

SUMMARY
Utilizing subcutaneous tumor models, we previously validated SPARC (secreted protein acidic and rich in cysteine) as a key component of the stromal response, where it regulated tumor size, angiogenesis and extracellular matrix deposition. In the present study, we demonstrate that pancreatic tumors grown orthotopically in Sparc-null (Sparc−/−) mice are more metastatic than tumors grown in wild-type (Sparc+/+) littermates. Tumors grown in Sparc−/− mice display reduced deposition of fibrillar collagens I and III, basement membrane collagen IV and the collagen-associated proteoglycan decorin. In addition, microvessel density and pericyte recruitment are reduced in tumors grown in the absence of host SPARC. However, tumors from Sparc−/− mice display increased permeability and perfusion, and a subsequent decrease in hypoxia. Finally, we found that tumors grown in the absence of host SPARC exhibit an increase in alternatively activated macrophages. These results suggest that increased tumor burden in the absence of host SPARC is a consequence of reduced collagen deposition, a disrupted vascular basement membrane, enhanced vascular function and an immune-tolerant, pro-metastatic microenvironment.
INTRODUCTION
Pancreatic cancer is the fourth leading cause of cancer-related death in the USA and little improvement has been seen over the last 20 years in the 5-year survival rate, which remains at 5% (Surveillance, Epidemiology, and End Results, SEER, http://seer.cancer.gov). Historically, studies have focused on cell-autonomous behavior or the molecular biology of cancer cells. However, focus is shifting to the interaction of cancer cells with their microenvironment. In particular, desmoplasia (or stromal response) is prominent in pancreatic adenocarcinoma (Korc, 2007). Crosstalk between malignant epithelial cells and the stromal compartment can promote extracellular matrix (ECM) remodeling, angiogenesis, immune cell recruitment and metastasis (Desmouliere et al., 2004; Liotta and Kohn, 2001; Wernert, 1997; Zalatnai, 2006).
Matricellular proteins are a functional family of extracellular proteins involved in the regulation of ECM deposition and remodeling. Although primarily nonstructural, they define and contribute to the structural integrity and composition of the ECM. A dominant feature of matricellular proteins is the capacity to influence ECM assembly and turnover, a function typified by their expression at sites of tissue remodeling and their increased synthesis during wound healing (Bornstein, 2001; Bornstein and Sage, 2002). In addition, by functioning as adaptors between the ECM and the cell surface, matricellular proteins can direct cell fate, survival, adhesion and motility (Bornstein, 2001; Bornstein and Sage, 2002; Brekken and Sage, 2001).
SPARC (secreted protein acidic and rich in cysteine), also known as osteonectin and BM-40, is a multifunctional glycoprotein that exemplifies the matricellular class of proteins (Framson and Sage, 2004). Post-development, SPARC expression is limited to tissues with high ECM turnover, such as bone and gut (Bornstein, 2002). Moreover, increased production of SPARC has been shown in wound healing, at sites of angiogenesis and during tumor progression (Bornstein, 2002; Mendis et al., 1998; Pen et al., 2007; Podhajcer et al., 2008; Reed et al., 1993). Mice lacking SPARC exhibit early cataractogenesis, lax skin, progressive osteopenia and a characteristic curly tail reminiscent of ECM defects (Framson and Sage, 2004). Indeed, collagen deposition and fiber assembly was found to be altered in the lens capsule and dermis of Sparc-null (Sparc−/−) mice (Bradshaw et al., 2003; Yan et al., 2002). Furthermore, SPARC binds directly to fibrillar collagens I, III and V, and to basement membrane collagen IV (Sage et al., 1989; Sasaki et al., 1998; Sasaki et al., 1999). These data support the claim that SPARC functions as a mediator of tissue remodeling. In vitro, SPARC has been shown to induce cell rounding, or a semi-adhesive state, by disrupting focal adhesions (Bradshaw et al., 1999; Sage et al., 1989). SPARC regulates the interaction of ECM structural proteins with cell surface receptors such as integrins. In fact, SPARC was reported recently to bind to integrin β1 (Nie et al., 2008; Weaver et al., 2008). SPARC also interacts with, or indirectly regulates, a variety of growth factors including fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), platelet-derived growth factor and transforming growth factor β (Francki et al., 2004; Hasselaar and Sage, 1992; Kupprion et al., 1998; Raines et al., 1992).
By directing ECM deposition, cell-ECM interactions and growth factor signaling, SPARC would be predicted to regulate several aspects of tumorigenesis including angiogenesis, migration, proliferation and survival. Not surprisingly, many cancers exhibit altered SPARC expression. Several cancers including glioma, melanoma, tongue and oral, head and neck, esophageal, and breast show an increased level of SPARC, relative to that of their respective normal tissue, which correlates positively with invasion and metastasis (Chin et al., 2005; Choi et al., 2008; Framson and Sage, 2004; Kato et al., 2005; Xue et al., 2006). Conversely, loss of SPARC in colorectal cancer is correlated with a worse prognosis (Cheetham et al., 2008; Yang et al., 2007). In this regard, SPARC also functions as a tumor suppressor in ovarian carcinoma (Mok et al., 1996). Loss of SPARC expression owing to promoter hypermethylation has been reported in pancreatic, cervical, colorectal and prostate cancers (Cheetham et al., 2008; Kahn et al., 2008; Sato et al., 2003; Sova et al., 2006; Wang et al., 2005; Wong et al., 2008; Yang et al., 2007). In pancreatic cancer, paradoxical compartmentalization of SPARC expression is observed, whereby 52% of tumors display strong SPARC expression in the tumor-associated stroma, but lack SPARC expression in the malignant epithelial cells (Infante et al., 2007). This contradictory compartmentalized expression pattern of SPARC is also seen in lung and endometrial cancers (Rodriguez-Jimenez et al., 2007; Suzuki et al., 2005). Moreover, positive stromal SPARC expression is associated with decreased survival of patients with pancreatic ductal adenocarcinoma, regardless of tumor-derived SPARC production (Infante et al., 2007). Therefore, the effect of SPARC on tumor growth and metastasis is both tumor-type dependent and context dependent. In other words, the source or location of SPARC in the tumor microenvironment contributes to the complexity of how SPARC influences tumor progression.
We have demonstrated previously that growth of subcutaneous tumors is augmented significantly in the absence of host-derived SPARC (Brekken et al., 2003; Puolakkainen et al., 2004). Furthermore, tumors grown in Sparc−/− animals exhibit a diminished deposition of ECM compared with those grown in wild-type (Sparc+/+) counterparts (Brekken et al., 2003; Puolakkainen et al., 2004). These findings indicate that SPARC governs stromal response to tumorigenesis and thereby regulates tumor progression. In the current study, to further assess the function of stromal-derived SPARC in pancreatic tumor growth and metastasis, we utilized an orthotopic model of pancreatic cancer in which the murine pancreatic adenocarcinoma cell line PAN02 is injected directly into the pancreas of Sparc+/+ and Sparc−/− mice. We now report that Sparc−/− mice exhibited a significant increase in metastasis compared with Sparc+/+ controls. In addition, tumors grown in the absence of host-derived SPARC showed a marked reduction in the deposition of fibrillar collagens I and III, basement membrane collagen IV and the collagen-associated proteoglycan decorin. Surprisingly, although the tumors grown in Sparc−/− mice were more invasive, they displayed a significant decrease in microvessel density (MVD) and pericyte recruitment. Alterations in the vascular basement membrane led to enhanced vascular permeability and perfusion, as well as a subsequent decrease in hypoxia in tumors established in the absence of host-derived SPARC. Finally, we found that tumors grown in Sparc−/− mice demonstrated increased fibroblast recruitment and increased activation of alternatively activated (M2) macrophages. These results validate the function of stromal-derived SPARC in angiogenesis, metastasis and the immune response during tumor development.
RESULTS
Tumors are more invasive and metastatic in the absence of host SPARC
We characterized the function of stromal-derived SPARC in tumor growth and metastasis by utilizing an orthotopic model of pancreatic cancer. Tumors were established by injecting the murine pancreatic adenocarcinoma cell line PAN02 directly into the pancreas of wild-type (Sparc+/+) and Sparc−/− mice. Previously, we showed that both subcutaneous and orthotopic PAN02 tumors grew larger in Sparc−/−mice compared with Sparc+/+ controls (Arnold et al., 2008; Puolakkainen et al., 2004). In the preceding orthotopic PAN02 study, animals were sacrificed at an endpoint that was predetermined to have solid tumor growth and that was regardless of animal morbidity (~4 weeks). In the current study, orthotopic tumors were allowed to grow for an extended time, until a majority of the mice appeared moribund, to permit the study of metastatic progression and expansion of secondary tumors (6–8 weeks). The results revealed that, although final primary tumor weight at this later time point was not significantly different (data not shown), tumors grown in Sparc−/−mice were more locally invasive than those in Sparc+/+ counterparts (Fig. 1A,B). In Sparc+/+ mice, tumors were solid with well-defined, encapsulated borders and preserved significant residual pancreas (Fig. 1A, 5× magnification). However, tumors grown in the absence of host SPARC were malleable with little encapsulation and revealed sparse pancreatic tissue (Fig. 1A, 5×magnification). Tumors grown in Sparc−/− mice had more irregular margins than those grown in Sparc+/+ mice and were characterized by invasive finger-like projections into adjacent structures, leading to greater tissue destruction and enhanced local invasion (Fig. 1A,B, 100× magnification).
Fig. 1.

