

SUMMARY
KRIT1, also called CCM1, is a member of a multiprotein complex that contains the products of the CCM2 and PDCD10 (also known as CCM3) loci. Heterozygous loss of any of the genes that encode these proteins leads to cerebral cavernous malformations (CCM), which are vascular lesions that are found in around 0.5% of humans. KRIT1 mediates the stabilization of β-catenin-containing endothelial cell-cell junctions downstream of the Rap1 GTPase. Here, we report that Rap1 and KRIT1 are negative regulators of canonical β-catenin signaling in mice and that hemizygous Krit1 deficiency exacerbates β-catenin-driven pathologies. Depletion of endothelial KRIT1 caused β-catenin to dissociate from vascular endothelial (VE)-cadherin and to accumulate in the nucleus with consequent increases in β-catenin-dependent transcription. Activation of Rap1 inhibited β-catenin-dependent transcription in confluent endothelial cells; this effect required the presence of intact cell-cell junctions and KRIT1. These effects of KRIT1 were not limited to endothelial cells; the KRIT1 protein was expressed widely and its depletion increased β-catenin signaling in epithelial cells. Moreover, a reduction in KRIT1 expression also increased β-catenin signaling in vivo. Hemizygous deficiency of Krit1 resulted in a ~1.5-fold increase in intestinal polyps in the ApcMin/+ mouse, which was associated with increased β-catenin-driven transcription. Thus, KRIT1 regulates β-catenin signaling, and Krit1+/− mice are more susceptible to β-catenin-driven intestinal adenomas.
INTRODUCTION
KRIT1 was first identified as a binding partner of the GTPase Rap1a (Serebriiskii et al., 1997), a regulator of cell-cell adhesion in many cell types (Price et al., 2004; Cullere et al., 2005). KRIT1, also called CCM1, is a member of a multiprotein complex that contains CCM2 and CCM3 (PDCD10) (Zawistowski et al., 2005; Voss et al., 2007). There are similar vascular malformations in KRIT1, CCM2 and PDCD10 heterozygous humans and similar lethal phenotypes in homozygous null animals (Whitehead et al., 2004; Plummer et al., 2005; Mably et al., 2006; Gore et al., 2008; Boulday et al., 2009; Kleaveland et al., 2009; Voss et al., 2009; Whitehead et al., 2009). These genetic relationships, combined with the physical association of these proteins, lend credence to their interdependence of function. Heterozygous loss of CCM1 is associated with the development of cerebral cavernous malformations (CCM) (Laberge-le Couteulx et al., 1999; Sahoo et al., 1999), a rare (0.1–0.5% incidence), autosomal dominant disorder characterized by the development of multiple vascular dysplasias within the brain. CCM lesions consist of beds of dilated, leaky capillary vessels. The vessels are also marked by a lack of accessory cells and altered gene expression (Kilic et al., 2000; Clatterbuck et al., 2001; Revencu and Vikkula, 2006). However, little is known about the mechanism(s) that underlie development of the disease. We previously reported that KRIT1 is a Rap1 effector that is required for the stabilizing effect of Rap1 on endothelial cell-cell junctions, where KRIT1 associates with junctional proteins including β-catenin and vascular endothelial (VE)-cadherin (Glading et al., 2007).
Cadherin-based structures (adherens junctions) regulate diverse cellular behaviors, including proliferation and migration (Ivanov et al., 2001), and play a dominant role in endothelial barrier function (Dejana, 2004). β-Catenin participates in the formation and stabilization of cadherin-based adhesions by forming a connection to the actin cytoskeleton (Aberle et al., 1996). β-Catenin is also a key element of the canonical Wnt (wingless and Int-1) signaling pathway, which promotes the nuclear localization of β-catenin by disrupting the axin–adenomatous polyposis coli (APC)–glycogen synthase kinase 3β (GSK3β)–β-catenin complex that normally targets cytoplasmic β-catenin for degradation (Clevers, 2006). The Wnt–β-catenin signaling pathway is crucial during development; dysregulation of this pathway has been implicated in the development of multiple tumors of epithelial origin, including colon adenocarcinoma and breast cancer. Binding of β-catenin to cadherins can antagonize Wnt signaling by sequestering β-catenin at the membrane (Sanson et al., 1996; Sadot et al., 1998; Orsulic et al., 1999). Disruption of adherens junctions is accompanied by the release of β-catenin from the cytoplasmic tail of the cadherin (Potter et al., 2005) and concomitant changes in gene expression owing to the increased nuclear localization of β-catenin and the subsequent activation of T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcriptional complexes (Solanas et al., 2008; Taddei et al., 2008).
We hypothesized that, because loss of KRIT1 disrupts adherens junctions, loss of KRIT1 could induce the nuclear localization of β-catenin, thereby increasing its transcriptional activity. Here, we show that KRIT1 depletion inhibits the association of VE-cadherin with β-catenin, and causes a concomitant increase in the presence and function of β-catenin in the nucleus. KRIT1 is a Rap1 effector and we found that Rap1, a tumor suppressor (Kitayama et al., 1989), inhibits canonical β-catenin signaling in confluent cells that have sufficient levels of KRIT1 (KRIT1-sufficient). However, depletion of KRIT1 blocked the ability of active Rap1 to inhibit β-catenin-driven transcription. Furthermore, we find that the KRIT1 protein is expressed in many cell types and that KRIT1 depletion or hemizygous deletion increases nuclear β-catenin signaling in several cell types. KRIT1 hemizygosity increased intestinal adenoma formation and β-catenin-dependent gene expression in the ApcMin/+ model of colon cancer. Thus, we identify Rap1 and KRIT1 as inhibitors of β-catenin signaling in multiple cell lineages and show that hemizyogus deficiency of Krit1 leads to exacerbation of a β-catenin-driven epithelial tumor. These studies reveal the function of the CCM signaling complex in maintaining endothelial junctional integrity, and provide new insight into how mutations in CCM proteins can affect vascular development and how they may lead to increased intestinal tumorigenesis.
