mutations in LFS genes that give rise to tumors in adults. As proof of principle, this screen identified an isoleucine (I) to threonine (T) mutation at codon 166, analogous to codon 195 in humans, in the highly conserved DNA-binding domain of p53. The resulting p53I166T mutant displayed highly penetrant tumorigenesis in both the heterozygous and homozygous states, and displayed a high rate of LOH, demonstrating conservation of this fundamental mechanism that contributes to human cancer. This mutant has the dominant phenotypes of human LFS: sarcomas, autosomal dominant tumor formation and DN functional activity. Utilizing the Mdm2 knockdown lethal phenotype, we show that this mutant p53 is a LOF allele with DN activities. This LFS zebrafish mutant will serve to uncover novel modifier genes and drugs that are capable of modulating the evolutionarily conserved LFS pathway.
RESULTS
Conservation of the p53 pathway in zebrafish
To determine whether zebrafish p53 can induce similar target genes to those induced by p53 in the mammalian system, we tested whether the zebrafish homologues of the genes encoding p21, cyclin G, Gadd45 and Mdm2 are induced following IR. Northern blot analysis of total RNA collected at 6 hours post-irradiation (hpi) from 36 hours post-fertilization (hpf) embryos treated with 100 gray (Gy) of IR indicated that all of these target genes were indeed induced (supplementary material Fig. S1A). To determine whether this induction was p53 dependent, a p53 splice-blocking morpholino oligonucleotide (MO) was utilized to knockdown p53 expression. Real time, reverse transcription (RT)-PCR and western blot analyses revealed that the p53 MO effectively knocked down wild-type (WT) p53 mRNA (data not shown) and protein levels. Embryos injected with the p53 MO were refractory to IR-dependent induction, indicating that these target genes are bona fide targets of the p53 pathway in zebrafish (supplementary material Fig. S1A). The induction kinetics of p53 target genes with a low dose of 10 Gy (supplementary material Fig. S1B) or a high dose of 100 Gy (supplementary material Fig. S1C) were assayed. At either dose, expression of p53 target genes was induced within 2 hours, with higher induction levels in the 100 Gy samples, and maintained through to 24 hpi. To determine the optimal dose for transcriptional induction, analysis was performed on embryos at 36 hpf and 6 hpi at different IR doses (supplementary material Fig. S1D). For p21 and Mdm2, significant induction was detected at 20 Gy, however maximal induction was at 60 Gy.
Apoptosis is one of the key mechanisms by which p53 prevents cancer. In vivo studies in the mouse have shown that p53 null embryos are deficient in IR-induced apoptosis in neural tissues (Lee et al., 2001). To determine whether similar apoptosis occurs in zebrafish, we irradiated embryos and then used Acridine Orange (AO) to stain apoptotic cells. Initially embryos were treated with 100 Gy of IR at 30 hpf, and strong apoptotic staining was found in the neural tube (NT) at 6 hpi. Doses of IR and the time post-IR were optimized for apoptosis detection. Very little apoptosis was seen in untreated embryos at 32, 36 and 50 hpf (analogous to 2, 6 and 20 hpi) (supplementary material Fig. S2A-C) or at 2 hpi in embryos treated with 5, 30 or 100 Gy of IR (supplementary material Fig. S2A,D,G,J). IR with 5 Gy resulted in low levels of apoptosis at 6 and 20 hpi (supplementary material Fig. S2E,F). By contrast, 30 and 100 Gy produced very strong AO staining at 6 hpi that persisted through to 20 hpi (supplementary material Fig. S2H,I,K,L). Interestingly, a curled-up tail phenotype was observed in 100% (n=50) of embryos treated with 100 Gy of IR (supplementary material Fig. S2L, inset) and 32% (n=50) of embryos treated with 30 Gy of IR (supplementary material Fig. S2I, inset).
To determine whether apoptosis in the NT and the curled tail phenotype are p53 dependent, p53 MO-injected embryos that were irradiated at 30 hpf with 30 Gy or 100 Gy were stained with AO at 6 hpi, or scored for the curly tail phenotype at 20 hpi. Both phenotypes were rescued in p53 MO-injected embryos (supplementary material Fig. S3E,H). In mammalian tissue culture, p53-dependent apoptosis can be inhibited by overexpression of Bcl2 (Cadwell and Zambetti, 2001). To test whether p53-dependent apoptosis can be inhibited by Bcl2 overexpression in zebrafish, embryos were injected with bcl2 mRNA at the 1–2-cell stage, and then treated with 30 Gy or 100 Gy of IR at 30 hpf. Unirradiated bcl2-injected embryos had no gross morphological phenotypes (supplementary material Fig. S3C), indicating that Bcl2 overexpression does not cause development defects. Embryos injected with bcl2 mRNA were resistant to the apoptosis normally induced by 30 Gy of IR (supplementary material Fig. S3F) and the curly tail phenotype that was invariably induced by 100 Gy (supplementary material Fig. S3I). This indicates that the curly tail phenotype is a useful indicator of IR-induced p53-dependent apoptosis.
Genetic screen for Li-Fraumeni syndrome in zebrafish
Based on the observation made above, we designed an F3 recessive genetic screen in zebrafish to identify components of the LFS/p53 pathway, using the phenotypic response in embryos to IR as an assay (Fig. 1A). Ionizing radiation mimics the genotoxic stress to which the p53 pathway responds during tumorigenesis. Thus, mutations that alter p53-dependent response pathways in embryos might also alter p53-dependent pathways leading to tumorigenesis in adults. The resulting screen scored for loss of NT apoptosis in zebrafish embryos irradiated at 30 hpf with 30 Gy and assessed at 8 hpi. Overall, 1800 F3 clutches from 300 F2 (F1×F1) families, or approximately 489 genomic equivalents, were screened, yielding one mutant with an IR-resistant phenotype (Fig. 1A) that segregated with mendelian inheritance. Homozygous mutants were completely viable and fertile.
Fig. 1.

Identification of the p53I166T mutation. (A) F3 screen for LFS mutants. Males were treated with the mutagen N-ethyl-N-nitrosourea (ENU) (G0) and bred to generate F1 fish, each of which is genetically unique and heterozygous for many mutations. To propagate heterozygotes, F1 fish were intercrossed to generate F2 fish, which are also heterozygous for a particular mutation. F2 fish were intercrossed to generate F3 offspring, a quarter of which are homozygous for a particular mutation. These F3 clutches were irradiated at 30 hpf and scored for AO staining (50 μg/ml) at 8 hpi. Approximately 1800 F3 clutches from 300 F2 (F1×F1) families were screened, yielding a family in which a quarter of the embryos (n=33/130) displayed resistance to IR-induced apoptosis. (B) Sequence analyses of the genomic PCR products from two WT and one mutant embryo from F2 heterozygous crosses. Mutants were homozygous for a thymine to cytosine transition in codon 166. (C) Amino acid sequence comparison of the human, mouse, chicken and zebrafish around codon 166 in zebrafish (codon 195 in human; * marks the mutated Ile codon). (D) Melting curve analysis of PCR products from p53+/+ (blue), p53I166T/+ (black) and p53I166T/I166T (red) embryos.
Sequencing of p53 cDNA and genomic DNA from mutant embryos revealed a thymine (T) to cytosine (C) transition at codon 166 (Fig. 1B), causing an isoleucine (I) to threonine (T) change in the DNA-binding domain of p53. Among vertebrates, this mutation occurs at a conserved amino acid position in p53 (Fig. 1C) and the exact mutation, in the analogous codon 195 (Fig. 1C), has been found in many human sporadic cancers. We employed a modified PCR-based high resolution melting analysis (HRMA) to genotype embryos (Fig. 1D) and adult zebrafish (Parant et al., 2009).