Orthotopic tumor local invasion and metastasis. A total of 1×106 PAN02 cells were injected into the tail of the pancreas of Sparc+/+ and Sparc−/− mice. Tumors were allowed to grow for 6–8 weeks. (A-C) Hematoxylin and eosin (H&E) stained tissue sections reveal the invasive nature of tumor growth in Sparc−/− mice. Dotted lines demarcate the normal pancreas (P) and adjacent tissue from the primary tumor (T), invasive lesions (I) or metastatic lesions (M). (A) Low (5×) magnification images of the primary tumor sections show the residual pancreas, medium (100×) magnification images show the primary tumor border, and high (400×) magnification images show the tumor cell morphology in Sparc+/+ mice compared with Sparc−/− mice. (B) These images reveal the finger-like invasion of tumors grown in the absence of host SPARC at both the primary pancreatic site (P) and into the adjacent tissue (muscle), in direct contrast to the well-defined borders of the tumors grown in Sparc+/+ controls. (C) These images display PAN02 local invasion into the spleen, abdominal muscle and visceral fat (adipose), as well as metastasis (M) to the liver. (D) Survival curve of Sparc+/+ and Sparc−/− mice challenged with orthotopic PAN02 tumors. n=10/group; P=0.018, Gehan-Breslow-Wilcoxon test. Bars, 5 mm (A, left panels); 250 μm (A, center panels; B; C, four panels on the left); 50 μm (A, right panels; C, right panel).
Fig. 5.

Collagen deposition and maturation in PAN02 orthotopic tumors. The amount of collagen production, deposition and maturation was assessed in orthotopic tumors from Sparc−/− and Sparc+/+ mice. (A) Immunohistochemistry was performed on tumors with the antibodies rabbit anti-collagen I (α1) (LF-67, green) (Bernstein et al., 1996; Bernstein et al., 1995) and rabbit anti-collagen I (α2) (46425, green). Nuclei are stained with DAPI (blue). (B) Picrosirius Red staining reveals the maturity of the fibrillar collagen within tumors. Sparc−/− mice show less mature and/or smaller collagen fibrils (yellow and orange) compared with tumors grown in Sparc+/+ mice, which display more mature and/or larger collagen fibrils (orange and red) under polarized light. (C) Masson’s trichrome staining shows the amount of fibrillar collagen (blue) deposited in and around tumors. Arrowheads point to the collagen capsule at the tumor border. Collagen deposition at the tumor edge and center was quantified on trichrome-stained tissue and was recorded as the percentage of thresholded area. (D) Immunohistochemistry with a rabbit anti-collagen type IV antibody (red), and quantification of the percentage of thresholded area. (E) The amount of collagen produced and secreted within tumors was quantified by hydroxyproline analysis. ns, not significant; *P<0.05, ***P<0.001; Student’s t-test. Bars, 100 μm (A; B, right panels; D); 500 μm (B, left panels); 250 μm (C).
In Sparc+/+ and Sparc−/− mice, PAN02 tumors invaded adjacent tissues such as the spleen, adipose, abdominal muscle, intestine and stomach (Fig. 1C, spleen, muscle, adipose). Orthotopic PAN02 tumors also exhibited macroscopic metastasis to peritoneal organs including the liver, mesenteric lymph nodes, peritoneal wall and diaphragm (Fig. 1C, liver). Therefore, we counted both adjacent tissue invasion and distant organ dissemination as metastatic events. Although Sparc+/+ and Sparc−/− mice had a similar metastatic incidence, the total tumor burden was increased significantly in Sparc−/− mice (Table 1, P<0.0001). Most striking was the increase in metastasis to the liver, where Sparc−/− mice displayed ten times the number of events compared with their Sparc+/+ counterparts (Table 1, P<0.0001). Accelerated tumor progression in the absence of host SPARC was highlighted by a survival study that revealed that Sparc−/− mice bearing orthotopic PAN02 tumors had significantly reduced survival compared with Sparc+/+ controls, with a median survival of 28 days versus 36 days, respectively (Fig. 1D, P<0.018). These data indicated that tumors were more invasive and metastatic in the absence of host SPARC. However, acceleration of tumor progression was not the result of changes in the proliferation or apoptosis of PAN02 cells (Arnold et al., 2008). Therefore, we aimed to characterize angiogenesis and the composition of the ECM in PAN02 tumors to delineate a mechanism by which the absence of host SPARC augments invasion and metastasis.
Table 1.
Metastatic incidences and events

Angiogenesis is diminished in tumors grown in Sparc-null mice
We analyzed MVD and endothelial cell-pericyte association by immunohistochemistry. Quantifying MVD with three vascular markers confirmed that tumors grown in Sparc−/− mice displayed decreased angiogenesis relative to tumors grown in Sparc+/+ controls (Fig. 2A–C). Meca-32, a panvascular marker, showed a significant reduction in the number of blood vessels per field in tumors collected from Sparc−/− mice compared with Sparc+/+ littermates (Fig. 2A, P<0.05). Immunohistochemistry for the detection of endomucin also revealed a significant reduction in MVD in tumors grown in the absence of host SPARC, as measured by the percentage of thresholded area (Fig. 2C, P<0.05). Endomucin is a sialomucin that is expressed by proliferating endothelial cells, especially those stimulated by tumor-conditioned media, and is found constitutively on venous and capillary endothelium, but not arterial endothelium (Liu et al., 2001; Morgan et al., 1999). Finally, CD31 (Pecam-1) staining showed reduced vascularity in tumors grown in Sparc−/− mice compared with Sparc+/+ controls, although the difference did not reach statistical significance (Fig. 2B).
Fig. 2.

Tumor MVD and maturity. Fluorescence immunohistochemistry was utilized to quantify MVD, pericyte association, and blood vessel activation in PAN02 orthotopic tumors grown in Sparc+/+ and Sparc−/− mice (A-D). (A) The number of Meca-32-positive vessels, and the number of Meca-32 and SMA colocalized vessels, was quantified in methyl Carnoy’s-fixed tumor sections stained with rat anti-mouse endothelial cell Meca-32 (Hallmann et al., 1995) and rabbit anti-SMA. The percentage of mature vessels was calculated as the number of Meca-32 and SMA colocalized vessels divided by the total number of Meca-32-positive vessels in each image. (B) The percentage of thresholded area was quantified for CD31, NG2, and CD31 and NG2 colocalization in frozen tumor sections stained with rat anti-mouse CD31 and rabbit anti-NG2. (C) Quantification of rat anti-endomucin, another blood vessel marker, was used to confirm MVD in methyl Carnoy’s-fixed tumor sections. Representative images of endomucin staining in tumors from Sparc+/+ and Sparc−/− mice are shown. (D) The levels of VEGFR2 and VEGF:VEGFR complex were quantified in frozen tumor sections stained with rat anti-VEGFR2 (RAFL-2) (Ran et al., 2003) and biotinylated mouse anti-VEGF:VEGFR complex (Gv39M) (Brekken et al., 1998). The total VEGFR2 and activated VEGFR (VEGF:VEGFR) staining was quantified as the percentage of thresholded area in an entire field. The amount of activated vasculature (Active/Total) was calculated by dividing the colocalized area of RAFL-2 and Gv39M by the total RAFL-2 area and was recorded as a percentage. *P<0.05, **P<0.01, ***P<0.001; Student’s t-test. Bars, 50 μm.
In addition to angiogenesis, we also characterized the extent of pericyte association of blood vessels within the tumors. We colocalized the aforementioned vascular markers with markers of smooth muscle cells/pericytes (Fig. 2A,B). Quantification of Meca-32 colocalized with smooth muscle actin (SMA) demonstrated that blood vessel maturity is compromised in tumors grown in Sparc−/−mice relative to Sparc+/+ counterparts (Fig. 2A, P<0.01). In addition, colocalization of CD31 with NG2 chondroitin sulfate validated that tumors grown in the absence of host SPARC exhibit reduced pericyte recruitment (Fig. 2B, P<0.05).
Finally, the decrease in MVD and pericyte recruitment in tumors grown in Sparc−/− mice is not a result of decreased levels of vascular endothelial growth factor receptor 2 (VEGFR2) or of a deficiency in vascular endothelial growth factor signaling (Fig. 2D). The amount of VEGFR2 on vascular endothelium was assessed by immunohistochemistry with the rat monoclonal antibody RAFL-2 (Ran et al., 2003). Surprisingly, quantification of RAFL-2 revealed that tumors grown in the absence of host SPARC have significantly increased levels of VEGFR2 (Fig. 2D, P<0.001). Therefore, in the absence of host-derived SPARC, tumor vascular endothelium showed elevated expression of VEGFR2; however, this did not correspond to increased MVD. In addition, staining with an antibody that specifically recognizes VEGF bound to a cognate receptor (Gv39M) indicated that the reduction in angiogenesis in Sparc−/− mice during tumorigenesis was not caused by a loss of VEGF binding to its receptors (Fig. 2D) (Brekken et al., 1998). This is displayed as both total Gv39M (VEGF:VEGFR) staining and the ratio of activated vasculature (Gv39M colocalized to RAFL-2) to total vasculature (total RAFL-2) (Fig. 2D, Active/Total). In either case, there was no difference in the amount of activated VEGFR2 on the vasculature of tumors grown in Sparc−/− mice compared with Sparc+/+ mice.
Vascular basement membrane is disrupted in tumors grown in the absence of host SPARC
Utilizing transmission electron microscopy (TEM), we examined the vascular basement membranes in organs from tumor-bearing Sparc+/+ and Sparc−/− mice. The tumor vasculature in Sparc−/− mice appeared altered compared with Sparc+/+ controls (Fig. 3A). There was a noticeable reduction in the density of the vascular basement membrane in tumors grown in the absence of host SPARC (Fig. 3A, arrows). Possibly owing to the lack of sufficient basement membrane, the endothelial cell layer was discontinuous and somewhat separated from the underlying tissue (Fig. 3A, arrowheads). The liver vasculature was also disrupted in tumor-bearing Sparc−/− mice relative to Sparc+/+ littermates (Fig. 3B). The hepatic endothelial cells were detached from the underlying hepatocytes (Fig. 3B, arrows), and the endothelial cell junctions appeared as gaps rather than as fenestrations (Fig. 3B, arrowheads). However, these vascular differences in tumor-bearing mice did not extend to the blood-brain barrier. Endothelial cell layers in the brain of Sparc+/+ and Sparc−/− mice were continuous and closely associated with an underlying dense vascular basement membrane (Fig. 3C, arrows).