RESULTS
Loss of KRIT1 regulates β-catenin nuclear localization and reduces its association with cadherins
We previously found that KRIT1 mediates the capacity of Rap1a to stabilize endothelial cell-cell junctions; notably, loss of KRIT1 led to a loss of β-catenin from cell-cell junctions and an overall decrease in junction integrity (Glading et al., 2007). Endothelial cell-cell adhesion can be modulated by internalization of the cadherin complex (Reynolds and Carnahan, 2004; Gavard and Gutkind, 2006); however, depletion of KRIT1 did not reduce surface expression of VE-cadherin in primary human arterial endothelial cells (HPAECs) (Fig. 1A). As internalization does not occur to the same extent in all types of endothelium (Alcaide et al., 2008), we examined the binding of β-catenin to the cadherin cytoplasmic domain, which also regulates cell-cell adhesion and homophilic cadherin binding (Carmeliet et al., 1999; Reynolds and Carnahan, 2004). KRIT1 depletion reduced the association of β-catenin with VE-cadherin, while having no effect on total catenin or cadherin protein levels (Fig. 1B,C).
Fig. 1.

Loss of KRIT1 reduces β-catenin association with VE-cadherin. (A) Depletion of KRIT1 does not reduce the surface expression of VE-cadherin. Staining of the extracellular domain of VE-cadherin was compared with isotype control staining (red line) by FACS analysis of negative control small interfering RNA (siRNA)-transfected cells (black line) or anti-KRIT1 siRNA-transfected cells (blue line). Histograms are representative, n=3. Knockdown of KRIT1 was confirmed by western blot (right panel) in cells used for FACS analysis and co-immunoprecipitation. NC, negative control siRNA; 530, anti-KRIT1 siRNA; IP, immunoprecipitation; IB, immunoblot; mab, monoclonal antibody; pab, polyclonal antibody. n=3 separate determinations. (B) β-Catenin co-immunoprecipitates with VE-cadherin in cells expressing negative control siRNA or untreated cells (not shown); this association is diminished in cells expressing anti-KRIT1 siRNA. RbIgG, rabbit IgG. Blots are representative, n=3. (C) Densitometric quantification of co-immunoprecipitation western blots. Data shown are the relative expression of VE-cadherin and β-catenin in both immunoprecipitation (IP) and input samples, normalized to negative control siRNA expressing cells ± standard deviation (S.D.). *P<0.001 by Student’s t-test, n=3. The far right panel shows quantification of KRIT1 expression in these experiments ±S.D. *P<0.001 by Student’s t-test, n=3.
Increased nuclear localization of β-catenin activates β-catenin-dependent transcription in KRIT1-deficient cells
Depletion of KRIT1 displaced β-catenin from cell-cell junctions; however, loss of KRIT1 did not result in a reduction of total β-catenin (Fig. 1B), rather it led to relocalization of β-catenin to the nucleus, as shown by an increase in nuclear immunofluorescent intensity (Fig. 2A) and by subcellular fractionation (Fig. 2B; supplementary material Fig. S1A). The co-expression of siRNA-resistant KRIT1 reversed the increase in β-catenin nuclear localization, which excluded off-target effects of the siRNA (Fig. 2A). These data indicate that the expression of KRIT1 stabilizes the association of β-catenin with VE-cadherin, and suggest a link between the effect of KRIT1 depletion on cell-cell adhesion and the nuclear localization of β-catenin.
Fig. 2.

Loss of KRIT1 regulates β-catenin nuclear localization and activatesβ-catenin-dependent transcription. (A) β-Catenin and KRIT1 staining of confluent BAECs. Top panels, negative control siRNA; center panels, anti-KRIT1 siRNA; lower panels, anti-KRIT1 siRNA co-transfected with siRNA-resistant KRIT1. Confocal images are representative, n=6. Bar, 50 μm. (B) Densitometric quantification of subcellular fractionation. The data shown are the representative expression of β-catenin in cytoplasmic, nuclear and membrane fractions of siRNA-transfected HPAECs, and are relative to total β-catenin ±S.D. *P<0.05 by Student’s two-tailed t-test, n=3. Representative blots for β-catenin and KRIT1 expression, as well as fractionation control blots, can be found in supplementary material Fig. S1A. (C) TOPFlash reporter activity in KRIT1-depleted cells. Data are shown relative to negative control siRNA-transfected cells (NC). β-cat, FLAG–β-catenin expressing cells; 530, anti-KRIT1 siRNA expressing cells; 530+HA-KRIT1, anti-KRIT1 siRNA and hemagglutinin-tagged (HA)-KRIT1 expressing cells. Overall P=0.002 by analysis of variance (ANOVA), *P<0.05 by Bonferroni post-hoc comparison to negative control siRNA-transfected cells, n=5.
Nuclear β-catenin stimulates transcription through its interaction with TCF and LEF, thereby activating gene expression (Clevers, 2006). The transcriptional output of β-catenin signaling can be monitored using the TOPFlash reporter, which includes six TCF binding sites (Korinek et al., 1997). KRIT1 depletion in bovine aortic endothelial cells (BAECs) increased nuclear β-catenin signaling, as judged by a ~2.5-fold increase in TOPFlash luciferase activity (Fig. 2C). Specificity of this effect was confirmed by the lack of change in FOPFlash activity, a TCF binding-deficient reporter (Korinek et al., 1997). Re-expression of KRIT1 in depleted cells reversed the increase in TOPFlash activity (Fig. 2C; supplementary material Fig. S1B). For comparison, a similar magnitude of increase was induced by the overexpression of FLAG–β-catenin (Fig. 2C; supplementary material Fig. S1B). Thus, depletion of KRIT1 leads to increased nuclear β-catenin and β-catenin-dependent transcription in endothelial cells in vitro.