The p53I166T protein is stabilized following DNA damage but does not transactivate p53 target genes
Stabilization of the p53 protein is important in the regulation of p53 activity in mammals. In wild-type zebrafish, a strong induction of p53 protein accumulation was observed at 2 hpi, which continued through until 20 hpi (Fig. 2A). Thus, p53 protein stabilization in response to IR-induced DNA damage is conserved in zebrafish. Therefore, we tested whether the p53I166T mutant protein was stabilized with kinetics similar to those of the wild-type p53 protein. Strikingly, the mutant p53 protein was induced by IR and accumulated with similar kinetics as the wild-type p53 protein (Fig. 2A), suggesting that the mutant phenotype is not the result of altered p53 protein stabilization.
Fig. 2.
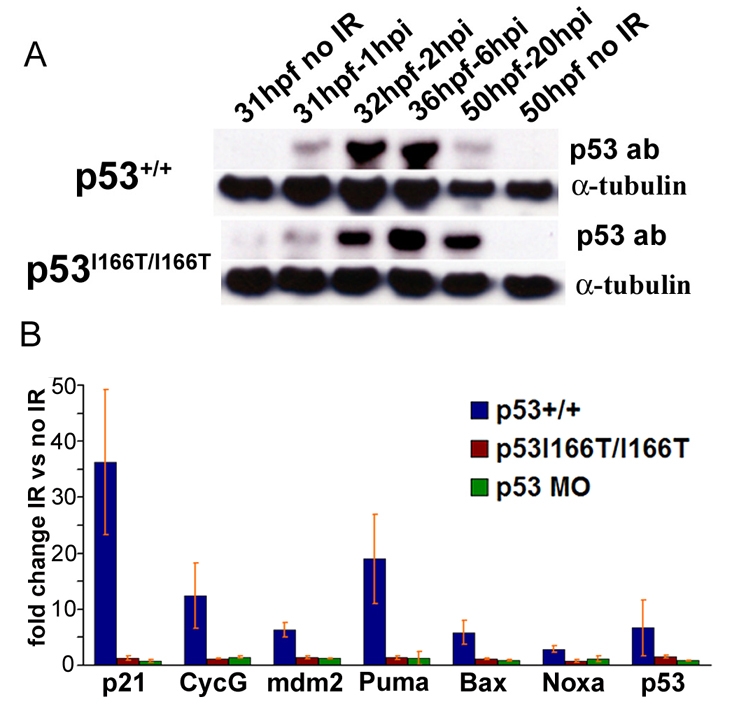
p53I166T protein has defects in transcriptional transactivation but not protein accumulation. (A) Western blots of protein extracts that were collected from WT and p53I166T/I166T mutant embryos at 1, 2, 6 and 20 hours following 0 Gy (mock IR) or 30 Gy of IR at 30 hpf. Blots were probed with p53 and α-tubulin antibodies. Note that the p53+/+ 50 hpf 20 hpi lane is underloaded. (B) RT-PCR assays for p21, cyclin G (CycG), mdm2, p53, puma, noxa and bax were performed in triplicate on WT, p53I166T/I166T and p53 MO-injected embryos. The fold induction reflects the comparison between IR-induced samples and matched non-IR samples. Error bars show the standard deviation (S.D.).
To determine whether the mutant protein could induce p53 target genes, we compared the expression of the cell cycle genes p21 (also known as cdkn1a) and cyclin G (also known as ccng1 and ccng2), the p53 regulatory gene mdm2, and the apoptotic genes bax (also known as baxa and baxb), puma (also known as pma) and noxa (also known as pmaip1) in IR conditions with non-IR conditions, in p53+/+, p53I166T/I166T and p53 MO-injected (p53-morphant) embryos (Fig. 2B). The p53 target genes were induced in wild-type samples following IR, but were not induced in p53 mutant or p53 morphants. The p53 morphant results indicate that induction of this battery of genes is dependent on the presence of functional p53 protein. Although mutant p53 protein is stabilized with normal kinetics following IR, it has lost the functional ability to induce transcription of p53 target genes in the cell cycle, autoregulatory and apoptotic pathways.
p53I166T/I166T mutants have high rates of tumorigenesis
To determine whether, similar to Li-Fraumeni patients, p53I166T mutant fish are susceptible to increased tumor incidence and penetrance, genotyped cohorts of p53I166T/I166T adult fish (n=144) were raised and compared with p53+/+ adult siblings (n=60) that had been generated from a heterozygous-heterozygous mutant cross. The first incidence of tumor formation in p53I166T/I166T fish occurred at 269 days (8.8 months), and the last occurred at 679 days (22.3 months), with a 50% survival of 465 days (15.2 months) (Fig. 3A), indicating that the p53I166T allele results in a fully penetrant tumor phenotype. One advantage of this zebrafish model is that every fish developed a tumor that was readily observable in the tank. Tumors predominantly occurred in three locations: the abdomen (n=64/134), eye (n=26/134) or flank (n=22/134), with less frequent occurrences in the gills (n=4/134), rectum (n=6/134) and skin (n=2/134). There was no difference in the onset of abdominal and flank tumors, but there was a significantly later onset of eye tumors (Fig. 3B).
Fig. 3.
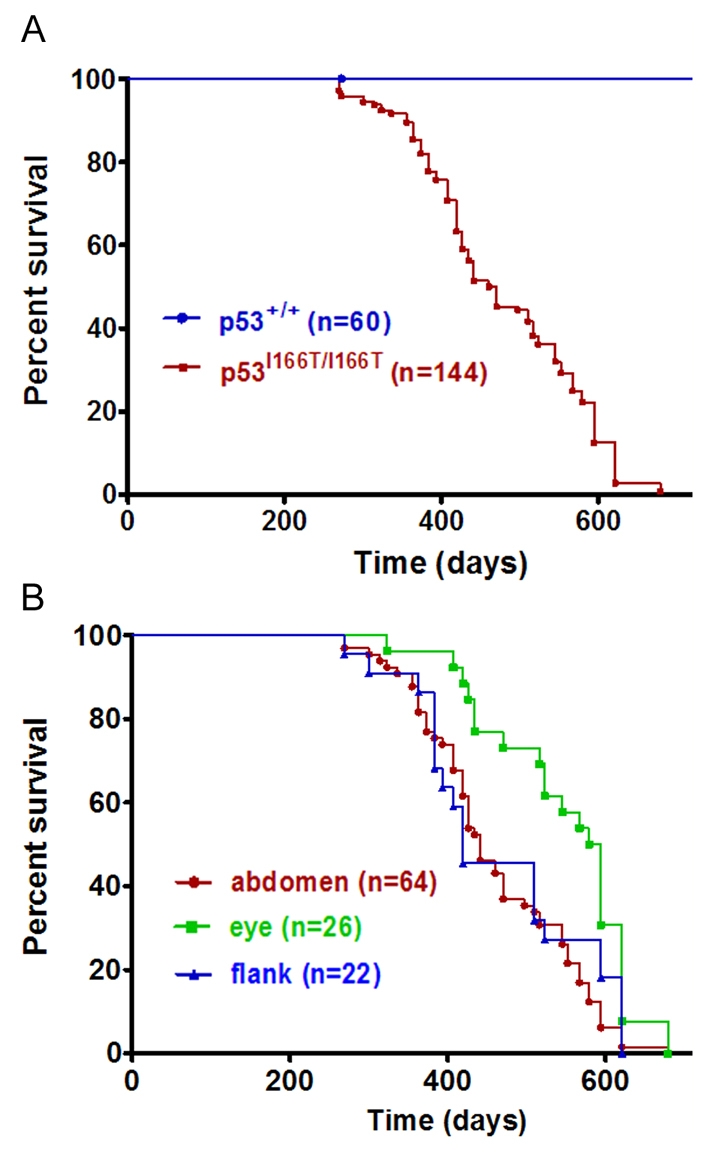
p53I166T/I166T survival curves. (A) Using Kaplan-Meier analysis, p53I166T/I166T adult fish (n=144) had a significantly increased tumor incidence (P<0.0001) compared with p53+/+ fish (n=60). The first tumor was identified at 269 days (8.8 months) of age, and the last tumors were identified at 679 days (22.3 months), with a 50% incidence at 465 days (15.2 months). (B) Tumors predominantly occurred in the abdomen, eye or flank. There was a significant difference between the later onset of eye tumor development (P<0.0086) versus flank or abdomen tumor development.