Fig. 3.

Microvessel structure in organs from tumor-bearing Sparc+/+ and Sparc−/− mice. TEM images of blood vessels within the tumor (A), liver (B) and brain (C) of Sparc+/+ and Sparc−/− animals bearing orthotopic PAN02 tumors. Red blood cells (RBC) and blood vessel lumens are labeled. (A) Note the reduction of ECM deposition under (arrows), and gaps between (arrowheads), endothelial cells (*) in tumors from Sparc−/− mice compared with Sparc+/+ animals. The lower panels are magnified regions taken from the upper panels, as indicated. (B) Note the differences in how the sinusoidal endothelial cells (*) associate with the underlying hepatocytes (arrows) and each other (arrowheads) in the liver of tumor-bearing Sparc−/− mice compared with their Sparc+/+ counterparts. The lower panels are magnified regions taken from the upper panels, as indicated. (C) Tumor-bearing Sparc−/− mice present a normal blood-brain barrier and normal endothelial cell (*) attachment to the vascular basement membrane (arrows). Insets are regions enlarged by approximately fourfold to aid in the visualization of the vascular basement membrane within the brain. Bars, 500 nm (A, top panels; C); 193 nm (A, bottom panels); 1000 nm (B, top panels); 344 nm (B, bottom panels).
Vascular function is enhanced during tumor progression in Sparc-null mice
We surmised that vascular function would be altered as a result of the disruptions in the vascular basement membrane in tumors grown in the absence of host SPARC. To test this hypothesis, we measured functional parameters such as permeability, perfusion and hypoxia. The amount of Evans Blue dye (EBD) entering the tumors after intravenous injection was quantified by fluorescence microscopy and found to be increased significantly in tumors grown in Sparc−/− mice (Fig. 4A, P<0.05, red). Next, we quantified the perfusion and permeability of fluorescently labeled dextrans (Fig. 4B). The permeability of fluorescein isothiocyanate (FITC)-labeled dextran (2×106 kDa) in tumors grown in Sparc−/− mice was increased significantly (Fig. 4B, P<0.001, green). In addition, there was a greater than fourfold increase in the perfusion of the smaller molecular weight rhodamine-dextran (10,000 kDa) in tumors grown in Sparc−/−mice relative to Sparc+/+ mice (Fig. 4B, P<0.01, red). Concomitant with the increase in permeability and perfusion, the degree of hypoxia within the tumors was found to be decreased significantly in Sparc−/−mice relative to Sparc+/+ counterparts (Fig. 4C, P<0.05, green).
Fig. 4.

Permeability, perfusion and vascular function. Mice bearing orthotopic PAN02 tumors were injected intravenously with EBD (A), fluorescent dextrans (B) or a hypoxia marker (Hypoxyprobe-1) (C). Tissue was snap-frozen, sectioned (10μm) and analyzed by fluorescence microscopy. (A) Quantification of EBD permeability within tumors was recorded as the percentage of thresholded area. Representative images of EBD fluorescence (red) in tumors grown in Sparc+/+ and Sparc−/− mice are shown. (B) FITC-dextran (2 million MW) (green) and rhodamine-dextran (10,000 MW) (red) permeability in tumors. The percentage of thresholded area was quantified. (C) The amount of hypoxia within tumors was quantified using an antibody, FITC-conjugated mouse anti-pimonidazole, directed against an adduct that forms when Hypoxyprobe-1 enters hypoxic tissue. The percentage of thresholded area was quantified and two separate experiments were combined by normalization to data from Sparc+/+ mice. Images display hypoxia (green) near the vasculature (red) stained with the rat anti-mouse endothelial cell (Meca-32) antibody within tumors. Nuclei are marked with DAPI (blue). (D) Liver permeability was also affected by the lack of host SPARC, as measured by both EBD and the fluorescent dextrans. *P<0.05, **P<0.01, ***P<0.001; Student’s t-test. Bars, 100 μm (A,C); 250 μm (B).
Consistent with alterations in the vascular basement membrane, tumor-bearing Sparc−/− mice also displayed enhanced liver permeability and perfusion relative to Sparc+/+ mice, as measured by EBD (P<0.05), FITC-dextran (P<0.001) and rhodamine-dextran (P<0.001) (Fig. 4D). It is important to note that there was a trend towards increased EBD permeability in the pancreas and liver of non-tumor-bearing Sparc−/− mice (supplementary material Fig. S1A). However, by TEM, the vasculature appeared comparable between the two non-tumor-bearing groups (supplementary material Fig. S1B). It appears as though the vasculature of Sparc−/−animals is more compliant or sensitive to perturbation; thereby, vascular permeability is exaggerated when challenged by the tumor. Therefore, it is possible that the compromised vascular basement membrane in tumor-bearing Sparc−/− mice not only contributes to primary tumor growth and survival, but also to enhanced metastasis, especially to the liver.
Collagen deposition and fibrillogenesis are decreased in tumors grown in the absence of host SPARC
Previous studies have shown the importance of stromal SPARC for collagen deposition and fibrillogenesis in subcutaneous tumors (Brekken et al., 2003; Puolakkainen et al., 2004). We wanted to validate this observation in the orthotopic model of pancreatic cancer. We first utilized immunohistochemistry to stain for collagen I (Fig. 5A). Staining with antibodies LF-67 and 46425 showed no significant change in the overall expression of collagen I (α1) or collagen I (α2) within the tumors when the percentage of thresholded area was quantified (data not shown). However, the pattern of staining was different in tumors grown in Sparc−/− versus Sparc+/+ animals. Most of the collagen I in the tumors of Sparc−/−mice was cell-associated and not organized into collagen fibrils (S.A.A. and R.A.B., unpublished), whereas collagen I in Sparc+/+ mice was found organized into large tracks of aligned fibrils with minimal cell association (Fig. 5A). Therefore, when looking specifically at tracks of fibrillar collagen I, it became apparent that tumors grown in the absence of host SPARC showed a decrease in collagen fibrillogenesis, especially at the tumor border (Fig. 5A). The images in Fig. 5A display typical collagen I fiber tracks found in tumors grown in Sparc−/− mice, relative to their Sparc+/+ counterparts, and do not represent total collagen production. Notice that in the absence of host SPARC, fibrillar collagen I tracks were more sparse and had significantly less organization.
Staining with Masson’s trichrome, which reacts with all fibrillar collagens (blue), confirmed that tumors grown in Sparc−/− mice displayed a significant reduction in fibril-associated collagen (Fig. 5C, center P<0.05, edge P<0.001). This was most apparent at the tumor edge where there was a greater than fourfold decrease in collagen deposition in tumors grown in Sparc−/− mice compared with Sparc+/+ controls (Fig. 5C, 1.4% vs 6.4%, arrowheads and graph). The size or maturity of collagen fibers in tumors from Sparc+/+ and Sparc−/− mice was assessed with Picrosirius Red, a dye that stains and enhances the birefringence of fibrillar collagens (Fig. 5B). Under polarized light, mature or larger fibers appear orange and red, whereas smaller, less mature fibers appear yellow and green. Tumors grown in Sparc−/− mice had fewer collagen fibers by Picrosirius Red staining than tumors grown in their Sparc+/+ counterparts. Furthermore, the collagen fibers were smaller and less mature, as depicted by the yellow and green staining, compared with the red and orange staining in the control tumors (Fig. 5B).
Not only are the fibrillar collagens affected, but tumors grown in Sparc−/− mice also displayed a significant reduction in the deposition of the basement membrane-associated collagen IV compared with their Sparc+/+ counterparts (Fig. 5D, P<0.05). The decrease in collagen deposition and fibrillogenesis in the tumors of Sparc−/− mice was not a result of changes in collagen production or secretion because we found no significant difference in the levels of hydroxyproline in tumor lysates, a post-translational modification that occurs almost exclusively on all collagens (Fig. 5E). Therefore, the absence of stromal-derived SPARC does not diminish the expression or secretion of collagen; rather, SPARC is indispensable for proper collagen maturation and fibrillogenesis during malignant progression, a role that is consistent with its participation in collagen deposition and fibrillogenesis during development and wound healing.
Noncollagenous ECM deposition in tumors grown in Sparc-null mice
We demonstrated that the absence of host SPARC during tumor progression leads to a reduction in collagen deposition and fibrillogenesis. However, it is not clear whether a lack of stromal-derived SPARC contributes to alterations in the deposition of noncollagenous ECM proteins during tumorigenesis. Decorin, a small leucine-rich proteoglycan, binds to fibrillar collagens and is vital for collagen fibrillogenesis (Reed and Iozzo, 2002). The quantity of decorin in orthotopic tumors was assessed by immunohistochemistry and found to be substantially decreased in Sparc−/− mice compared with Sparc+/+ littermates (Fig. 6A). Because collagen fibers serve, in part, as a storage compartment for decorin, the reduction in collagen deposition in Sparc−/− mice might lead to a concomitant loss of decorin retention in tumors.
Fig. 6.

ECM deposition and composition in PAN02 orthotopic tumors. Immunohistochemistry for decorin (A), fibronectin (B) and laminin (C) was performed on methyl Carnoy’s-fixed, paraffin-embedded tumor sections. (A) Immunohistochemistry with a goat anti-mouse decorin antibody (brown). (B) Immunohistochemistry with a rabbit anti-fibronectin antibody (red). Insets are higher magnification images of the tumor center. (C) Immunohistochemistry with a rabbit anti-laminin antibody (red). Images show the tumor center and edge. Arrowheads in B and C specify the tumor capsule. Bars, 100 μm (A,C); 500 μm (B).