Activation of Rap1 inhibits β-catenin transcriptional activity via KRIT1
Activated Rap1 increases the localization of KRIT1 to cell-cell junctions and its association with junctional proteins, such as β-catenin; the ability of active Rap1 to stabilize cell-cell junctions requires KRIT1 (Glading et al., 2007). Having found that KRIT1 regulates β-catenin signaling, we suspected that this function might lie downstream of Rap1 signaling, particularly since Rap1 has been identified as a growth inhibitory tumor suppressor (Kitayama et al., 1989). Treatment of BAECs with 50 μM of 8′pCPT-2OMe-cAMP (OMe-cAMP), a specific activator of the Rap1 exchange factor Epac1, or expression of a constitutively active form of Rap1a, RapV12, inhibited β-catenin-driven transcription in β-catenin overexpressing cells (Fig. 3A; supplementary material Fig. S2A). Rap1 activation also reversed β-catenin-dependent transcription induced by thrombin (Fig. 3B), supporting a role for Rap1 in inhibiting the redistribution of β-catenin to the nucleus (Beckers et al., 2008). Furthermore, activation of Rap1 did not inhibit β-catenin reporter activity in cells depleted of KRIT1 (Fig. 3C). Importantly, KRIT1 depletion had no effect on Rap1 expression levels (supplementary material Fig. S2C), suggesting that Rap1 acts via KRIT1 to maintain the association of β-catenin with cadherins, thereby preventing β-catenin-driven transcription. We suspected that Rap1 activation blocked β-catenin-driven transcription by stabilizing cell-cell adhesion, thus promoting β-catenin localization at junctions. To test this hypothesis, we treated cells plated at low density (sparse) or high density (confluent) with OMe-cAMP to determine whether the effect of Rap1 activation was dependent on the presence of cell-cell adhesion. In cells plated sparsely, which had few or no cell-cell contacts (supplementary material Fig. S2B), activation of Rap1 did not inhibit β-catenin reporter activity. However, the effect of Rap1 activation was rescued in sparse cells when the cells were adhered to plates coated with Fc-linked VE-cadherin (Fig. 3D), although the effect of β-catenin overexpression was muted, probably owing to an increase in cadherin adhesion complex formation. Thus, we conclude that Rap1 activation can negatively regulate β-catenin signaling by promoting the stability of cell-cell junctions through a mechanism involving KRIT1.
Fig. 3.

Rap1 activation inhibits β-catenin reporter activity via KRIT1. (A) β-Catenin-dependent reporter activity in cells treated with 50 μM of OMe-cAMP, a membrane-permeant activator of Rap1, or that express constitutively active RapV12. TOPFlash activity is shown relative to the vector-transfected vehicle-treated (vec/veh) cells, n=3, P=0.0001 by ANOVA. *P<0.001 by Bonferroni post-hoc test compared with vector-transfected vehicle-treated cells; **P<0.001 compared with vehicle-treated, β-catenin expressing cells. (B) Rap1 inhibits β-catenin-dependent reporter activity induced by thrombin. TOPFlash activity is shown relative to vehicle-treated cells, n=3, P=0.001 by ANOVA. *P<0.01 by Bonferroni post-hoc test compared with vehicle-treated cells; **P<0.01 compared with thrombin-treated cells. (C) Activation of Rap1 does not inhibit reporter activity in KRIT1-depleted cells, n=3, P=0.0001 by ANOVA, *P<0.001 by Bonferroni post-hoc test compared with vector-transfected vehicle-treated cells. (D) Inhibition of nuclear β-catenin reporter activity requires the presence of cell-cell adhesion. Endothelial cells overexpressing β-catenin were plated at high (confluent) and low (sparse) cell densities on tissue culture-treated plastic, or sparsely in wells coated with an Fc-VE-cadherin extracellular domain fusion protein, in the presence or absence of 50 μM of OMe-cAMP. Control wells were blocked with excess non-immune mouse IgG and soluble cadherin-Fc in the presence of calcium to assess background binding. TOPFlash activity is normalized to vector-transfected vehicle-treated cells, n=3, P=0.001 by ANOVA, *P<0.01 by Bonferroni post-hoc test compared with vector-transfected vehicle-treated cells, **P<0.01 compared with β-catenin overexpressing cells.
KRIT1 protein is expressed in multiple cell types
KRIT1 is required for the stabilizing effects of Rap1 on endothelial cell junctions; however, Rap1 also stabilizes cell-cell junctions in epithelial cells (Price et al., 2004), suggesting that KRIT1 may also regulate cell junctions and β-catenin signaling in this cell type. The KRIT1 protein is expressed in aortic and venous endothelial cells, however, KRIT1 mRNA is expressed in non-endothelial tissues (Denier et al., 2002); therefore, we examined the expression of KRIT1 in endothelial cells and other cell types using a sensitive quantitative tandem ELISA. BAECs expressed approximately 4 nanograms of KRIT1 human protein equivalents per milligram of total cell protein. We found similar levels of KRIT1 protein in multiple human and murine cell types including those representing the T-cell lineage (Jurkat), monocyte/macrophage lineage (RAW), mammary epithelial cells (MCF10A) and vascular smooth muscle cells (SMC). Consistent with our previous work (Glading et al., 2007), we detected endogenous KRIT1 expression in Chinese hamster ovary (CHO) cells, and increased expression by transfection with hemagglutinin-tagged (HA) KRIT1 (Fig. 4A). Western blotting confirmed the presence of KRIT1 protein at the characteristic molecular weight of ~80 kD in these cell types (supplementary material Fig. S3A).
Fig. 4.

KRIT1 protein is expressed widely and regulates β-catenin signaling in epithelial cells. (A) KRIT1 protein was quantified based on a standard curve of recombinant human glutathione S-transferase (GST)-KRIT1 FERM (protein 4.1, ezrin, radixin, moesin) domain protein. Data are expressed as nanograms of human protein equivalents per milligram of total cell protein, ± two standard errors (S.E.). Cell types: BAEC, bovine endothelial; FLS, mouse primary synoviocyte; RAW, mouse macrophage cell line; Jurkat, human T-cell line; SMC, mouse primary aortic smooth muscle cells; MCF10 (MCF10A), normal human mammary epithelial cell line; CHO, Chinese hamster ovary epithelial cell line. (B) β-Catenin reporter activity in MDCK cells after KRIT1 depletion (black bars) and in the presence of constitutively active Rap1 (white bars). TOPFlash activity is given as in Fig. 1C, P<0.0001 by ANOVA, *P<0.05 by Bonferroni post-hoc test compared with negative control siRNA-transfected cells, **P<0.05 compared with β-catenin expressing cells, n=4.