Histological analyses of p53I166T/I166T tumors from the abdomen (Fig. 4A,E,I), eye (Fig. 4B,F,J), flank (Fig. 4C,G,K) or gills (Fig. 4D,H,L) indicated that they were predominantly spindle cell tumors, most often a subclass of soft tissue sarcomas (STS) (n=27/29), similar to the majority of sarcomas in LFS (Varley, 2003). To give a definitive subtype diagnosis would require additional immunohistochemical evidence; however, many of the reagents used for immunohistochemistry in mammals do not appear to react appropriately in zebrafish. For example, there was no evidence of staining in the neoplastic cells for either actin (a muscle marker) or keratin (a marker of epithelial differentiation), suggesting that the malignant neoplasms do not appear to be carcinomas, nor do they show differentiation towards sarcomas with muscle differentiation (rhabdomyosarcoma and leiomyosarcoma). The tumors did not stain with an anti-S-100 protein (for nerve sheaths) or with protein gene product (PGP) 9.5 (used in human tumor histopathology to mark peripheral nerves); thus, there is not sufficient evidence that these lesions are differentiating towards malignant peripheral nerve sheath tumors (MPNST). Other zebrafish research groups have named these tumors zMPNSTs, a subclass of STS (Berghmans et al., 2005). However, we were reluctant to make this further distinction owing to the lack of reactive diagnostic antibodies available for the zebrafish and because these tumors might include synovial sarcomas, fibrosarcomas, or other STSs.
Fig. 4.

Gross and histological analysis of tumors. (A-D) p53I166T/I166T fish with abdominal (A), eye (B), flank (C) and gill (D) tumors. Sections of abdominal (E,I), eye (F,J), flank (G,K) and gill (H,L) tumors stained with hematoxylin and eosin (H&E), shown at low magnification (E-H) and high magnification (I-L). Histologically, seven of the nine abdominal tumors, all ten eye tumors, all seven flank tumors, and both gill tumors (in total, n=27/29 tumors) were spindle cell sarcomas. Arrows indicate tumors.
p53I166T/+ heterozygotes are tumor prone and exhibit LOH
To determine whether the p53I166T/+ heterozygous mutants develop tumors, cohorts of heterozygous (n=190) and wild-type (n=60) siblings were monitored for tumor burden. Similar to Li-Fraumeni patients, heterozygous mutant zebrafish developed tumors at significantly increased rates compared with wild-type siblings (Fig. 5A). As in the p53I166T/I166T homozygotes, the tumors in the p53I166T/+ heterozygotes were predominantly in the abdomen (n=15/33), eye (n=2/33), flank (n=4/33), gills (n=3/33), rectum (n=5/33) and skin (n=1/33).
Fig. 5.
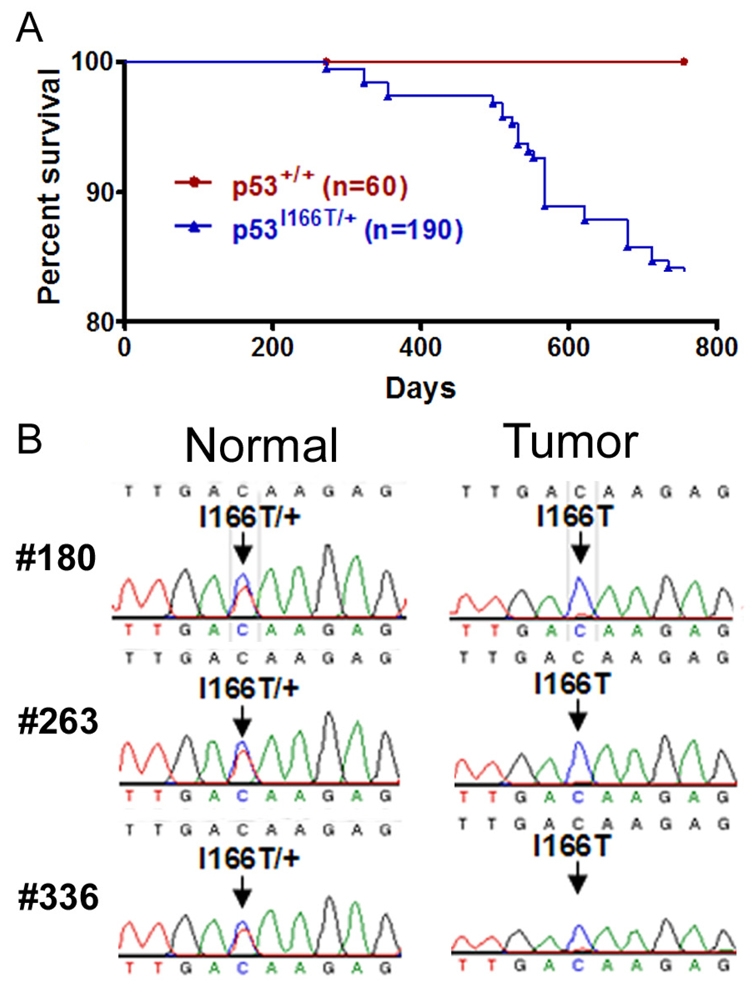
p53I166T/+ heterozygotes develop tumors and display LOH. (A) p53I166T/+ fish show a significant increase (P<0.0006) in tumor incidence compared with the p53+/+ cohorts. By 23 months, 33 of 190 p53I166T/+ fish had developed tumors. (B) Sequence analysis of the PCR products from three heterozygous fish (#180, #263 and #336) comparing normal (tail) and tumor genomic DNA. A compounded red and blue trace at codon position 166 (arrow) indicates heterozygosity, whereas a trace with blue only indicates LOH in tumor tissue samples.
To determine whether LOH occurred in tumors in p53I166T/+ fish, pair-matched tumor and non-tumorigenic genomic DNA samples were obtained from individual fish and PCR products containing codon 166 were amplified and sequenced. There was clear LOH in 87.5% (n=7/8) of cases, with loss of the wild-type allele in tumors but not in the other tissues from the same individual fish (Fig. 5B). This example of LOH in zebrafish indicates that the p53I166T mutant recapitulates human LFS and opens the possibility of applying genetics to elucidate the regulatory mechanisms of LOH.
p53I166T/I166T is a LOF allele that rescues Mdm2 knockdown lethality
Mdm2 is a transcriptional target of p53 and an E3 ubiquitin ligase that binds to p53 and promotes its degradation, thereby providing a negative feedback loop in the p53 response pathway. Mdm2 null mice are embryonic lethal, but are viable if they are also p53 null. In contrast to the IR response, in which p53 maintains its regulatory feedback control mechanism, loss of Mdm2 results in unrestrained activation of p53. Thus, loss of Mdm2 provides a stringent assay for testing whether there are remnants of functionality in mutant p53 proteins in multiple tissues and pathways, including apoptosis, cell cycle arrest, and others (Chavez-Reyes et al., 2003; Iwakuma et al., 2004; Jones et al., 1995; Lang et al., 2004; Liu et al., 2007; Montes de Oca Luna et al., 1995; Toledo et al., 2006).