Fibronectin and laminin are both noncollagenous glycoproteins that have been shown to promote cell adhesion and migration, particularly in tumors (Kaplan et al., 2005; McCarthy and Furcht, 1984; Ohnishi et al., 1997; Ohnishi et al., 1998). Fibronectin is also a provisional matrix that is laid down during wound healing and augmented during tumor formation. We found no remarkable difference in the amount of fibronectin deposited within (Fig. 6B, inset), or encapsulating, the tumor (Fig. 6B). However, it was surprising that the tumor capsule in Sparc−/− mice is pronounced when assessed by fibronectin staining, but indiscernible by collagen staining (compare Fig. 5C with Fig. 6B).
Laminin is a constituent of the mature basement membrane and enhances tumor cell motility and invasion (Kim et al., 1999; Wang et al., 2004). By immunohistochemistry, there was no significant difference in laminin deposition within the tumors (Fig. 6C, tumor center). However, the level of laminin staining was noticeably increased at the tumor border in Sparc−/− mice relative to Sparc+/+ mice (Fig. 6C, tumor edge). Increased laminin surrounding the tumors in Sparc−/− mice might be a compensatory reaction to the reduction in collagen deposition. Collectively, these data indicate that SPARC is involved predominantly in collagen synthesis and fibrillogenesis. In addition, the similar levels of fibronectin and increased accumulation of laminin, both proven stimulators of tumor cell invasion, leads to the proposal that these alternative matrices might contribute to the accelerated tumor growth in Sparc−/− mice.
Tumors grown in the absence of host SPARC have alterations in the infiltration of host cells
The stromal compartment is an important component of the ECM and is capable of influencing tumor growth and metastasis. It consists of endothelial cells, pericytes, fibroblasts and immune cells. Fibroblasts are a rich source for ECM secretion and deposition. Consequently, we assessed fibroblast infiltration into orthotopic PAN02 tumors to determine whether the diminished collagen deposition and fibrillogenesis in the absence of host SPARC is the result of a deficiency in fibroblast recruitment and/or activation. Staining for SMA, a marker of activated mesenchymal cells, and with a pan-reticular fibroblast marker confirmed that tumors grown in Sparc−/− mice did not have a deficit in fibroblast recruitment or activation relative to Sparc+/+ mice (Fig. 7A). In fact, by anti-SMA immunoreactivity, there was a significant increase in the number of activated fibroblasts infiltrating tumors grown in Sparc−/− mice compared with Sparc+/+ littermates (Fig. 7A, P<0.05). Thus, fibroblasts are recruited and activated in the absence of SPARC but are unable to elicit a proper fibrotic response.
Fig. 7.

Host-cell infiltration in PAN02 orthotopic tumors. Fluorescence immunohistochemistry was performed to evaluate the stromal response in tumors grown in Sparc+/+ and Sparc−/− mice (A,B). (A) Methyl Carnoy’s-fixed tumors were stained with rabbit anti-SMA (red) and rat anti-pan-reticular fibroblast marker (fibroblasts, green) antibodies, and the percentage of thresholded area was quantified. (B) Snap-frozen tumors were stained with a rabbit anti-MPO antibody (red) and the number of MPO-positive cells per field was quantified. Nuclei are stained with DAPI (blue). *P<0.05; Student’s t-test. Bars, 50 μm (A); 250 μm (B).
A cell population previously shown to be involved in angiogenesis and tumor progression are neutrophils (Murdoch et al., 2008). Therefore, we stained tumor sections for myeloperoxidase (MPO), a marker of granulocytes, to assess neutrophil infiltration into orthotopic tumors. The number of MPO-positive cells within tumors grown in Sparc−/− mice compared with Sparc+/+ controls was similar (Fig. 7B, red). These results introduce the possibility that monocytic cell populations other than neutrophils might be contributing to the accelerated metastasis in the absence of host-derived SPARC.
Macrophage recruitment and activation of the M2 phenotype is increased in tumors grown in Sparc-null mice
Tumor-associated macrophages have been implicated in tumor progression, metastasis, angiogenesis and immunosuppression (Sica et al., 2008). In a previous study that focused on the subcutaneous growth of PAN02 tumors in the absence of host SPARC, we found that macrophage distribution was altered such that, at the tumor border, tumors grown in Sparc+/+ mice displayed a concentrated macrophage population which was diminished in tumors grown in Sparc−/− mice (Puolakkainen et al., 2004). However, it was noted that macrophages were distributed evenly throughout the tumors grown in the absence of host SPARC. Therefore, we sought to quantify and define the macrophage population in our orthotopic PAN02 model by fluorescence immunohistochemistry. The total macrophage infiltration was determined by staining for F4/80, a general macrophage marker, and found to be significantly increased in tumors grown in Sparc−/−mice compared with Sparc+/+ littermates (Fig. 8A, P<0.01). Staining for an activated macrophage marker, Mac-3, also displayed augmented macrophage infiltration and activation in the absence of host SPARC (Fig. 8A, P<0.001). To further define the macrophage population within these tumors, immunohistochemistry was performed against markers that have been shown to delineate classically-activated, immunostimulatory macrophages (M1) from alternatively-activated, immunosuppressive macrophages (M2) (Mantovani et al., 2002; Murdoch et al., 2008). Colocalization of CD11b (green) with inducible nitric oxide synthase (iNOS, red) revealed that tumors grown in Sparc−/− mice exhibit similar activation of M1 macrophages relative to Sparc+/+ mice (Fig. 8B). Conversely, M2 macrophage activation is significantly enhanced in tumors grown in the absence of host SPARC, as measured by the colocalization of the macrophage mannose receptor (MMR, green) with CD163 (red) (Fig. 8C, P<0.001). Therefore, the absence of host-derived SPARC increases macrophage recruitment/activation and polarizes the macrophage population within tumors towards an M2 phenotype. This suggests that the microenvironment in tumors grown in Sparc−/− mice is immunosuppressive and, thus, pro-tumorigenic and metastatic.
Fig. 8.

Macrophage recruitment and activation in PAN02 orthotopic tumors. Fluorescence immunohistochemistry was utilized to assess macrophage recruitment and activation in tumors grown in Sparc+/+ and Sparc−/− mice (A-C). (A) Methyl Carnoy’s-fixed tumors were stained with a rat anti-F4/80 or a rat anti-Mac-3 antibody and the percentage of thresholded area was quantified. (B) Snap-frozen tumors were stained with rat anti-CD11b (green) and rabbit anti-iNOS (red) antibodies. The percentage of thresholded area showing CD11b colocalized with iNOS was quantified. (C) Snap-frozen tumors were stained with rat anti-mannose receptor (MMR, green) and rabbit anti-CD163 (red) antibodies. The percentage of thresholded area showing MMR alone, CD163 alone, and MMR and CD163 colocalization was quantified. Nuclei are stained with DAPI (blue). *P<0.05, **P<0.01, ***P<0.001; Student’s t-test. Bars, 50 μm (A,B); 100 μm (C).
DISCUSSION
At first glance, it is counterintuitive to imagine how tumor growth and metastasis can be augmented in Sparc−/− animals in the face of reduced angiogenesis and pericyte recruitment. However, owing to the importance of SPARC for appropriate ECM deposition and collagen fibrillogenesis, we speculated that the absence of SPARC could weaken the vascular basement membrane and alter the tumor microenvironment such that the tumors were able to overcome this diminution in angiogenesis. Indeed, we found that the vascular basement membrane was disrupted and that vascular function was enhanced in tumors grown in Sparc−/− mice compared with controls. Ultimately, enhanced vascular function in the absence of host SPARC is probably a consequence of a combination of factors including decreased pericyte recruitment, a deficient vascular basement membrane, reduced MVD, and/or altered chemokine and growth factor signaling.
Alterations in the ECM, as well as enhanced vascular function, could account for the accelerated tumor progression and metastasis in Sparc−/− mice. Structural and functional abnormalities of the tumor vasculature have been attributed to decreases in pericyte coverage and stabilization (Baluk et al., 2005; Morikawa et al., 2002). Consistent with these observations, tumors grown in Sparc−/− mice display a reduction in pericyte recruitment and a concomitant alteration in vascular structure and function. Furthermore, it has been shown that the integrity of the vasculature is vital to the control of metastasis (Gerhardt and Semb, 2008; Xian et al., 2006). In our orthotopic model of pancreatic cancer, the enhancement of metastatic progression and burden in the absence of host SPARC might be a result of hematological tumor cell dissemination augmented by the loss of pericyte coverage and the concomitant disruption of the vascular basement membrane. In tumor-bearing Sparc−/− mice, the endothelial cell layer is compromised, not only in the primary tumor, but also at metastatic sites such as the liver. These changes could facilitate intravasation and the extravasation of tumor cells. In addition, the enhanced vascular permeability and perfusion in tumor-bearing Sparc−/− mice are expected to contribute to tumor progression by, presumably, improving the delivery of oxygen and nutrients.
Tumors grown in Sparc−/− mice also exhibit deficits in collagen fibrillogenesis and alterations in noncollagenous ECM protein deposition. The ECM is not just a passive bystander. In addition to providing structural support, the ECM influences cell adhesion, migration, differentiation, proliferation and survival by binding to, and signaling through, adhesion receptors such as integrins (Juliano, 2002). Several studies have shown that the ECM contributes substantially to tumor progression and metastasis (Engbring and Kleinman, 2003; Ioachim et al., 2002; Timar et al., 2002). Our studies further highlight the importance of proper deposition and organization of the ECM in the control of pancreatic tumor cell dissemination.