The presence of KRIT1 in epithelial cells led us to ask whether the effects of KRIT1 on the canonical Wnt signaling pathway might extend to non-endothelial tissues. Transfection of Madin-Darby canine kidney (MDCK) epithelial cells with anti-KRIT1 siRNA reduced KRIT1 levels by 50–60% (supplementary material Fig. S3B) and was associated with an approximate twofold increase in TOPFlash reporter activity (Fig. 4B). Expression of constitutively active Rap1 was also able to block β-catenin-driven reporter activity in KRIT1-sufficient cells; hence, the KRIT1-Rap1 pathway is a regulator of cell junctions and β-catenin signaling in multiple cell types.
KRIT1 deficiency leads to increased intestinal adenoma formation and decreased survival in ApcMin/+ mice
Dysregulation of the Wnt–β-catenin signaling pathway is implicated in the development of many tumors of epithelial origin; epithelial cancers of the colon are particularly associated with increased β-catenin-driven transcription (Fuchs et al., 2005). Epithelial cells from Krit1+/− mice manifested an approximate 50% reduction in Krit1 mRNA (Fig. 5A), similar to that induced in our siRNA-mediated depletion experiments, indicating a gene dosage effect in KRIT1 expression. Although we have not detected spontaneous adenoma/adenocarcinoma formation in Krit1+/− mice to date, given that KRIT1 is widely expressed and regulates β-catenin-dependent transcription in epithelial cells, we hypothesized that the hemizygous Krit1 null state would exacerbate β-catenin-driven disease.
Fig. 5.

Hemizygous Krit1 deficiency exacerbates β-catenin-driven tumors. (A) Krit1 mRNA expression in primary intestinal epithelium. Data are shown as the average amount of target mRNA +/−S.E.M., n=4 for each tissue type. *P=0.0095 by Student’s two-tailed t-test compared with wild type. (B) Quantification of intestinal adenomas in 90-day-old Krit1+/− ApcMin/+ and Krit1+/+ ApcMin/+ mice. Data shown are the mean±S.E.M., n=9, *P=0.0153 by Student’s two-tailed t-test compared with wild type. (C) Size distribution of intestinal polyps in 90-day-old Krit1+/− ApcMin/+ and Krit1+/+ ApcMin/+ mice. Data shown are the mean±S.E.M., n=9, P=0.034 by two-way ANOVA, *P<0.001 by Bonferroni post-hoc test compared with wild type. Right panels, representative images of Methylene Blue-stained sections of the ileum; upper panel, Krit1+/+ ApcMin/+; lower panel, Krit1+/− ApcMin/+. Bar, 1.5 mm. (D) Quantification of intense nuclear β-catenin staining in intestinal adenomas. Data are shown as the mean±S.E.M., n=9/group, *P=0.009 by Student’s t-test. (E) Kaplan-Meier survival analysis of Krit1+/− ApcMin/+ and Krit1+/+ ApcMin/+ mice, n=8/group, P=0.0237 by log-rank test. (F) Representative images of β-catenin staining in intestinal adenomas. Black arrowheads indicate densely stained β-catenin-positive nuclei; white arrowheads show β-catenin staining in non-involved intestinal epithelium.
To test the effect of hemizygous KRIT1 deficiency on the development of β-catenin-driven cancers, we bred Krit1 hemizygous null mice onto the ApcMin/+ model of familial APC. At 90 days of age, the small intestines of the Krit1+/− ApcMin/+ mice had significantly more polyps than Krit1+/+ ApcMin/+ mice (44±4 versus 29±1.6, respectively) (Fig. 5B), although these polyps were generally smaller in size than those found in wild-type mice (Fig. 5C). This difference was also observed in older animals (120 days of age) (supplementary material Fig. S4A). As expected (Yamada et al., 2002), occasional colonic polyps were observed, but the trend towards an increased number of colonic polyps in Krit1+/− mice did not reach statistical significance (Krit1+/− 2.6±0.5 versus Krit1+/+ 1.6±0.3) (supplementary material Fig. S4B).
APC serves as an essential component of a complex that targets cytoplasmic β-catenin for degradation. The oncogenic effect of the ApcMin/+ mutation can be ascribed to increased levels of nuclearβ-catenin (Oyama et al., 2008). As the loss of KRIT1 results in increased nuclear β-catenin accumulation in vitro (Fig. 2A), we analyzed β-catenin localization in the tumors of Krit1+/− ApcMin/+ and Krit1+/+ ApcMin/+ mice. Although adenomas from both genotypes exhibited cytoplasmic and nuclear β-catenin staining in a range of intensities, Krit1+/− ApcMin/+ adenomas contained a larger number of nuclei with intense nuclear β-catenin staining compared with histologically matched Krit1+/+ ApcMin/+ adenomas (Fig. 5D,F), as well as some low-level staining of nuclei in adjacent non-involved tissue (Fig. 5F, white arrowheads). Krit1+/− ApcMin/+ mice also exhibited reduced survival (134 days median survival), compared with Krit1+/+ ApcMin/+ mice (164 days median survival) (Fig. 5E). No significant difference in the number of adenocarcinomas was observed by histopathological analysis of moribund animals (data not shown). The earlier death of the Krit1+/− ApcMin/+ mice may, therefore, simply reflect greater numbers of adenomas thereby increasing the likelihood of intestinal obstruction or bleeding, both of which are common causes of morbidity in this model (Moser et al., 1995). These effects may be enhanced by underlying defects in endothelial permeability, such as those reported by Whitehead et al. in Ccm2 heterozygous mice (Whitehead et al., 2009). Thus, loss of one allele of Krit1 exacerbates β-catenin signaling, resulting in the formation of small intestinal adenomas and reduced survival in a mouse model of human colon cancer.