We have shown (Fig. 2B) that the induction of Mdm2 expression after IR is dependent on p53 and is abrogated in p53I166T/I166T embryos. To assess whether Mdm2 is required to regulate p53 activity in zebrafish, an Mdm2 splice-blocking MO that effectively knocks down mdm2 mRNA accumulation (data not shown) was used to induce Mdm2-dependent embryonic lethality. The earliest gross observable Mdm2 MO phenotypes occurred at 14 hpf, but were more apparent at 26 hpf and 50 hpf (Fig. 6B,H). We found that Mdm2 knockdown lethality was the result of both increased apoptosis, indicated by AO staining (Fig. 6D), and cell cycle arrest, indicated by decreased staining with the M phase indicator phospho-histone-H3 (PH3) (Fig. 6F). Rescue by injection of mdm2 mRNA (Fig. 6J) indicated that this phenotype was not the result of MO off-target effects. Rescue of the Mdm2 knockdown lethality by co-injection with the p53 MO (Fig. 6L) indicated that the p53-Mdm2 regulatory loop that has been described in mammals is conserved in zebrafish.
Fig. 6.

Mdm2 knockdown embryonic lethality is rescued in p53 morphants and p53I166T/I166T mutants. (A-F) Embryos injected with water (A,C,E) or Mdm2 MO (B,D,F) were assessed at 26 hpf by transmitted-light microscopy (A,B) AO staining for apoptotic cells (C,D) and PH3-specific antibody staining for cells in mitosis (E,F). Mdm2 morphants displayed embryonic lethality (B), which was probably the result of both increased apoptosis (D) and decreased cell cycling (F). With mock (water) injections as controls (G,I,K,M), Mdm2 MO injections (H) yield a readily discernable lethal phenotype at 50 hpf. (J) As a control for non-specific MO effects, co-injection of 150 pg of mdm2 mRNA rescues the Mdm2 MO phenotype. (L) Co-injection of p53 MO rescues Mdm2 MO lethality, indicating that loss of p53 function abrogates loss of the Mdm2 phenotype. (N) Injection of Mdm2 MO into p53I166T/I166T embryos did not yield the Mdm2 lethal phenotype, indicating that the p53 mutant has LOF similar to the p53 MO. (O) p53 protein levels were assessed using an anti-p53 antibody (ZFp53-9.1) on western blots of extracts from p53+/+ and p53I166T/I166T embryos that were unirradiated (no IR), irradiated with 30 Gy (IR) or injected with the Mdm2 MO (M2 MO). Extracts of wild-type embryos injected with the p53 MO are shown as controls for antibody specificity.
Within the context of this epistatic approach, a p53 LOF mutant would be predicted to rescue Mdm2 knockdown lethality, whereas a p53 hypomorphic allele would be predicted to have residual p53 function that would be sufficient to confer lethality in Mdm2-deficient embryos. To test whether the p53I166T mutant has a LOF or a hypomorphic allele, p53I166T/I166T embryos were injected with the Mdm2 MO and were found to be completely viable, providing functional evidence that p53I166T is a LOF mutation (Fig. 6N). In mammalian tissue culture, loss of Mdm2 results in stabilization (or lack of degradation) of the p53 protein (Iwakuma and Lozano, 2003). Western blot analysis revealed that wild-type and mutant p53 proteins accumulated to comparable levels in wild-type and p53I166T/I166T mutant embryos, respectively, following Mdm2 MO injection (Fig. 6O), indicating there is no in vivo defect in the Mdm2-dependent arm of the regulation of p53I166T mutant protein stability.
p53I166T has DN activity for IR-induced apoptosis
If the mutant p53 protein has DN activity, it would be capable of reducing the functional activity of the co-expressed wild-type p53 protein. In order to test this, we designed several sensitized assays of p53 function in p53I166T/+ heterozygotes. We predicted that, if the p53 mutant protein had strong DN activity, the heterozygotes would have a phenotype similar to the homozygous mutants or, if the p53 mutant protein had partial/weak DN activity, they would have an intermediate phenotype. Conversely, if a p53 mutant protein did not have DN activity, the heterozygotes would display a wild-type phenotype.
Our results from multiple in vivo assays indicate that the p53I166T protein has DN activity. In the first assay, embryos were treated with a low dose (5 Gy) of IR that was sufficient to cause mild apoptosis in wild-type embryos (Fig. 7B) but no apoptosis in p53I166T/I166T homozygotes (Fig. 7D). There was also no increase in apoptosis in p53I166T/+ embryos, indicating that the heterozygous phenotype (Fig. 7C) was similar to the homozygous mutant phenotype.
Fig. 7.

p53I166T/+ DN phenotypes. (A) The NT in the trunk and in the tail of non-irradiated p53+/+ embryos has a low level of apoptosis, detected with AO. (B) Apoptosis is increased in embryos 8 hours after they were irradiated with 5 Gy of IR. By contrast, p53I166T/+ (C) and p53I166T/I166T (D) embryos did not display increased apoptosis at 8 hours after 5 Gy of IR. (E-G) Wild-type embryos (E) have a curled tail phenotype at 20 hours after 100 Gy of IR, whereas both p53I166T/+ (F) and p53I166T/I166T (G) embryos did not display a curled tail phenotype. (H-J) Confocal analysis of EGFP expression at 9 dpf in p53+/+; Lck-EGFP/+ (H), p53I166T/+; Lck-EGFP/+ (I) and p53I166T/I166T; Lck-EGFP/+ (J) fish at 1 day after 30 Gy of IR. Insets are higher magnification views of EGFP expression. As another test for DN phenotypes, embryos from a p53I166T/+ intercross were injected with the Mdm2 MO and sorted by phenotype at 24 hpf: 25.4% had a severe lethal phenotype (K), 52.7% had a partially rescued phenotype (L) and 21.9% had a rescued phenotype (M). (N) Melting curve analysis of PCR products from individual genomic DNA samples from embryos with severe (black), partial (red) and rescued (blue) phenotypes, which correspond to the p53+/+, p53I166T/+ and p53I166T/I166T genotypes, respectively.
In the second assay, we observed that p53 mutants were resistant to the curled tail phenotype that was normally induced by 100 Gy of IR (Fig. 7E,G). In addition, irradiated p53I166T/+ heterozygotes (Fig. 7F) did not have a curled tail phenotype. The similar phenotypic response observed in heterozygotes and homozygous mutants indicates DN activity in the heterozygotes.
In a third assay, to assess whether DN activity also occurs in late-stage fry, we developed an in vivo radiation resistance thymocyte paradigm using transgenic fish that express enhanced green fluorescent protein (EGFP) in mature T cells, under the regulation of the Lck promoter Tg(lck:lck-GFP) (Langenau et al., 2004). Eight-day-old p53+/+; Tg(lck:lck-GFP)/+ and p53I166T/I166T; Tg(lck:lck-GFP)/+ fry were irradiated with 0 or 30 Gy and imaged the following day for EGFP expression. Untreated p53+/+; Tg(lck:lck-GFP)/+ and p53I166T/I166T; Tg(lck:lck-GFP)/+ fry were identical in their EGFP expression (data not shown), indicating that the p53I166T allele did not alter T-cell development or Lck-GFP expression. When irradiated, p53+/+; Tg(lck:lck-GFP)/+ fry had severely reduced EGFP expression (Fig. 7H). By contrast, p53I166T/I166T; Tg(lck:lck-GFP)/+ fry retained EGFP expression (Fig. 7J) similar to non-irradiated controls, indicating that the IR-induced T-cell apoptosis is p53 dependent. To test DN activity, the thymocyte assay was repeated with p53I166T/+; Tg(lck:lck-GFP)/+ fry. When irradiated, p53I166T/+; Tg(lck:lck-GFP)/+ fry retained some EGFP expression (Fig. 7I), indicating that IR-induced apoptosis in thymocytes was at least partially abrogated in p53I166T heterozygotes, suggesting that the mutant allele has DN activity at older stages as well. Thus, in each of these three IR-response assays, the mutant p53I166T protein appeared to have DN activity that was sufficient to block the function of the wild-type p53 protein.
p53I166T has DN activity that partially rescues Mdm2 knockdown lethality
To assess whether the p53I166T mutant has DN activity in other cellular processes besides the response to IR, we used Mdm2 knockdown lethality as an assay for p53 function. Injection of the Mdm2 MO into embryos from a p53I166T/+ intercross resulted in three phenotypes in a mendelian ratio (Fig. 7K–M), suggesting that the p53 wild-type, heterozygous and homozygous mutants each had a distinct phenotype. Embryos were segregated into each of these three phenotypic categories and then individually genotyped. Mdm2 morphant embryos with a lethal phenotype were wild type for p53 (p53+/+, n=43). Morphants with a partially rescued phenotype were heterozygous mutants (p53I166T/+, n=89). Completely rescued embryos were homozygous mutants (p53I166T/I166T, n=37) (Fig. 7N).