The immune cell compartment of the tumor microenvironment participates in tumor progression. The balance between immunostimulatory and immunosuppressive cell recruitment and activation is a crucial factor in the fate of malignant lesions. Tumor-associated macrophages can elicit opposing effects (Mantovani et al., 2002; Murdoch et al., 2008). Classically-activated macrophages (M1) are important for tumor clearance because they release inflammatory cytokines that stimulate the adaptive immune response, whereas alternatively-activated macrophages (M2) are associated with tumor angiogenesis, immunosuppression and metastasis (Mantovani et al., 2002; Murdoch et al., 2008). It is the M2 macrophage that contributes to immune tolerance by subverting adaptive immune responses. Furthermore, an increased number of M2 macrophages correlates with a poor prognosis in several human cancers (Bingle et al., 2002). In our murine model of pancreatic carcinoma, macrophage recruitment and polarization to the M2 phenotype is augmented in the absence of host-derived SPARC. This provides another explanation for accelerated tumor progression and enhanced metastasis in Sparc−/− mice.
A study of human pancreatic adenocarcinoma has shown that increased SPARC expression by the tumor stroma correlates with decreased patient survival (Infante et al., 2007). Nevertheless, correlation does not always translate to causation. Indeed, Sato et al. found that SPARC expression is increased in fibroblasts isolated from human pancreatic tumors compared with noncancerous tissue and that exposure of the noncancerous-derived fibroblasts to human pancreatic cancer cells upregulated their expression of SPARC (Sato et al., 2003). However, this same group, as well as others, have demonstrated that exogenous administration of SPARC inhibits the proliferation of human pancreatic cell lines (Guweidhi et al., 2005; Sato et al., 2003). Therefore, we suspect that SPARC expression is a guide for how responsive the stroma is to the tumor microenvironment and may even be a protective mechanism by which the stroma attempts to control aggressive tumor growth. In other words, stromal SPARC expression may correlate with a worse prognosis simply because it is upregulated in response to, and acts as an indicator of, invasive disease. Similar to SPARC, transforming growth factor-β (TGF-β) isoforms have been shown to have increased expression in pancreatic cancer; to correlate with increased invasion and decreased survival in late-stage disease; and to have paradoxical effects on pancreatic tumor progression (Pardali and Moustakas, 2007; Truty and Urrutia, 2007). Moreover, TGF-β isoforms activate pancreatic stellate cells and increase SPARC expression to induce a desmoplastic response that is characteristic of pancreatic ductal adenocarcinoma (Mahadevan and Von Hoff, 2007; Podhajcer et al., 2008; Zhou et al., 2005). A common occurrence in the metastatic progression of pancreatic carcinoma is epithelial-to-mesenchymal transition (EMT). Therefore, orthotopic tumors derived from PAN02 cells, which display mesenchymal characteristics in vitro and in vivo, model late-stage, invasive pancreatic cancer. Interestingly, TGF-β functions as a tumor promoter in late-stage pancreatic adenocarcinoma by stimulating EMT and invasion. Therefore, the effect of SPARC on malignant progression may depend on TGF-β and other growth factor signaling events. Indeed, preliminary data from our laboratory suggest that TGF-β contributes substantially to the phenotypic alterations observed during tumor progression and metastasis in the absence of host SPARC.
Although one predicts that, from studies of SPARC expression in human pancreatic cancer, loss of SPARC expression from stromal cells would diminish tumor growth and metastasis, in the present model, the absence of stromal-derived SPARC accelerates tumor progression. Our study indicates that this is the result of alterations in the ECM and vascular basement membrane, which enhance vascular function, specifically, in the tumor microenvironment and at metastatic sites. Although this aspect of the model may not precisely mimic human disease, it does raise concerns related to the use of SPARC as a therapeutic target. A recent review featured SPARC as a protumorigenic, prometastatic protein and proposed that it should be inhibited as a means of tumor therapy (Podhajcer et al., 2008). Our data suggest that inhibiting SPARC could have unforeseen consequences on the vasculature and tumor microenvironment, which could result in an altered therapeutic response and increased metastasis. In other words, targeting SPARC might enhance drug delivery but also pose an increased risk for metastatic dissemination. In addition, several labs have shown that SPARC expression by tumor cells in human pancreatic adenocarcinoma is a prognostic indicator of better outcome, which is in direct opposition to how SPARC expression by the stromal compartment correlates with survival (Brune et al., 2008; Hong et al., 2008; Sato et al., 2003). However, these studies are consistent with the aforementioned data and show that SPARC inhibits pancreatic tumor cell proliferation (Guweidhi et al., 2005; Sato et al., 2003). These examples highlight the necessity for studies aimed at clarifying what and how distinct cell populations are effected by the loss of SPARC in the tumor microenvironment.
We propose that the function of SPARC in malignant progression is not trivial and is dependent on a variety of factors such as the epithelial/mesenchymal state or stage of the tumor; integrin expression and surface localization; ECM composition; chemokine/growth factor profiles and bioavailability; and immune modulation. Our study emphasizes the complexity of SPARC function during tumorigenesis and underscores the necessity for comprehensive studies in the pursuit of a SPARC mechanism(s).
METHODS
Tissue culture
The murine pancreatic adenocarcinoma cell line (PAN02, also known as Panc02) was purchased from the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Frederick, MD), and grown in high glucose Dulbecco’s modified Eagle’s medium (DMEM plus GlutaMAX-1) (Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum (FBS) (Life Technologies, Grand Island, NY). The PAN02 cell line was tested (Impact III PCR profiles) (MU Research Animal Diagnostic Laboratory, Columbia, MO) and was found to be pathogen free.
Orthotopic tumor model
B6;129S-Sparctm1Hwe mice, which can be obtained from The Jackson Laboratory (Stock #003728), were generated as described previously (Norose et al., 1998) and backcrossed into the C57BL/6J background for a minimum of ten generations. The mice were housed in a pathogen-free facility and experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee of UT Southwestern Medical Center (Dallas, TX). All experiments were performed with Sparc-null (Sparc−/−) and wild-type (Sparc+/+) littermates. For injections, confluent cultures of PAN02 cells (>90% viable) were treated with trypsin, pelleted in DMEM 5% FBS, washed twice in phosphate buffered saline (PBS) and resuspended in 0.9% sterile saline (Sigma, St Louis, MO). Tumor cells (1×106) were injected directly into the tail of the pancreas to establish orthotopic tumors, as described previously (Arnold et al., 2008; Bruns et al., 1999). The mice were evaluated for changes in body weight and signs of discomfort or morbidity, whereas bulk tumor growth was monitored through abdominal palpation. Mice were euthanized 6–8 weeks after tumor cell injection, when the majority of mice showed signs of morbidity, and screened visually for locally invasive and metastatic lesions. The liver, heart, lung, kidney, brain, spleen and pancreas, including the tumor, were removed and weighed. Metastasis was assessed macroscopically by visual examination. Suspected metastases were fixed, stained with H&E, and verified as metastatic lesions histologically. Therefore, only the visible macroscopic metastatic and locally invasive lesions were counted towards metastatic incidence and burden. Five independent experiments were performed. For the survival study, individual mice were monitored daily and were euthanized when they displayed signs of tumor-associated morbidity such as excessive weight gain or loss, ascites, lethargy and/or distress.
Immunohistochemistry
Fixed tissue
Tissues were fixed in either methyl Carnoy’s solution (60% methanol, 30% chloroform, 10% glacial acetic acid) or 10% formalin and sent to the Molecular Histopathology Laboratory at UT Southwestern Medical Center (Dallas, TX) for paraffin embedding and sectioning. The Molecular Histopathology Laboratory also performed staining with H&E, Masson’s trichrome or Picrosirius Red according to standard protocols. Tissue sections were deparaffinized and rehydrated in PBS containing 0.2% Tween-20 (PBSt) prior to staining.
Frozen tissue
Tissues were snap-frozen in liquid nitrogen, embedded in optimal cutting temperature compound (OCT) (Tissue-Tek), cut into 10μm-thick sections, air-dried overnight, fixed for 2 minutes in acetone, and washed in PBSt prior to staining.
Chromagen detection
For sections that were developed with either diaminobenzidine (DAB) (Research Genetics, Huntsville, AL) or 3-amino-9-ethylcarbazole (AEC) (Sigma), endogenous peroxidases were blocked by incubating the samples in a 3% H2O2/methanol solution. Sections were then blocked in 20% AquaBlock (East Coast Biologics, North Berwick, ME), incubated with primary antibody overnight at 4°C, then incubated with peroxidase-conjugated secondary antibody (1:500) (Jackson Immunoresearch, West Grove, PA) for 1 hour at room temperature. Lastly, sections were developed with DAB or AEC for 5–20 minutes and counterstained with Meyer’s hematoxylin solution for 3–5 minutes. DAB-developed slides were mounted in Permount (Fisher Scientific, Pittsburgh, PA), whereas AEC-treated sections were mounted in Crystal Mount (Biomeda Corporation, Foster City, CA) and dried for 1 hour at 60°C.
Fluorescence detection
Deparaffinized and rehydrated sections were blocked in 20% AquaBlock (East Coast Biologics), incubated with primary antibody overnight at 4°C, incubated with fluorophore-conjugated (FITC or Cy3) secondary antibody (1:500) (Jackson Immunoresearch) for 1 hour at room temperature, and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen).
Antibody specifics
Primary antibodies used for immunohistochemistry are listed in supplementary material Table S1 (Bernstein et al., 1996; Bernstein et al., 1995; Brekken et al., 1998; Hallmann et al., 1995; Ran et al., 2003). Antigen retrieval was performed, as indicated, by either boiling in citrate buffer (Lab Vision/Thermo Scientific, Fremont, CA) in a pressure cooker for 20 minutes or by digesting with 20 μg/ml proteinase K for 5 minutes at room temperature. Biotinylated Gv39M staining required blocking with avidin-biotin block (Cell Marque, Rocklin, CA) according to the manufacturer’s protocol.