KRIT1 depletion increases β-catenin-dependent gene expression
β-Catenin regulates a number of genes involved in transformation, proliferation and tumor progression, and it is these changes that are thought to drive tumor formation in the ApcMin/+ mouse model. Thus, altered gene expression downstream of increased nuclear β-catenin could contribute to the differences observed between wild-type and hemizygous animals. Indeed, Krit1+/− ApcMin/+ mice exhibited increased expression of β-catenin-driven mRNAs relative to their Krit1+/+ ApcMin/+ littermates. Quantitative real-time (RT)-PCR revealed increased expression of Mmp3, Axin2, Fn1, Ccnd1 and Myc, known targets of the β-catenin–TCF–LEF complex, in the normal small intestine of Krit1+/− ApcMin/+ mice compared with their Krit1+/+ ApcMin/+ littermates. We observed around a fourfold increase in the expression of all five genes in adenomas versus the normal small intestine. However, Krit1+/− ApcMin/+ adenomas showed significant increases in expression relative to adenomas from Krit1+/+ ApcMin/+ littermates (Fig. 6A).
Fig. 6.
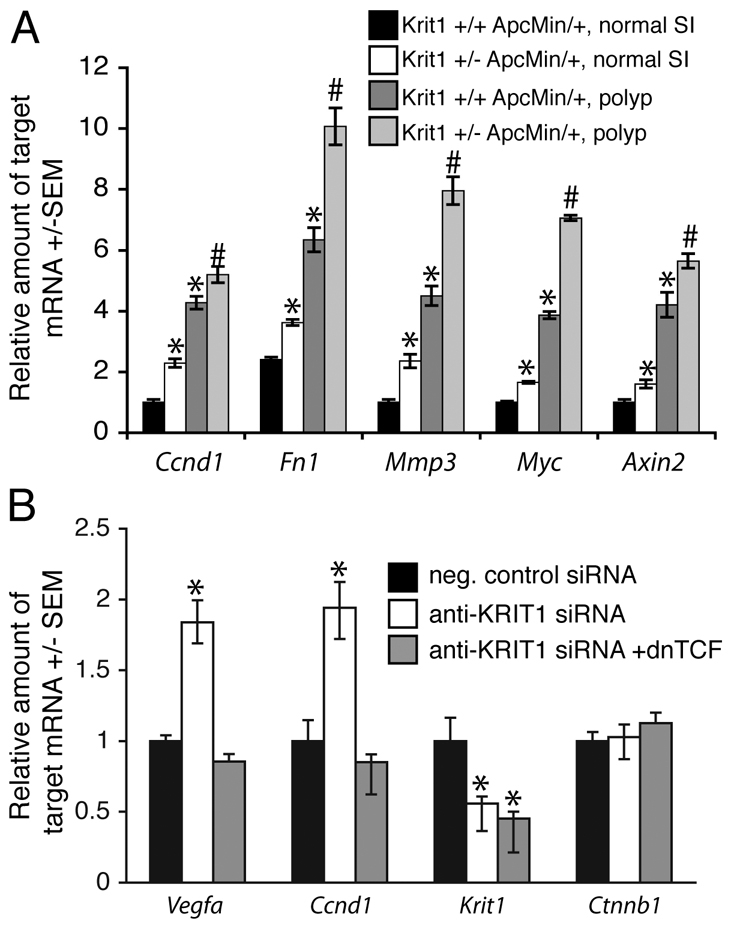
Loss of KRIT1 increases β-catenin-mediated gene expression. (A) Expression of β-catenin-regulated genes in normal non-involved small intestine (SI) and intestinal adenomas of Krit1+/− ApcMin/+ and Krit1+/+ ApcMin/+ mice. Data shown are the average expression from six mice per group ±S.E.M. Overall P<0.03 by ANOVA, *P<0.05 by Bonferroni post-hoc test compared with Krit1+/+ ApcMin/+ normal SI, #P<0.05 by Bonferroni post-hoc test compared with Krit1+/+ ApcMin/+ adenomas. (B) β-Catenin-regulated gene expression in KRIT1-depleted BAECs. dnTCF, dominant-negative TCF. Data shown are the average expression from five experiments ±S.E.M., P<0.0001 by ANOVA, *P<0.01 by Bonferroni post-hoc testing when compared with negative control siRNA-transfected cells.
Deficiency of KRIT1 is associated with the development of CCMs, which are vascular dysplasias that exhibit increased expression of multiple growth factors, including vascular endothelial growth factor (VEGF), and altered expression of matrix components (Rothbart et al., 1996; Kilic et al., 2000). Furthermore, embryonic aortic endothelial cells from Krit1 null mice are highly proliferative (Whitehead et al., 2004). As β-catenin transcriptional activity is implicated in the expression of VEGF (Skurk et al., 2005) and endothelial cell proliferation (Goodwin et al., 2006), we asked whether the expression of VEGFA (Vegfa) or cyclinD1 (Ccnd1) is enhanced in KRIT1-depleted endothelial cells. Anti-KRIT1 siRNA-expressing BAECs exhibited greater than a 50% reduction in KRIT1 (Krit1) mRNA by quantitative RT-PCR and around a twofold increase in Vegfa and Ccnd1 mRNA expression (Fig. 6B). The increase in Vegfa and Ccnd1 gene expression in KRIT1-depleted cells was reversed by co-expression of dominant-negative TCF (Leung et al., 2002), demonstrating that KRIT1 regulates the expression of these genes by inhibiting nuclear β-catenin signaling.β-Catenin (Ctnnb1) mRNA levels were unchanged by KRIT1 siRNA treatment, confirming that loss of KRIT1 does not affect the expression of β-catenin (Fig. 6B).