The intermediate phenotype of the p53I166T/+ heterozygotes injected with the Mdm2 MO is strikingly distinct from that of the p53 wild-type embryos that were injected with the Mdm2 MO. In p53I166T/+ Mdm2 morphants, the opaque, necrotic head phenotype is similar to the phenotype in the p53+/+ Mdm2 morphants or IR-treated embryos, suggesting that apoptosis contributes to the intermediate phenotype. However the phenotype could be the result of a delay in p53-dependent apoptosis. Alternatively, the eye, head, tail and somite appear to develop normally in p53I166T/+ Mdm2 morphants. These tissues also do not appear to undergo apoptosis following IR, suggesting that p53 may have alternative effects, such as cell cycle arrest or other outcomes, in these tissues and that these p53 activities are inhibited by the p53 mutant DN activity, thereby making these tissues appear normal in p53I166T/+ Mdm2 morphants but not in p53+/+ Mdm2 morphants. Together, this suggests that p53I166T has DN activity that is capable of rescuing some, but not all, aspects of the lethal Mdm2 knockdown phenotype. In addition, the fact that p53I166T heterozygous fish develop tumors, unlike p53M214K heterozygous fish, suggests that tumor induction might occur in part through the DN activity of the p53I166T allele.
DISCUSSION
We performed a zebrafish forward genetics screen using a specific IR-induced phenotype in embryos to successfully identify mutations that give rise to tumors in adult fish. We show that, similar to human LFS patients, heterozygous p53I166T fish have a high rate of tumor development and follow Knudson’s two-hit hypothesis, in which the tumors display LOH at the p53 locus. These results demonstrate the evolutionary conservation of the vertebrate LFS pathway and suggest that zebrafish genetics will accelerate the discovery of other LFS genes. In addition, we have shown that, at a molecular level, regulation of the p53 pathway is conserved between zebrafish and mammals. Cell cycle target genes (p21, cyclin G and gadd45), a regulatory gene (mdm2) and apoptosis genes (bax, puma and noxa) are all induced in a p53-dependent manner in zebrafish. Loss of Mdm2 in zebrafish results in embryonic lethality owing to both increased apoptosis and cell cycle arrest, which can be rescued in p53I166T mutants. As in mammalian systems, the wild-type and mutant p53 proteins are stabilized, and IR-induced apoptosis is p53 dependent in the embryonic NT and in thymocytes. Here, we use whole-animal studies to show that the zebrafish p53I166T mutation is a LOF allele with DN activity, analogous to the roles of the mutant p53 in LFS patients.
Loss of function (LOF)
Within LFS families, 80% of p53 mutations are missense mutations. However, not all p53 mutations are complete LOF mutations; some mutations have been shown to lose apoptosis but preserve growth arrest function (Aurelio et al., 2000). Here, multiple assays indicate that p53I166T is a LOF allele. First, it is deficient in the induction of p53 target genes, including cell cycle, regulatory and apoptotic pathway genes (Fig. 2B). Interestingly, IR-induced p53 mRNA expression was diminished in p53 morphants and mutants, suggesting that p53 is a p53 target gene, as has been implicated previously in murine cell culture (Deffie et al., 1993). It is not known whether this apparent p53 autoregulation is direct or the result of indirect feedback regulation. Second, the defect in IR response is not the result of altered stabilization of the mutant protein, but instead lies in its inability to transactivate target genes. Third, correlating with its failure to induce p53 apoptotic genes, the p53I166T mutant is deficient in p53-dependent apoptosis in embryonic NT and in thymocytes (Fig. 1A; Fig. 7J). Fourth, in a more global assay, Mdm2 knockdown lethality is prevented in the p53I166T/I166T background. Following IR, p53 continues to maintain its feedback regulatory control mechanism. By contrast, loss of Mdm2 results in unrestrained activation of p53. Thus, loss of Mdm2 provides a stringent assay for testing whether there are remnants of functionality in the mutant p53 protein in multiple tissues and many pathways, including apoptosis, cell cycle arrest, and others. Finally, in contrast to the low penetrance of the p53M214K allele (Berghmans et al., 2005), the 100% penetrance of tumor formation in the p53I166T mutants suggests that p53I166T is a LOF allele. In genetics, the formal definition of a full LOF in a missense allele is that the missense allele over a deficiency (deletion; protein null) allele has the same phenotype as a homozygous deletion mutant. However, in the absence of a p53 null allele in zebrafish, all of these results suggest that p53I166T is a LOF allele.
Dominant-negative (DN) activity
Distinct LFS missense mutations in the DNA-binding domain result in an earlier cancer onset compared with truncation mutations or LFS mutations that lie outside of the DNA-binding domain (Birch et al., 1998). One possibility is that the increased tumor potential is the result of DN activity. Milner and Medcalf (Milner and Medcalf, 1991) showed that some, but not all, human missense mutations had DN activity in cell culture. Here, multiple in vivo assays indicate that the p53I166T allele has DN activity. p53I166T heterozygous fish were partially resistant to both IR-induced embryonic neural apoptosis and thymocyte apoptosis, and were able to partially rescue the loss of Mdm2. Interestingly, unlike p53M214K heterozygous fish, p53I166T heterozygous fish develop tumors, suggesting that this might occur in part through the DN activity of the p53I166T allele. Although not the case in the mouse model, it is possible that partial rescue in some of the zebrafish phenotypes could be the result of haploinsufficiency, which currently cannot be excluded owing to the lack of a p53 null allele in zebrafish.
Protein stability
Human tumor and cell culture studies have shown that mutant p53 proteins are typically very stable. However, we show that in the absence of stress the p53I166T mutant protein is not abundant and that, following IR-induced DNA damage or Mdm2 loss, the mutant protein becomes stable and more abundant, at levels that are comparable to the wild-type p53 protein. This suggests that the higher levels of p53 protein in human tumor and cell culture studies may be the result of constitutively active DNA damage signals in tumors and ongoing cell stress in culture, which provide signals that stabilize and activate the p53 protein. This difference may confound studies that are meant to address DN or GOF activities, which might depend on high protein levels that are increased by DNA damage, lack of negative regulation (loss of Mdm2) or tumorigenesis. This is consistent with the context in which we detect DN activity and the circumstances in which the LFS mice display DN and GOF activities.
Studies by Terzian et al. (Terzian et al., 2008) have shown that Mdm2−/−; p53R172H/R172H mice develop tumors much faster than p53R172H/R172H animals and that mutant protein is stabilized in non-tumor tissues of Mdm2−/−; p53R172H/R172H mice. Together, these observations suggest that additional activities and phenotypes may manifest when mutant p53 protein is stabilized. The observation of similar p53 mutant protein stability in zebrafish implies that zebrafish may be a useful model to explore the mechanisms of p53 stability, either through genetic or pharmacological perturbation in vivo. Furthermore, this emphasizes the importance of careful evaluation of p53 mutations in cancer patients when considering treatment with p53-activating compounds. For example, the use of nutlin and nutlin-like compounds that work by inhibiting Mdm2, and thereby stabilizing p53, might be therapeutic if wild-type p53 is present but could be detrimental if particular mutants in p53 are present. This same theme might be applicable to considerations of chemotherapeutics, such as doxorubicin, or DNA-damaging agents that are known to stabilize p53.