Imaging and quantification
Tissue sections were analyzed with a Nikon Eclipse E600 microscope (Nikon, Lewisville, TX). Color images were captured with a Nikon Digital Dx1200me camera and Act1 software (Universal Imaging Corporation, Downington, PA). Collagen deposition in Masson’s trichrome-stained tissue was performed with NIS Elements AR 2.30 software (Nikon) by thresholding images for the characteristic blue hue that is associated with fibrillar collagens. Sections stained with Picrosirius Red were visualized under polarized light. Fluorescence images were captured with a Photometric Coolsnap HQ camera and NIS Elements. Fluorescent images were captured randomly throughout the entire tumor, including the center and border, and under identical conditions, including magnification and exposure time, to allow quantification of signal intensities, object counts and the percentage of thresholded area with NIS Elements AR 2.30 software (Nikon). Images were thresholded to exclude any background signal from the secondary antibody alone. An average of ten images per tumor and a minimum of three tumors per group were used for each target.
Transmission electron microscopy (TEM)
Mice were anesthetized with isoflurane and perfused with 20 mls of TEM buffer (4% paraformaldehyde and 1% glutaraldehyde in 0.1 M cacodylate buffer). The organs were collected, weighed and placed in TEM buffer at 4°C. The tissues were processed for TEM by the Molecular and Cellular Imaging Facility (MCIF) at UT Southwestern Medical Center (Dallas, TX). Images were taken with the FEI Tecnai G2 Spirit Biotwin (FEI Co., Hillsboro, OR), which was housed in the MCIF.
Evans Blue dye (EBD) permeability
Prior to sacrifice, mice were injected intravenously (in the tail vein) with 5 mg/ml EBD (Sigma) in 0.9% sterile saline at a dose of 30 mg/kg. The EBD was allowed to circulate for 30 minutes before the mice were anesthetized with isoflurane and perfused with sterile PBS at a constant rate for 8 minutes. Organs were removed, weighed, snap-frozen, embedded in OCT and cut into 10 μm-thick sections. EBD permeability was visualized under fluorescence microscopy with excitation and emission wavelengths of 580 nm (green) and 680 nm (red), respectively. Images were taken for both the EBD (red channel) and the autofluorescence (green channel). A minimum of six random pictures were taken of each tissue and an average of three animals per group were used to quantify EBD permeability. Using NIS Elements software, images were corrected for background autofluorescence on a pixel-by-pixel basis, as described previously (Murphy and Lever, 2002). Briefly, the red autofluorescence of images from injected mice was calculated by multiplying the green autofluorescence image by the mean ratio of red to green autofluorescence intensity of uninjected control tissue (ratio=0.37). The resulting image was subtracted from the corresponding red image to yield the autofluorescence-corrected EBD permeability. Corrected images were then thresholded and the percentage of thresholded area was recorded.
Fluorescent dextran permeability
Prior to sacrifice, mice were injected intravenously (in the tail vein) with a mixture of FITC-conjugated dextran (FITC-dextran) (25 mg/ml; 2×106 kDa; D7137) (Molecular Probes/Invitrogen, Eugene, OR) and rhodamine B-conjugated dextran (rhodamine-dextran) (12.5 mg/ml; 1×104 kDa; D1824) (Molecular Probes) in 0.9% sterile saline at a dose of 200 μl/mouse. The fluorescent dextrans were allowed to circulate for 10 minutes before the mice were anesthetized with isoflurane. Organs were removed, weighed, snap-frozen, embedded in OCT and cut into 10 μm-thick sections. FITC-dextran and rhodamine-dextran permeability were assessed by fluorescence microscopy. A minimum of six random photographs were taken of each tissue, with an average of three animals per group. Results were recorded as the mean percentage of thresholded area.
Tumor hypoxia analysis
Prior to sacrifice, mice were injected intravenously (in the tail vein) with 60 mg/kg of Hypoxyprobe-1 (HP2–100; Chemicon, Temecula, CA) that had been resuspended at a concentration of 30 mg/ml in 0.9% sterile saline. The solution was allowed to circulate for 90 minutes before the mice were anesthetized with isoflurane. Organs were removed, weighed, snap-frozen, embedded in OCT and cut into 10 μm-thick sections. Next, the sections were fixed, blocked and stained with the primary antibodies rat anti-mouse endothelial cell (Meca-32) and FITC-conjugated mouse anti-pimonidazole (Chemicon), followed by Cy3-conjugated donkey anti-rat secondary IgG (Jackson Immunoresearch). A minimum of six random photographs were taken of each tissue and an average of three animals per group were used to quantify hypoxia.
Hydroxyproline analysis
Mice were anesthetized with isoflurane and their organs were removed and weighed as described before. Organs were then snap-frozen, lyophilized, weighed (dry weight), pulverized and subjected to complete acid hydrolysis with 6 N HCl for 18 hours at 120°C. Each sample was then neutralized to pH 7 with 4 N NaOH. One milliliter of chloramine T was added to 2 ml volumes of collagen sample and incubated at room temperature for 20 minutes. One milliliter of Ehrlich’s reagent (60% perchloric acid, 15 ml 1-propanol, 3.75 g p-dimethylaminobenzaldehyde in 25 ml) was added and samples were incubated at 60°C for 20 minutes. Absorbance was read at a wavelength of 558 nm on a spectrophotometer. Collagen was quantified as μg of hydroxyproline per mg dry weight of starting material.
Statistical analyses
Metastatic incidence rates (the number of mice with metastasis divided by the total number of mice in the group) in Sparc−/− mice were compared with Sparc+/+ mice by logistic regression model, assuming a binomial distribution. Metastatic event rates (the number of metastatic events divided by the total number of mice) in Sparc−/−mice were compared with Sparc+/+ mice by Poisson regression model, assuming a Poisson distribution. We assume that metastatic events in different organs were independent. Both the logistic regression and Poisson regression were implanted in R software (R Development Core Team, 2005) (http://www.R-project.org). The unpaired Student’s t-test of immunohistochemistry quantification was carried out in GraphPad Prism (GraphPad Software, San Diego, CA) where a P value of less than 0.05 was considered significant. The survival curves were analyzed in GraphPad Prism with the Gehan-Breslow-Wilcoxon test.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr Tom Wight and members of the Brekken laboratory for helpful discussion, the Molecular and Cellular Imaging Facility at UT-Southwestern for assistance with TEM, Dr Shane E. Holloway for assistance with surgical implantation of tumor cells, and Dr Larry Fisher for providing the anti-collagen I antibody LF-67. The hybridoma MECA-32, developed by Dr Eugene C. Butcher, was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of NICHD and maintained by The University of Iowa (Iowa City, IA 52242). This study was supported in part by The Effie Marie Cain Scholarship in Angiogenesis Research (R.A.B.) and the NIH (R01CA118240 to R.A.B. and R01GM40711 to E.H.S.). S.A.A. was supported by an NIH training grant (GM007062). D.H.C. was supported by an NIH/NCRR grant (K26RR024196). P.P. was supported by Helsinki and Turku University Central Hospital Research grants (EVO) and a grant from the Sigrid Juselius Foundation. Deposited in PMC for release after 12 months.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
S.A.A. contributed to the design and execution of all experiments, performed the data analysis and wrote the manuscript. L.B.R. assisted with animal studies. A.F.M. contributed to the initial design and optimization of the permeability and perfusion studies. J.G.C. assisted in the maintenance of the animal colony and contributed to the animal studies. S.P.D. performed surgical implantation of tumor cells and assisted in animal studies. D.H.C. reviewed the pathology of tumor/tissue sections. Y.X. performed the statistical analysis on the metastatic incidence and events. E.H.S. provided reagents, contributed to the interpretation of results and aided in the drafting of the manuscript. P.P. performed the tumor studies, contributed to the interpretation of results and aided in the drafting of the manuscript. A.D.B. performed hydroxyproline analysis, contributed to the interpretation of results and aided in the drafting of the manuscript. R.A.B. oversaw the design, execution and interpretation of the experiments and the writing of the manuscript.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.003228/-/DC1