DISCUSSION
We previously demonstrated that loss of KRIT1 decreased endothelial junction stability resulting in increased permeability and stress fiber formation. Here, we show that loss of KRIT1 decreased the interaction of β-catenin with VE-cadherin (Fig. 1B,C), an interaction thought to be required for stabilization of VE-cadherin at the cell surface (Lilien and Balsamo, 2005). However, the decreased β-catenin–VE-cadherin interaction we observed was not sufficient to cause a reduction in cell surface expression of VE-cadherin (Fig. 1A). β-Catenin supports cadherin adhesive function by acting as an adaptor protein connecting cadherins with the actin cytoskeleton. β-Catenin is also a component of the canonical Wnt signaling pathway; this commonality has suggested a relationship between cadherin and Wnt signaling that is not completely understood (for a review, see Jeanes et al., 2008). Overexpression of cadherins antagonizes Wnt–β-catenin signaling (Sadot et al., 1998; Orsulic et al., 1999); conversely, a reduction in cadherin levels or a disruption of cell-cell adhesion enhances β-catenin signaling events (Gottardi et al., 2001; Kuphal and Behrens, 2006; Taddei et al., 2008). This suggests that cadherins may serve as a sink for β-catenin signals, such that the level of cadherin expression or stabilized cadherin adhesion in a cell sets a threshold over whichβ-catenin protein levels must rise in order to gain access to the nucleus. In cell culture, KRIT1 depletion inhibited the association of VE-cadherin with β-catenin, and promoted the nuclear localization of β-catenin, without affecting the total cellular level of β-catenin (Fig. 2A,B). Furthermore, loss of junctional stability in KRIT1-depleted cells is sufficient to activate β-catenin-dependent transcription (Fig. 2C) in the absence of exogenous Wnt addition or GSK3β inhibition. This effect is similar to that seen in endothelial cells expressing a form of VE-cadherin that cannot bind to β-catenin (Taddei et al., 2008). Thus, our data suggest that loss of KRIT1 deregulates β-catenin nuclear signaling by destabilizing cadherin-mediated adhesion.
KRIT1 is a Rap1 effector and we found that Rap1, a tumor suppressor (Kitayama et al., 1989), inhibits β-catenin signaling in confluent cells that are KRIT1-sufficient (Fig. 3). Whereas activation of Rap1, either through activation of Epac or by expression of constitutively active RapV12, did not significantly affect basal β-catenin-dependent transcription, it strongly inhibited β-catenin-driven transcription in β-catenin overexpressing cells. Activation of Rap1 increases β-catenin localization to cell-cell junctions and its association with cadherins (Sato et al., 2006), thus we hypothesized that Rap1 inhibits nuclear β-catenin signaling by promoting the association of β-catenin with cadherins and reducing the availability of β-catenin for nuclear signaling. Supporting this hypothesis, activation of Rap1 was unable to inhibit β-catenin signaling in cells that did not have intact cell-cell junctions, but was able to inhibit the nuclear β-catenin signaling induced by thrombin, which disrupts endothelial cell junctions and causes a redistribution of β-catenin to the nucleus (Beckers et al., 2008). Moreover, KRIT1 is required for Rap1 to stabilize cell-cell adhesion in endothelial cells and our data show that it is also required for Rap1 to suppress β-catenin signaling, as activation of Rap1 had no effect in KRIT1-depleted cells. Thus, Rap1 requires the presence of cell-cell contact and KRIT1 to inhibit nuclear β-catenin signaling. Taken together, these data indicate that Rap1 inhibits β-catenin signaling by promoting stable cell-cell adhesion, thus limiting the amount of β-catenin that is available to translocate into the nucleus. Because Rap1 has many cellular effectors, it is possible that it could use alternative mechanisms to regulate β-catenin signaling. Nevertheless, our data point directly to the ability of Rap1 to interact with KRIT1 in order to stabilize cell-cell junctions as an important component of its ability to antagonize β-catenin signaling.
Rap1 regulates the formation and stability of cell-cell adhesion in epithelial cells (Price et al., 2004). Our documentation of KRIT1 protein expression in non-endothelial cells suggests that KRIT1 could mediate some of the functions of Rap1 in this cell type. Depletion of KRIT1 in kidney epithelial cells increased β-catenin-dependent transcription and blocked the ability of active Rap (RapV12) to block β-catenin-dependent transcription (Fig. 4B). Thus, we propose that the signaling pathway described here functions in both epithelial and endothelial cells. Our discovery of a role for KRIT1 in epithelial cells bears similarities to the C. elegans KRIT1 ortholog, kri-1. C. elegans lacks a vasculature; however, kri-1 is expressed in intestinal cells and regulates organism life span by altering transcription (Berman and Kenyon, 2006).
The potential consequence of KRIT1 depletion in epithelial cells is underscored by our findings using the ApcMin/+ mouse model. Although Krit1+/− Apc+/+ mice do not have a higher incidence of spontaneous tumors, Krit1+/− ApcMin/+ mice developed more intestinal adenomas than their wild-type littermates and exhibited higher levels of nuclear β-catenin staining (Fig. 5). The relevance of our results to human colonic adenomas is uncertain since mutations or deletions of the region of chromosome 7 (7q21.2) that contains the KRIT1 gene have not been reported in human colon cancer patients, and unpublished data suggest that KRIT1/CCM1 has a wide range of expression in benign and malignant colon tumors compared with normal colon (S. F. Giardina, D. Notterman, P. Paty and F. Barany, pers. commun.). Nevertheless, this region is a hotspot for translocations and deletions in other cancers, including leukemias, lymphomas and thyroid tumors (Fischer et al., 1997; Basirico et al., 2003; Trovato et al., 2004); loss of heterozygosity has been observed on 7q21 in breast cancer and this region also has putative tumor suppressor function in prostate cancer (Kristjansson et al., 1997; Ichikawa et al., 2000). Thus, analysis of a potential role of KRIT1 as a tumor suppressor seems warranted.
Tumor formation is a complex process and it is possible that disrupting KRIT1/Rap1 signaling could have effects on many pathways; however, our data point strongly to increased β-catenin signaling as a major factor. The mechanisms of tumor formation in the ApcMin/+ mouse model are well characterized and are driven primarily by increased nuclear β-catenin signaling (Kongkanuntn et al., 1999; Ray et al., 2003; Oyama et al., 2008). Although the exact mechanism(s) underlying increased adenoma formation in Krit1+/−ApcMin/+ mice remain undefined, we observed changes in the expression of a number of β-catenin-driven genes in non-involved intestine ex vivo, including Myc, which is known to be essential for tumor formation in this model (Ignatenko et al., 2006; Yekkala and Baudino, 2007). We also observed a proportionally larger number of small adenomas in Krit1+/− ApcMin/+ mice (Fig. 5C), but not an increase in progression to adenocarcinoma. As Rap1 was originally identified as Krev (Kirsten ras revertant 1), a GTPase whose activity reversed Ras-induced transformation (Sakoda et al., 1992) and inhibited the development of multiple forms of adenocarcinoma (Burney et al., 1994; Damak et al., 1996; Leach et al., 1998), and because KRIT1 is upregulated during cAMP-stimulated reversal of Ras-mediated cell transformation (Bachrati et al., 1999), it will be of interest in future studies to examine whether the increase in adenoma number is the result of an effect on tumor initiation/transformation downstream of nuclear β-catenin signaling.