A zebrafish LFS model
Tumors from LFS families are diverse; however, sarcomas are the hallmark that defines the syndrome. Correspondingly, the p53I166T fish develop primarily sarcomas. Owing to the absence of a full set of diagnostic antibodies in zebrafish, the exact subtype of these sarcomas has not been determined but, histologically, they appear to be STSs, similar to the majority of sarcomas in LFS (Varley, 2003).
Do p53I166T fish display the complete spectrum of tumors that are seen in LFS patients? After sarcomas, the most prevalent tumor in LFS patients is breast carcinoma. Given the absence of mammary glands in fish, it is not surprising that breast carcinoma was not observed in our p53 mutants. However, in LFS patients, the fifth and sixth most common cancers are brain tumors or hematological tumors, which were not detected in p53 mutant fish. This could be because of the time of onset. In LFS patients, the mean incidence of STSs is 15.5 years of age, however, the mean incidence of brain or hematological tumors is 25 years of age (Kleihues et al., 1997). Therefore, with the high penetrance of STSs that are seen in p53I166T fish, it is likely that the fish die (or are sacrificed) owing to the onset of sarcomas before the onset of brain or hematological tumors. Similar observations were made for p53 knockout mice: the predominant tumors in these mice were lymphomas (not found in LFS patients) and sarcomas, with very few other tumors.
Another possibility is that genetic background may influence the diversity of tumor types, as supported by studies of tumor spectrum changes in p53 null mice on different genetic backgrounds (Kuperwasser et al., 2000). Both mice and zebrafish have been bred to isogenic or partial isogenic backgrounds, whereas human LFS patients are genetically diverse.
One of the hallmarks of LFS tumorigenesis and Knudson’s two-hit hypothesis is loss of the wild-type allele (LOH). To date, other zebrafish cancer models have not exhibited LOH. The lack of LOH in ribosomal protein (RP) mutants (Amsterdam et al., 2004) may not be surprising in that partial loss of translational efficiencies may be tolerated and tumorigenic, but complete loss is probably cell lethal. However, the fact that adenomatous polyposis coli (APC) tumors do not exhibit LOH is surprising (Haramis et al., 2006). Here, tumors in p53I166T heterozygotes displayed high levels of LOH, which further demonstrates the similarity between p53I166T tumors and human LFS tumors. Utilizing the p53I166T mutant and the powerful genetics of zebrafish provides an opportunity to identify the regulatory mechanisms of LOH and their influence on cancer progression.
One confusing issue in all three LFS models, human, mouse and zebrafish, is that tumors from heterozygous individuals often display LOH even though the mutant allele has been shown to have DN activity. There are at least three potential explanations for this: (1) the DN activity is not against p53 but other interacting proteins (potential GOF activity) and, in order for a cell to be tumorigenic, it still needs to lose the wild-type p53 allele; (2) the DN activity does not inhibit all p53 functions, hence the partial rescue of the Mdm2 lethality; and (3) the DN activity depends on the mutant protein being stable, as discussed above, which does not occur until tumorigenesis has begun. Now that we have identified a LFS model in zebrafish, we can begin to address these questions using both molecular and genetic techniques.
Zebrafish: new directions in cancer genetics
Zebrafish are proving to be a very useful model for studying tumorigenesis (Feitsma and Cuppen, 2008; Goessling et al., 2007). With the ability to perform powerful genetics in zebrafish, future studies can use this mutant p53 allele to identify cooperative tumor suppressor genes, p53 modifiers and modifiers of genomic stability. Since most familial tumor suppressors are autosomal dominant and most homozygous mouse tumor suppressor knockouts are embryonic lethal, a dominant/heterozygous screen would be particularly rewarding. For example, dominant screens for tumor enhancers in the p53I166T/I166T background may identify tumor suppressors as well as regulators of p53 protein stability and modulators of mutant p53 DN activity. In addition, since tumor onset in p53I166T/+ mutants is largely dependent on LOH, an enhancer screen in the p53I166T/+ background could identify modifiers that influence the frequency or timing of LOH events.
Zebrafish provide an ideal organism for the discovery of novel cancer therapeutics. Drug screens can be performed on p53I166T/I166T embryos to identify compounds that either specifically kill p53 mutant embryos or restore apoptosis in these mutant embryos following DNA damage. Identified compounds could conceivably be invaluable in fighting cancer since 70% of all tumors have p53 mutations.
METHODS
Zebrafish maintenance, lines and mutagenesis
Zebrafish (Danio rerio) were maintained on an Aquatic Habitat system (FL, USA). Embryos were collected from natural matings and raised as described (Westerfield, 1995). The Tübingen (TU) wild-type strain was used for MO and mRNA injections. ENU mutagenesis was carried out in the AB wild-type background using standard protocols (Mullins et al., 1994). Adult male fish were exposed to between four and six weekly treatments of 3.0 mM ENU (Sigma), allowed to recover for 1 month, bred to golden (Mullins et al., 1994; Streisinger et al., 1981) to determine the mutation frequency, and subsequently bred with wild-type females to generate F1 fish. F2 families were obtained from F1 intercrosses. For the p53I166T cancer studies, fry were raised at a density of 30 fish per 3-liter tank until 8 months of age, and then maintained at 16 fish per 3-liter tank. Tg(lck:lck-GFP) fish (Langenau et al., 2004) were maintained on a WIK wild-type background.
Morpholino knockdown and mRNA injections
MO antisense oligonucleotides (Gene Tools, Oregon) and mRNA that was synthesized with an mMessage machine SP6 transcription kit (Ambion) were injected into the yolk, just under the nuclei, of 1–2-cell-stage embryos in a volume of 0.5 nl or 1.0 nl. The p53 splice-blocking morpholino (p53 MO) (5′-CCCTTGC-GAACTTACATCAAATTCT-3′) overlaps with the splice donor site of exon 2 and intron 2, effectively preventing the splicing out of intron 2; the Mdm2 splice-blocking morpholino (Mdm2 MO) (5′-TGTTAAGAGATTCAGTACGCACCGC-3′) overlaps with the splice donor site of exon 4 and intron 4. The working concentrations of the p53 MO and the Mdm2 MO were both 0.35 mM. The bcl2 and mdm2 open reading frames (ORFs) were cloned by RT-PCR using the following primers: Bcl2 cDNA f1, 5′-ACCATGGCTAACGAAATT-3′; Bcl2 cDNA r1, 5′-CGCAGAGGCTGTCACTTC-3′ and Mdm2 cDNA f1, 5′-CCAAAATGGCAACAGAGA-3′; Mdm2 cDNA r1, 5′-ATTTCAGTGCCTCAGCTC-3′, and subcloned into the CS2+ expression plasmid (Rupp et al., 1994; Turner and Weintraub, 1994). Capped RNAs were synthesized from these CS2+ plasmids using the mMessage machine SP6 transcription kit. The working mRNA concentration for both bcl2 and mdm2 was 300 ng/μl.
Irradiation and apoptotic detection
Embryos were placed at the closest position to the source of IR in a 137Cs source irradiator (Mark I-30 Irradiator, J.L. Shepherd and Associates, CA) in order to expose the embryos to 25 Gy/minute. Apoptosis was assayed following IR treatment or MO injection by soaking embryos in 50 μg/ml of Acridine Orange (Sigma) for 45 minutes and subsequently destaining for 15 minutes. Photographs were taken using a FITC filter.