REFERENCES
- Arnold S, Mira E, Muneer S, Korpanty G, Beck AW, Holloway SE, Manes S, Brekken RA. (2008). Forced expression of MMP9 rescues the loss of angiogenesis and abrogates metastasis of pancreatic tumors triggered by the absence of host SPARC. Exp Biol Med. 233, 860–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Hashizume H, McDonald DM. (2005). Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 15, 102–111 [DOI] [PubMed] [Google Scholar]
- Bernstein EF, Fisher LW, Li K, LeBaron RG, Tan EM, Uitto J. (1995). Differential expression of the versican and decorin genes in photoaged and sun-protected skin. Comparison by immunohistochemical and northern analyses. Lab Invest. 72, 662–669 [PubMed] [Google Scholar]
- Bernstein EF, Chen YQ, Kopp JB, Fisher L, Brown DB, Hahn PJ, Robey FA, Lakkakorpi J, Uitto J. (1996). Long-term sun exposure alters the collagen of the papillary dermis. Comparison of sun-protected and photoaged skin by northern analysis, immunohistochemical staining, and confocal laser scanning microscopy. J Am Acad Dermatol. 34, 209–218 [DOI] [PubMed] [Google Scholar]
- Bingle L, Brown NJ, Lewis CE. (2002). The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 196, 254–265 [DOI] [PubMed] [Google Scholar]
- Bornstein P. (2001). Thrombospondins as matricellular modulators of cell function. J Clin Invest. 107, 929–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P. (2002). Cell-matrix interactions: the view from the outside. Methods Cell Biol. 69, 7–11 [DOI] [PubMed] [Google Scholar]
- Bornstein P, Sage EH. (2002). Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 14, 608–616 [DOI] [PubMed] [Google Scholar]
- Bradshaw AD, Francki A, Motamed K, Howe C, Sage EH. (1999). Primary mesenchymal cells isolated from SPARC-null mice exhibit altered morphology and rates of proliferation. Mol Biol Cell 10, 1569–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw AD, Puolakkainen P, Dasgupta J, Davidson JM, Wight TN, Helene Sage E. (2003). SPARC-null mice display abnormalities in the dermis characterized by decreased collagen fibril diameter and reduced tensile strength. J Invest Dermatol. 120, 949–955 [DOI] [PubMed] [Google Scholar]
- Brekken RA, Sage EH. (2001). SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 19, 816–827 [DOI] [PubMed] [Google Scholar]
- Brekken RA, Huang X, King SW, Thorpe PE. (1998). Vascular endothelial growth factor as a marker of tumor endothelium. Cancer Res. 58, 1952–1959 [PubMed] [Google Scholar]
- Brekken RA, Puolakkainen P, Graves DC, Workman G, Lubkin SR, Sage EH. (2003). Enhanced growth of tumors in SPARC null mice is associated with changes in the ECM. J Clin Invest. 111, 487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune K, Hong SM, Li A, Yachida S, Abe T, Griffith M, Yang D, Omura N, Eshleman J, Canto M, et al. (2008). Genetic and epigenetic alterations of familial pancreatic cancers. Cancer Epidemiol Biomarkers Prev. 17, 3536–3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. (1999). In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1, 50–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham S, Tang MJ, Mesak F, Kennecke H, Owen D, Tai IT. (2008). SPARC promoter hypermethylation in colorectal cancers can be reversed by 5-Aza-2’deoxycytidine to increase SPARC expression and improve therapy response. Br J Cancer 98, 1810–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin D, Boyle GM, Williams RM, Ferguson K, Pandeya N, Pedley J, Campbell CM, Theile DR, Parsons PG, Coman WB. (2005). Novel markers for poor prognosis in head and neck cancer. Int J Cancer 113, 789–797 [DOI] [PubMed] [Google Scholar]
- Choi P, Jordan CD, Mendez E, Houck J, Yueh B, Farwell DG, Futran N, Chen C. (2008). Examination of oral cancer biomarkers by tissue microarray analysis. Arch Otolaryngol Head Neck Surg. 134, 539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmouliere A, Guyot C, Gabbiani G. (2004). The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol. 48, 509–517 [DOI] [PubMed] [Google Scholar]
- Engbring JA, Kleinman HK. (2003). The basement membrane matrix in malignancy. J Pathol. 200, 465–470 [DOI] [PubMed] [Google Scholar]
- Framson PE, Sage EH. (2004). SPARC and tumor growth: where the seed meets the soil? J Cell Biochem. 92, 679–690 [DOI] [PubMed] [Google Scholar]
- Francki A, McClure TD, Brekken RA, Motamed K, Murri C, Wang T, Sage EH. (2004). SPARC regulates TGF-beta1-dependent signaling in primary glomerular mesangial cells. J Cell Biochem. 91, 915–925 [DOI] [PubMed] [Google Scholar]
- Gerhardt H, Semb H. (2008). Pericytes: gatekeepers in tumour cell metastasis? J Mol Med. 86, 135–144 [DOI] [PubMed] [Google Scholar]
- Guweidhi A, Kleeff J, Adwan H, Giese NA, Wente MN, Giese T, Buchler MW, Berger MR, Friess H. (2005). Osteonectin influences growth and invasion of pancreatic cancer cells. Ann Surg. 242, 224–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmann R, Mayer DN, Berg EL, Broermann R, Butcher EC. (1995). Novel mouse endothelial cell surface marker is suppressed during differentiation of the blood brain barrier. Dev Dyn. 202, 325–332 [DOI] [PubMed] [Google Scholar]
- Hasselaar P, Sage EH. (1992). SPARC antagonizes the effect of basic fibroblast growth factor on the migration of bovine aortic endothelial cells. J Cell Biochem. 49, 272–283 [DOI] [PubMed] [Google Scholar]
- Hong SM, Kelly D, Griffith M, Omura N, Li A, Li CP, Hruban RH, Goggins M. (2008). Multiple genes are hypermethylated in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol. 21, 1499–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C, Goggins M. (2007). Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 25, 319–325 [DOI] [PubMed] [Google Scholar]
- Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis DL, Agnantis NJ, Pavlidis N. (2002). Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer 38, 2362–2370 [DOI] [PubMed] [Google Scholar]
- Juliano RL. (2002). Signal transduction by cell adhesion receptors and the cytoskeleton: functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members. Annu Rev Pharmacol Toxicol. 42, 283–323 [DOI] [PubMed] [Google Scholar]
- Kahn SL, Ronnett BM, Gravitt PE, Gustafson KS. (2008). Quantitative methylation-specific PCR for the detection of aberrant DNA methylation in liquid-based Pap tests. Cancer 114, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Nagashima Y, Baba Y, Kawano T, Furukawa M, Kubota A, Yanoma S, Imagawa-Ishiguro Y, Satake K, Taguchi T, et al. (2005). Expression of SPARC in tongue carcinoma of stage II is associated with poor prognosis: an immunohistochemical study of 86 cases. Int J Mol Med. 16, 263–268 [PubMed] [Google Scholar]
- Kim WH, Lee BL, Kim DK, Kleinman HK. (1999). Laminin-1-adherent cancer cells show increased proliferation and decreased apoptosis in vivo. Anticancer Res. 19, 3067–3071 [PubMed] [Google Scholar]
- Korc M. (2007). Pancreatic cancer-associated stroma production. Am J Surg. 194, S84–S86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupprion C, Motamed K, Sage EH. (1998). SPARC (BM-40, osteonectin) inhibits the mitogenic effect of vascular endothelial growth factor on microvascular endothelial cells. J Biol Chem. 273, 29635–29640 [DOI] [PubMed] [Google Scholar]
- Liotta LA, Kohn EC. (2001). The microenvironment of the tumour-host interface. Nature 411, 375–379 [DOI] [PubMed] [Google Scholar]
- Liu C, Shao ZM, Zhang L, Beatty P, Sartippour M, Lane T, Livingston E, Nguyen M. (2001). Human endomucin is an endothelial marker. Biochem Biophys Res Commun. 288, 129–136 [DOI] [PubMed] [Google Scholar]
- Mahadevan D, Von Hoff DD. (2007). Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 6, 1186–1197 [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. (2002). Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 [DOI] [PubMed] [Google Scholar]
- McCarthy JB, Furcht LT. (1984). Laminin and fibronectin promote the haptotactic migration of B16 mouse melanoma cells in vitro. J Cell Biol. 98, 1474–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendis DB, Ivy GO, Brown IR. (1998). SPARC/osteonectin mRNA is induced in blood vessels following injury to the adult rat cerebral cortex. Neurochem Res. 23, 1117–1123 [DOI] [PubMed] [Google Scholar]
- Mok SC, Chan WY, Wong KK, Muto MG, Berkowitz RS. (1996). SPARC, an extracellular matrix protein with tumor-suppressing activity in human ovarian epithelial cells. Oncogene 12, 1895–1901 [PubMed] [Google Scholar]