Our study has specifically investigated the role of KRIT1 in β-catenin-driven epithelial cancer. However, it also provides proof of principle that heterozygous loss of KRIT1 can influence β-catenin signaling in vivo. It is also conceivable that increased β-catenin signaling contributes to the formation of CCM. Although relatively little is known about the natural history of CCMs, ultrastructural analysis of mature lesions has shown a decrease in endothelial cell-cell junctions (Clatterbuck et al., 2001). Our findings therefore suggest that enhanced β-catenin signaling could occur within CCM lesions, where increased β-catenin-driven gene expression could contribute to the development of CCM lesions by altering the developmental program of the affected blood vessels. For example, increased expression of VEGF has been observed in human CCM lesions (Kilic et al., 2000); we now show that depletion of KRIT1 increases Vegfa mRNA in a β-catenin-dependent manner (Fig. 6). VEGF signaling contributes to endothelial proliferation, stimulates angiogenesis (Ferrara and Gerber, 2001), and regulates cadherin-based adhesion (Gavard and Gutkind, 2006). However, it remains to be seen whether VEGF signaling significantly contributes to the development of CCM lesions. In addition, CCM lesions express less laminin and more fibronectin, thus altering the composition of the basal lamina (Rothbart et al., 1996), and we observed increased fibronectin expression in the intestinal epithelium of Krit1+/− mice (Fig. 6). Fibronectin promotes endothelial cell survival and increased expression of fibronectin is common in the basal lamina of immature growing vessels (Hynes, 2007). These points illustrate that loss of KRIT1 could contribute to the formation of CCM lesions in humans by altering β-catenin-driven gene expression. In addition, it is important to note that a specific second hit at the KRIT1 locus in CCM lesions was reported recently (Akers et al., 2008). As our findings suggest that the basal heterozygous state exhibits increased β-catenin-driven transcription, this implies that complete loss of KRIT1 could cause an even greater increase in β-catenin signaling, thereby contributing to the development of CCM lesions.
In conclusion, we have identified a novel pathway that regulates cell-cell adhesion and β-catenin signaling. Depletion of KRIT1 increased the localization of β-catenin in the nucleus, increased β-catenin transcriptional activity, and increased β-catenin-dependent gene expression in many cell types. Conversely, activation of Rap1 inhibited β-catenin signaling, an effect that required the presence of cell-cell contact and KRIT1. Furthermore, hemizygous loss of Krit1 in mice exacerbates β-catenin-dependent intestinal adenoma formation. Thus, these studies reveal the function of Krit1 in endothelial cell biology, provide new insight into how Rap1 signaling can influence gene transcription, and suggest that KRIT1 may also function to regulate β-catenin signaling in non-endothelial tissues.
METHODS
Cell culture and transfection
HPAECs and BAECs were obtained from Lonza (Walkerville, PA), maintained in endothelial growth medium-2 (Lonza), and used prior to passage 7. MDCK epithelial cells were purchased from ATCC (Manassas, VA) and maintained in Eagle’s minimum essential medium with 10% FBS and 1% penicillin-streptomycin. Cells were transfected using an Amaxa nucleofection device (Amaxa GmbH, Germany) according to manufacturer’s instructions. Briefly, 0.5×106 cells/transfection were suspended in Amaxa basic endothelial cell solution (BAECs and HPAECs) or Solution L (MDCK) together with 25 nM of siRNA, with or without 2 μg of DNA. The cells were then nucleoporated using program M-003 (BAECs and HPAECs) or A-024 (MDCK). This method garnered transfection efficiencies ranging from 50% to 80%, dependent on cell type and the total amount of DNA transfected. KRIT1 knockdown in transfected cells was approximately 90% (Fig. 2A). Representative blots for KRIT1 expression in each set of experiments are shown in the supplementary material Figs S1–S3.
siRNA and plasmids
Non-targeting negative control siRNA and anti-KRIT1 siRNA (Ambion/Invitrogen) were used as reported previously (Glading et al., 2007). The anti-KRIT1 siRNA was found to have activity against bovine, murine and canine protein, as would be predicted by the 100% identity in the target sequence, and was used in all experiments. Cloning details for the HA-tagged full-length KRIT and HA-RapV12 were reported previously (Glading et al., 2007). Wobble mutations in the siRNA targeting sequence (four silent mutations) were introduced using the Quikchange II site-directed mutagenesis kit (Stratagene, La Jolla, CA). FLAG-tagged β-catenin and dominant-negative TCF constructs were obtained from Dr Eric Fearon (University of Michigan). The TOPFlash and FOPFlash reporter constructs were obtained from Dr Barry Gumbiner (University of Virginia).
Immunofluorescence and immunohistochemistry
Immunofluorescent detection of β-catenin and KRIT1 was performed as reported (Glading et al., 2007). Goat anti-mouse IgG Alexa 488 and goat anti-mouse IgG Alexa 568 were used as secondary antibodies (1:500, Invitrogen/Molecular Probes). Images were obtained using a Leica TCS SP2 AOBS confocal microscopy system (Leica DMRE microscope and HCX PL APO 63×/1.32 oil objective, Leica) and processed using Adobe PhotoShop software.