Genotyping using melting curve analysis
Initially, the I166T mutation was identified by RT-PCR of total RNA from pooled mutant embryos and pooled wild-type embryos using the following primers: p53 cDNA f1, 5′-ATGGCGCAAAACGACA-3′; p53 cDNA r1, 5′-TAGCATCCCATCACCTTA-3′, followed by single-embryo genomic PCR with the same primers. Genomic DNAs from embryos or fin clips were digested in 100 μl of ELB solution (0.01 M Tris, pH 8.3, 0.05 M KCl, 1.5 ml 0.3% Tween 20, 0.3% NP40) and 5 μl of proteinase K (Roche) at 55°C overnight, followed by 10 minutes at 95°C to inactivate the proteinase K. Melting curve analysis primers (5′-GCGCCTGCTGGTCA-3′ and 5′-CTGATTGCCCTCCACTCTT-3′) were designed to produce a 40 bp PCR product surrounding the point mutation. The PCR reactions were performed in 96-well plates (Biorad) with the following PCR conditions: 95°C for 10 seconds; 30 cycles of 95°C for 2 seconds, 60°C for 2 seconds and 72°C for 2 seconds; and a final cycle of 95°C for 2 seconds, then 45°C for 10 seconds. Each reaction contained 1μl of genomic DNA, 1× lcgreen+, 1× ExTaq buffer, 1× dNTP, 0.5 mM of primer1, 0.5 mM of primer2 and 0.25 units of ExTAQ. The melting curve analysis was performed on a Lightscanner (Idaho Technologies, UT).
Northern blot analysis of p53 target genes
Total RNA was obtained from pools of approximately 30 zebrafish embryos using Trizol reagent (Invitrogen). Zebrafish p21 and cyclin G were cloned by RT-PCR (Ambion) of irradiated embryos (30 Gy, 6 hpi), using the following primers: p21 cDNA f1, 5′-ATGGCGGCGCACAAGC-3′; p21 cDNA r1, 5′-CACT-AGACGCTTCTTGGC-3′ and cyclin G cDNA f1, 5′-CCACCATGATTGACCAGGTGACC-3′; cyclin G cDNA r1, 5′-TCTTAACAAGCATATTCAGG-3′. Gadd45 was a gift from Dr Majors. Probes were randomly labeled with γ-32P-dCTP using the prime-it II random primer kit (Stratagene). Northern blots and hybridization were performed as per the instructions of the NorthernMax-Gly kit (Ambion). Each lane was loaded with 15 μg of total RNA.
Protein analysis and real-time PCR
The quantity of the protein loaded on the western blots was assessed by hybridizing with an α-tubulin primary antibody (Santa Cruz Biotechnologies); subsequent gels were adjusted based on the α-tubulin results. p53 was detected by hybridization with the ZFp53-9.1 antibody (Lee et al., 2008). For PH3 staining, embryos were incubated with a rabbit polyclonal anti-phospho-histone H3 (ser10) antibody (1:1000 dilution) (Santa Cruz Biotechnology). Real time, RT-PCR was performed on a Lightcycler PCR machine (Roche). The following primer sets were used: p21: SG1, 5′-TGA-CATCAGCGGGTTTACAG-3′ and i102, 5′-TTCTGCT-GCTTTTCCTGACA-3′; cyclin G: C11, 5′-CCACCAT-GATTGACCAGGTGACC-3′ and C15, 5′-AGCAGCACAGACC-CACAC-3′; Mdm2: SG76, 5′-CTCGGTGCTGTTCTTGGAGT-3′ and SG77, 5′-CACTGCTTCTCCTCCTCCTG-3′; Bax: SG66, 5′-ACAGGGATGCTGAAGTGACC-3′ and SG67, 5′-GAAAAGCGC-CACAACTCTTC-3′; Puma: i97, 5′-ACGCTGTCTTCCTTCA-GAGG-3′ and i98, 5′-CCTGCAGAAAATTCCCAGAG-3′; Noxa: i91, 5′-ATGGCGAAGAAAGAGCAAAC-3′ and i92, 5′-CGCTTC-CCCTCCATTTGTAT-3′; p53: SG72, 5′-ACCCCGGATGGA-GATAACTT-3′ and SG73, 5′-CCCAGCAACTGACCTTCCTGAG-3′; and for β-actin: SG86, 5′-GGTATGGGACAGAAAGACAG-3′ and SG87, 5′-AGAGTCCATCACGATACCAG-3′. Experiments were performed in triplicate and normalized to β-actin.
Tumor analysis
Adult fish were screened weekly for tumors and/or missing/dead fish. Fish that were identified with a tumor burden were fixed in 10% neutral buffer formalin (VWR). A portion of each tumor from the abdomen of burdened fish was flash frozen along with a fin clip for future DNA-RNA analysis. Kaplan-Meier analysis was performed using GraphPad Prism 5 software. Tumor-burdened fish were sectioned and H&E stained for histological analysis. LOH analysis was performed by sequencing PCR products (primers: 5′-GTGCTGTTAAAGCCACCACA-3′ and 5′-GGTCCTACA-AAAAGGCTGTGA-3′) from the genomic DNA of the tumor and somatic tissues from the same heterozygous tumor-burdened fish.
Imaging
Light and fluorescent dissection images were taken on an Olympus SZX12 stereomicroscope with an Olympus S97809 color CCD camera. LCK-EGFP images were taken using an Olympus FluoView FV300 laser-scanning confocal microscope. The fluorescent images presented represent the sum of multiple focal planes through the embryo (z-series) assembled using ImageJ (NIH) and Photoshop (Adobe) software.