- Morgan SM, Samulowitz U, Darley L, Simmons DL, Vestweber D. (1999). Biochemical characterization and molecular cloning of a novel endothelial-specific sialomucin. Blood 93, 165–175 [PubMed] [Google Scholar]
- Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. (2002). Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 160, 985–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Muthana M, Coffelt SB, Lewis CE. (2008). The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8, 618–631 [DOI] [PubMed] [Google Scholar]
- Murphy CL, Lever MJ. (2002). A ratiometric method of autofluorescence correction used for the quantification of Evans blue dye fluorescence in rabbit arterial tissues. Exp Physiol. 87, 163–170 [DOI] [PubMed] [Google Scholar]
- Nie J, Chang B, Traktuev DO, Sun J, March K, Chan L, Sage EH, Pasqualini R, Arap W, Kolonin MG. (2008). IFATS collection: Combinatorial peptides identify alpha5beta1 integrin as a receptor for the matricellular protein SPARC on adipose stromal cells. Stem Cells 26, 2735–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norose K, Clark JI, Syed NA, Basu A, Heber-Katz E, Sage EH, Howe CC. (1998). SPARC deficiency leads to early-onset cataractogenesis. Invest Ophthalmol Vis Sci. 39, 2674–2680 [PubMed] [Google Scholar]
- Ohnishi T, Arita N, Hiraga S, Taki T, Izumoto S, Fukushima Y, Hayakawa T. (1997). Fibronectin-mediated cell migration promotes glioma cell invasion through chemokinetic activity. Clin Exp Metastasis 15, 538–546 [DOI] [PubMed] [Google Scholar]
- Ohnishi T, Hiraga S, Izumoto S, Matsumura H, Kanemura Y, Arita N, Hayakawa T. (1998). Role of fibronectin-stimulated tumor cell migration in glioma invasion in vivo: clinical significance of fibronectin and fibronectin receptor expressed in human glioma tissues. Clin Exp Metastasis 16, 729–741 [DOI] [PubMed] [Google Scholar]
- Pardali K, Moustakas A. (2007). Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta 1775, 21–62 [DOI] [PubMed] [Google Scholar]
- Pen A, Moreno MJ, Martin J, Stanimirovic DB. (2007). Molecular markers of extracellular matrix remodeling in glioblastoma vessels: microarray study of laser-captured glioblastoma vessels. Glia 55, 559–572 [DOI] [PubMed] [Google Scholar]
- Podhajcer OL, Benedetti LG, Girotti MR, Prada F, Salvatierra E, Llera AS. (2008). The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 27, 691–705 [DOI] [PubMed] [Google Scholar]
- Puolakkainen PA, Brekken RA, Muneer S, Sage EH. (2004). Enhanced growth of pancreatic tumors in SPARC-null mice is associated with decreased deposition of extracellular matrix and reduced tumor cell apoptosis. Mol Cancer Res. 2, 215–224 [PubMed] [Google Scholar]
- R Development Core Team (2005). A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing [Google Scholar]
- Raines EW, Lane TF, Iruela-Arispe ML, Ross R, Sage EH. (1992). The extracellular glycoprotein SPARC interacts with platelet-derived growth factor (PDGF)-AB and -BB and inhibits the binding of PDGF to its receptors. Proc Natl Acad Sci USA 89, 1281–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran S, Huang X, Downes A, Thorpe PE. (2003). Evaluation of novel antimouse VEGFR2 antibodies as potential antiangiogenic or vascular targeting agents for tumor therapy. Neoplasia 5, 297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed CC, Iozzo RV. (2002). The role of decorin in collagen fibrillogenesis and skin homeostasis. Glycoconj J. 19, 249–255 [DOI] [PubMed] [Google Scholar]
- Reed MJ, Puolakkainen P, Lane TF, Dickerson D, Bornstein P, Sage EH. (1993). Differential expression of SPARC and thrombospondin 1 in wound repair: immunolocalization and in situ hybridization. J Histochem Cytochem. 41, 1467–1477 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Jimenez FJ, Caldes T, Iniesta P, Vidart JA, Garcia-Asenjo JL, Benito M. (2007). Overexpression of SPARC protein contrasts with its transcriptional silencing by aberrant hypermethylation of SPARC CpG-rich region in endometrial carcinoma. Oncol Rep. 17, 1301–1307 [PubMed] [Google Scholar]
- Sage H, Vernon RB, Funk SE, Everitt EA, Angello J. (1989). SPARC, a secreted protein associated with cellular proliferation, inhibits cell spreading in vitro and exhibits Ca+2-dependent binding to the extracellular matrix. J Cell Biol. 109, 341–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Hohenester E, Gohring W, Timpl R. (1998). Crystal structure and mapping by site-directed mutagenesis of the collagen-binding epitope of an activated form of BM-40/SPARC/osteonectin. EMBO J. 17, 1625–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Miosge N, Timpl R. (1999). Immunochemical and tissue analysis of protease generated neoepitopes of BM-40 (osteonectin, SPARC) which are correlated to a higher affinity binding to collagens. Matrix Biol. 18, 499–508 [DOI] [PubMed] [Google Scholar]
- Sato N, Fukushima N, Maehara N, Matsubayashi H, Koopmann J, Su GH, Hruban RH, Goggins M. (2003). SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions. Oncogene 22, 5021–5030 [DOI] [PubMed] [Google Scholar]
- Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. (2008). Macrophage polarization in tumour progression. Semin Cancer Biol. 18, 349–355 [DOI] [PubMed] [Google Scholar]
- Sova P, Feng Q, Geiss G, Wood T, Strauss R, Rudolf V, Lieber A, Kiviat N. (2006). Discovery of novel methylation biomarkers in cervical carcinoma by global demethylation and microarray analysis. Cancer Epidemiol Biomarkers Prev. 15, 114–123 [DOI] [PubMed] [Google Scholar]
- Suzuki M, Hao C, Takahashi T, Shigematsu H, Shivapurkar N, Sathyanarayana UG, Iizasa T, Fujisawa T, Hiroshima K, Gazdar AF. (2005). Aberrant methylation of SPARC in human lung cancers. Br J Cancer 92, 942–948 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Timar J, Lapis K, Dudas J, Sebestyen A, Kopper L, Kovalszky I. (2002). Proteoglycans and tumor progression: janus-faced molecules with contradictory functions in cancer. Semin Cancer Biol. 12, 173–186 [DOI] [PubMed] [Google Scholar]
- Truty MJ, Urrutia R. (2007). Basics of TGF-beta and pancreatic cancer. Pancreatology 7, 423–435 [DOI] [PubMed] [Google Scholar]
- Wang H, Fu W, Im JH, Zhou Z, Santoro SA, Iyer V, DiPersio CM, Yu QC, Quaranta V, Al-Mehdi A, et al. (2004). Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J Cell Biol. 164, 935–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yu Q, Cho AH, Rondeau G, Welsh J, Adamson E, Mercola D, McClelland M. (2005). Survey of differentially methylated promoters in prostate cancer cell lines. Neoplasia 7, 748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver MS, Workman G, Sage EH. (2008). The copper binding domain of SPARC mediates cell survival in vitro via interaction with integrin beta1 and activation of integrin-linked kinase. J Biol Chem. 283, 22826–22837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernert N. (1997). The multiple roles of tumour stroma. Virchows Arch. 430, 433–443 [DOI] [PubMed] [Google Scholar]
- Wong SY, Crowley D, Bronson RT, Hynes RO. (2008). Analyses of the role of endogenous SPARC in mouse models of prostate and breast cancer. Clin Exp Metastasis 25, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xian X, Hakansson J, Stahlberg A, Lindblom P, Betsholtz C, Gerhardt H, Semb H. (2006). Pericytes limit tumor cell metastasis. J Clin Invest. 116, 642–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue LY, Hu N, Song YM, Zou SM, Shou JZ, Qian LX, Ren LQ, Lin DM, Tong T, He ZG, et al. (2006). Tissue microarray analysis reveals a tight correlation between protein expression pattern and progression of esophageal squamous cell carcinoma. BMC Cancer 6, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Clark JI, Wight TN, Sage EH. (2002). Alterations in the lens capsule contribute to cataractogenesis in SPARC-null mice. J Cell Sci. 115, 2747–2756 [DOI] [PubMed] [Google Scholar]
- Yang E, Kang HJ, Koh KH, Rhee H, Kim NK, Kim H. (2007). Frequent inactivation of SPARC by promoter hypermethylation in colon cancers. Int J Cancer 121, 567–575 [DOI] [PubMed] [Google Scholar]
- Zalatnai A. (2006). Molecular aspects of stromal-parenchymal interactions in malignant neoplasms. Curr Mol Med. 6, 685–693 [DOI] [PubMed] [Google Scholar]
- Zhou X, Tan FK, Guo X, Wallis D, Milewicz DM, Xue S, Arnett FC. (2005). Small interfering RNA inhibition of SPARC attenuates the profibrotic effect of transforming growth factor beta1 in cultured normal human fibroblasts. Arthritis Rheum. 52, 257–261 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
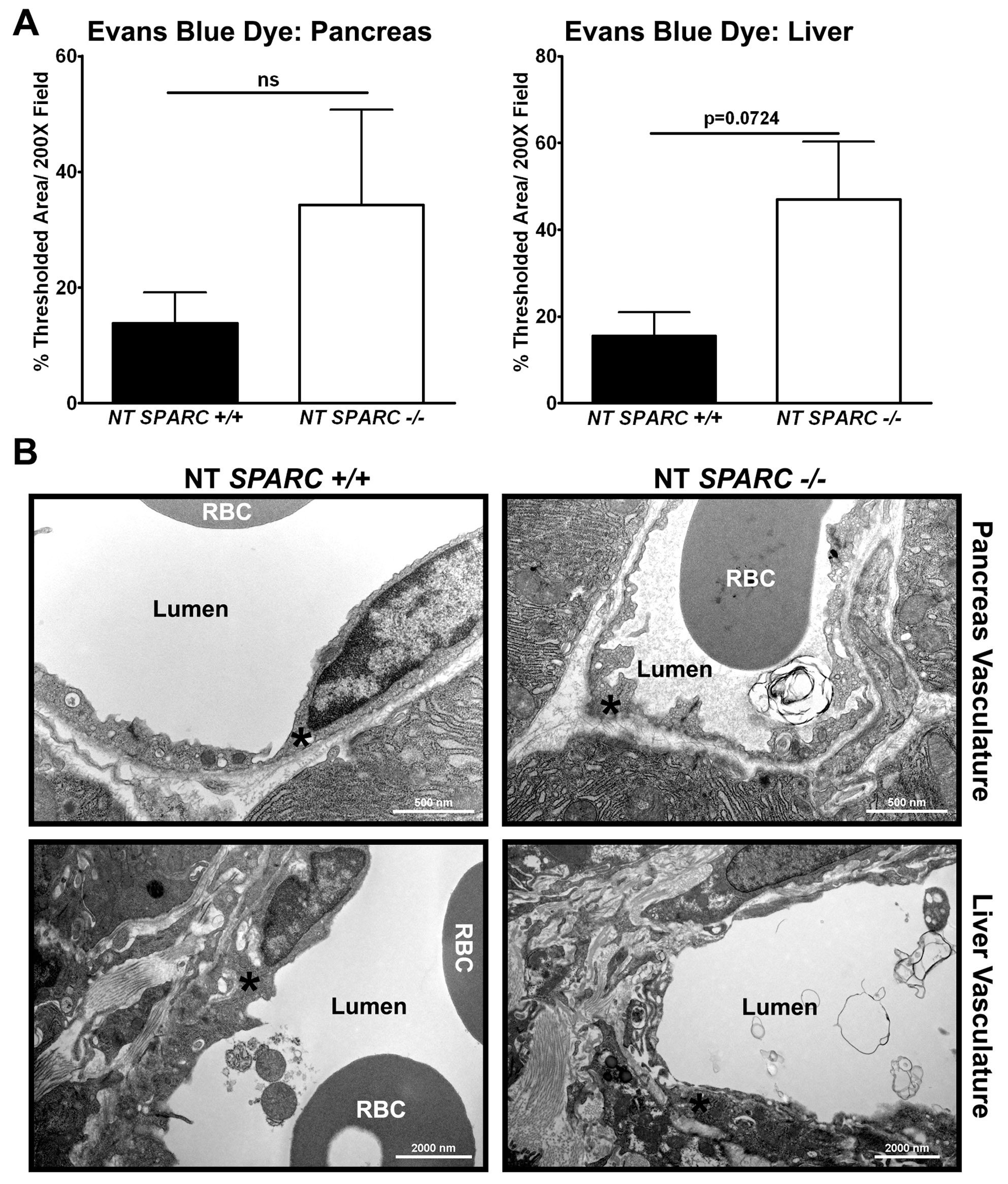{kind=link}