Supplementary Material
Acknowledgments
This paper is dedicated to the memory of Dr Joseph Holden who passed away before this article was published. We thank Lester Layfield and Allie Grossmann for consultation on pathology, Nick Trede for the Tg(lck:lck-GFP) fish, David Lane for the anti-p53 antibody, Ray Warters for use of the 137Cs irradiator, Bradley Demarest for assistance with statistical analyses, and Carl Wittwer and Rob Pryor for design and use of the Lightscanner. Owing to space constraints, significant primary literature is cited within review articles. We thank Alejandro Sánchez Alvarado, Jeff Amack, Maureen Condic, Kimble Frazer, Nathan Meeker, Bret Pearson, Nick Trede and anonymous reviewers for helpful comments on this manuscript. J.M.P. was funded in part by the NIH Multidisciplinary Cancer Research Training Program T32CA093247; H.J.Y. was funded by the Primary Children’s Medical Foundation, and NIH grants R01HL066292 and R01HL075472. Deposited in PMC for release after 12 months.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
H.J.Y., J.M.P. and S.A.G. designed the experiments and wrote the manuscript; J.M.P. and S.A.G. performed the zebrafish experiments; and J.A.H. performed histology and pathology analyses.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.003749/-/DC1
REFERENCES
- Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N. (2004). Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2, E139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurelio ON, Kong XT, Gupta S, Stanbridge EJ. (2000). p53 mutants have selective dominant-negative effects on apoptosis but not growth arrest in human cancer cell lines. Mol Cell Biol. 20, 770–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachinski LL, Olufemi SE, Zhou X, Wu CC, Yip L, Shete S, Lozano G, Amos CI, Strong LC, Krahe R. (2005). Genetic mapping of a third Li-Fraumeni syndrome predisposition locus to human chromosome 1q23. Cancer Res. 65, 427–431 [PubMed] [Google Scholar]
- Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, Lubratovich M, Verselis SJ, Isselbacher KJ, Fraumeni JF, et al. (1999). Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 286, 2528–2531 [DOI] [PubMed] [Google Scholar]
- Berghmans S, Murphey RD, Wienholds E, Neuberg D, Kutok JL, Fletcher CD, Morris JP, Liu TX, Schulte-Merker S, Kanki JP, et al. (2005). tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci USA 102, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch JM, Hartley AL, Tricker KJ, Prosser J, Condie A, Kelsey AM, Harris M, Jones PH, Binchy A, Crowther D, et al. (1994). Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res. 54, 1298–1304 [PubMed] [Google Scholar]
- Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, Varley JM. (1998). Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene 17, 1061–1068 [DOI] [PubMed] [Google Scholar]
- Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM, Abrams JM. (2000). Drosophila p53 binds a damage response element at the reaper locus. Cell 101, 103–113 [DOI] [PubMed] [Google Scholar]
- Cadwell C, Zambetti GP. (2001). The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of-function on cell growth. Gene 277, 15–30 [DOI] [PubMed] [Google Scholar]
- Chavez-Reyes A, Parant JM, Amelse LL, de Oca Luna RM, Korsmeyer SJ, Lozano G. (2003). Switching mechanisms of cell death in mdm2- and mdm4-null mice by deletion of p53 downstream targets. Cancer Res. 63, 8664–8669 [PubMed] [Google Scholar]
- Chompret A, Brugieres L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, Hua D, Ligot L, Dondon MG, Bressac-de Paillerets B, et al. (2000). P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer 82, 1932–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. (1993). Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362, 849–852 [DOI] [PubMed] [Google Scholar]
- Deffie A, Wu H, Reinke V, Lozano G. (1993). The tumor suppressor p53 regulates its own transcription. Mol Cell Biol. 13, 3415–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry WB, Putzke AP, Rothman JH. (2001). Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294, 591–595 [DOI] [PubMed] [Google Scholar]
- Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ. (1993). Gain of function mutations in p53. Nat Genet. 4, 42–46 [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 [DOI] [PubMed] [Google Scholar]
- Evans DG, Birch JM, Narod SA. (2008). Is CHEK2 a cause of the Li-Fraumeni syndrome? J Med Genet. 45, 63–64 [DOI] [PubMed] [Google Scholar]
- Feitsma H, Cuppen E. (2008). Zebrafish as a cancer model. Mol Cancer Res. 6, 685–694 [DOI] [PubMed] [Google Scholar]
- Field M, Shanley S, Kirk J. (2007). Inherited cancer susceptibility syndromes in paediatric practice. J Paediatr Child Health 43, 219–229 [DOI] [PubMed] [Google Scholar]
- Frebourg T, Barbier N, Yan YX, Garber JE, Dreyfus M, Fraumeni J, Jr, Li FP, Friend SH. (1995). Germ-line p53 mutations in 15 families with Li-Fraumeni syndrome. Am J Hum Genet. 56, 608–615 [PMC free article] [PubMed] [Google Scholar]
- Ganjavi H, Malkin D. (2002). Genetics of childhood cancer. Clin Orthop Relat Res. 401, 75–87 [DOI] [PubMed] [Google Scholar]
- Goessling W, North TE, Zon LI. (2007). New waves of discovery: modeling cancer in zebrafish. J Clin Oncol. 25, 2473–2479 [DOI] [PubMed] [Google Scholar]
- Haramis AP, Hurlstone A, van der Velden Y, Begthel H, van den Born M, Offerhaus GJ, Clevers HC. (2006). Adenomatous polyposis coli-deficient zebrafish are susceptible to digestive tract neoplasia. EMBO Rep. 7, 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakuma T, Lozano G. (2003). MDM2, an introduction. Mol Cancer Res. 1, 993–1000 [PubMed] [Google Scholar]
- Iwakuma T, Parant JM, Fasulo M, Zwart E, Jacks T, de Vries A, Lozano G. (2004). Mutation at p53 serine 389 does not rescue the embryonic lethality in mdm2 or mdm4 null mice. Oncogene 23, 7644–7650 [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. (1994). Tumor spectrum analysis in p53-mutant mice. Curr Biol. 4, 1–7 [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. (1995). Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208 [DOI] [PubMed] [Google Scholar]
- Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. (1997). Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 150, 1–13 [PMC free article] [PubMed] [Google Scholar]
- Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, Naber SP, Jerry DJ. (2000). Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice. A model for Li-Fraumeni syndrome. Am J Pathol. 157, 2151–2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, et al. (2004). Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872 [DOI] [PubMed] [Google Scholar]
- Langenau DM, Ferrando AA, Traver D, Kutok JL, Hezel JP, Kanki JP, Zon LI, Look AT, Trede NS. (2004). In vivo tracking of T cell development, ablation, and engraftment in transgenic zebrafish. Proc Natl Acad Sci USA 101, 7369–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KC, Goh WL, Xu M, Kua N, Lunny D, Wong JS, Coomber D, Vojtesek B, Lane EB, Lane DP. (2008). Detection of the p53 response in zebrafish embryos using new monoclonal antibodies. Oncogene 27, 629–640 [DOI] [PubMed] [Google Scholar]
- Lee Y, Chong MJ, Mckinnon PJ. (2001). Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status. J Neurosci. 21, 6687–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FP, Fraumeni JF., Jr (1969). Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome. J Natl Cancer Inst. 43, 1365–1373 [PubMed] [Google Scholar]
- Liu G, Terzian T, Xiong S, Van Pelt CS, Audiffred A, Box NF, Lozano G. (2007). The p53-Mdm2 network in progenitor cell expansion during mouse postnatal development. J Pathol. 213, 360–368 [DOI] [PubMed] [Google Scholar]
- Lu WJ, Abrams JM. (2006). Lessons from p53 in non-mammalian models. Cell Death Differ. 13, 909–912 [DOI] [PubMed] [Google Scholar]
- MacGeoch C, Turner G, Bobrow LG, Barnes DM, Bishop DT, Spurr NK. (1995). Heterogeneity in Li-Fraumeni families: p53 mutation analysis and immunohistochemical staining. J Med Genet. 32, 186–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner J, Medcalf EA. (1991). Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 65, 765–774 [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206 [DOI] [PubMed] [Google Scholar]
- Mullins MC, Hammerschmidt M, Haffter P, Nusslein-Volhard C. (1994). Large-scale mutagenesis in the zebrafish: in search of genes controlling development in a vertebrate. Curr Biol. 4, 189–202 [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. (2004). Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860 [DOI] [PubMed] [Google Scholar]
- Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, et al. (2000). Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 101, 91–101 [DOI] [PubMed] [Google Scholar]
- Parant JM, George SA, Pryor R, Wittwer CT, Yost HJ. (2009). A rapid and efficient method of genotyping zebrafish mutants. Dev Dyn. 238, 3168–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp RA, Snider L, Weintraub H. (1994). Xenopus embryos regulate the nuclear localization of XMyoD. Genes Dev. 8, 1311–1323 [DOI] [PubMed] [Google Scholar]
- Schumacher B, Hofmann K, Boulton S, Gartner A. (2001). The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol. 11, 1722–1727 [DOI] [PubMed] [Google Scholar]
- Sigal A, Rotter V. (2000). Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 60, 6788–6793 [PubMed] [Google Scholar]
- Streisinger G, Walker C, Dower N, Knauber D, Singer F. (1981). Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 291, 293–296 [DOI] [PubMed] [Google Scholar]
- Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, et al. (2002). Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 21, 5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G. (2008). The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 22, 1337–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo F, Krummel KA, Lee CJ, Liu CW, Rodewald LW, Tang M, Wahl GM. (2006). A mouse p53 mutant lacking the proline-rich domain rescues Mdm4 deficiency and provides insight into the Mdm2-Mdm4-p53 regulatory network. Cancer Cell 9, 273–285 [DOI] [PubMed] [Google Scholar]
- Turner DL, Weintraub H. (1994). Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 8, 1434–1447 [DOI] [PubMed] [Google Scholar]
- Varley JM. (2003). Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 21, 313–320 [DOI] [PubMed] [Google Scholar]
- Westerfield M. (1995). The Zebrafish Book. Eugene, OR: University of Oregon Press [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.