Immunohistochemical staining of β-catenin was performed on cross-sections of paraffin-embedded intestinal tissue. Antigen retrieval was performed by incubation in citrate buffer at 100°C for 20 minutes. Slides were treated with 3% H2O2 for 5 minutes to remove endogenous peroxidases, then blocked for 1 hour in a solution of 2% normal goat serum, 1% BSA and 0.01% Triton X-100. Rabbit anti-human β-catenin was added at a 1:100 dilution in blocking buffer and slides were incubated overnight at 4°C in a humidified chamber. Control serial sections were incubated with an equivalent concentration of normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Donkey anti-rabbit IgG-HRP (1:250, Jackson Immunoresearch, West Grove, PA) was used as a secondary antibody. Staining was developed using the diaminobenzidine (DAB)+ liquid stain reagent (Dako, Denmark); slides were counterstained for 30 seconds with Mayer’s hematoxylin (Sigma, St Louis, MO). β-Catenin nuclear staining of adenomas was quantified using bright-field microscopy by investigators who were blinded to the identity of each section. Intensely stained nuclei were counted in 8–12 representative fields per mouse under high magnification (40×). The number of positive nuclei per high-power field was averaged by genotype.
Immunoprecipitation and western blotting
KRIT1 and β-catenin expression was monitored, and VE-cadherin immunoprecipitation was performed, as reported previously (Glading et al., 2007). Mouse anti-FLAG was used for immunoblotting at a dilution of 1:10,000 (Sigma). Mouse anti-HA (Covance, Princeton, NJ) was used at a dilution of 1:1000. Rabbit anti-VE cadherin (Cayman Chemical, Ann Arbor, MI) was used for immunoprecipitation at a dilution of 1:100 and for western blotting at a dilution of 1:1000. Rabbit anti-β-catenin (Santa Cruz), rabbit anti-RhoGDI (cytosolic fraction marker) (Santa Cruz), rabbit anti-lamin A/C (nuclear fraction marker) (Cell Signaling Technology, Boston, MA) and mouse anti-β1 integrin (membrane marker) (Santa Cruz) were used for western blotting at a dilution of 1:1000. The secondary antibodies used were: goat anti-rabbit Alexa 680 (Invitrogen) and goat anti-mouse IRDye 800 (Rockland Immunochemicals, Gilbertsville, PA). Blots were imaged and densitometry was performed using a Li-Cor Odyssey infrared imaging system, and the processing for figures was performed using Adobe Photoshop; all lanes were adjusted equally.
FACS analysis
Cell surface expression of VE-cadherin was measured by antibody labeling of the extracellular domain of VE-cadherin, followed by FACS analysis. Briefly, at 24 hours post-transfection, cells transfected with negative control siRNA or anti-KRIT1 siRNA were dissociated from the tissue culture plate using dissociation buffer (137 mM NaCl, 4.2 mM NaHCO3, 5.4 mM KCl, 5.6 mM glucose, 0.5 mM EDTA, pH 7.2) at 37°C for 10 minutes. The cells were then aliquoted into tubes in triplicate and spun down in buffer containing 1.2 mM CaCl2, 135 mM NaCl, 1.2 mM MgCl2, 10 mM HEPES, 10 mM glucose and 0.05% BSA (pH 7.4), and then labeled with rabbit anti-VE-cadherin (Cayman) or a non-immune isotype control (Santa Cruz) for 1 hour on ice. The cells were washed, and incubated with secondary antibody (goat anti-rabbit FITC, Jackson Immunoresearch) for 30 minutes on ice. The cells were washed again, and fluorescent labeling was then detected using a FACScan flow cytometer and analyzed using CellQuest software (BD Biosciences, San Jose, CA).
Subcellular fractionation
Subcellular fractionation was performed as reported in Chou et al. (Chou et al., 2003). Briefly, HPAECs were transfected, as described above, with negative control siRNA or anti-KRIT1 siRNA. At 24 hours post-transfection, cells were harvested in 0.5 ml of lysis buffer (20 mM HEPES-KOH, pH 7.5, 1.5 mM MgCl2, 5 mM KCl, 0.2 mM Na3VO4, 10 g/ml leupeptin, 10 g/ml aprotinin, 1 mM phenylmethyl-sulfonyl fluoride). After a 10-minute incubation on ice, cells were disrupted using 35 strokes in a Dounce homogenizer. A fraction of the total cell lysate was saved prior to centrifugation at 450 g for 10 minutes to pellet the nuclei. The supernatant was subjected to additional centrifugation at 18,900 g for 30 minutes to pellet the membrane fraction. The resultant supernatant was the cytosolic fraction. The nuclear and membrane pellets were extracted at 4°C with lysis buffer containing 1% Nonidet P-40. The total lysate, and nuclear, membrane, and cytosolic fractions were analyzed for protein expression by immunoblot.
TOPFlash reporter assays
Reporter assays were performed using the Dual-Glo luciferase assay system (Promega, Madison, WI) according to the manufacturer’s instructions. Firefly luciferase activities produced by the TCF-dependent firefly luciferase reporter plasmid TOPFlash were measured using a Wallac Victor2 1420 multilabel counter. All transfections received the same number and total amount of plasmids, and included the constitutive Renilla luciferase plasmid pRL-TK. Cells were plated in at least triplicate per condition, and each experiment was performed independently (i.e. separate transfections) at least three times (n≥3). Firefly luciferase activities were standardized to constitutive Renilla luciferase activities and statistical analyses were performed using one-way ANOVA. When appropriate, cells were treated with 50 μM of 8′pCPT-2OMe-cAMP (Biolog, Bremen, Germany) or an equivalent volume of dH2O overnight prior to luciferase detection. Cells pre-treated ±OMe-cAMP were stimulated with 2 U/ml of thrombin (Sigma) or an equivalent volume of dH2O for 6 hours prior to luciferase detection. To assess the contribution of cadherin-based adhesion, VE-cadherin-Fc protein was isolated from stably transfected CHO cells (obtained from Dr Dietmar Vestweber, Max-Planck-Institute for Molecular Biomedicine, Germany) as reported previously (Gotsch et al., 1997). Experimental wells were coated with 30 μg/ml of VE-cadherin-Fc fusion protein, as reported previously (Winter et al., 2004), and blocked with 0.05% BSA. Control wells were pre-treated with non-immune mouse IgG (Sigma) followed by an excess of soluble cadherin-Fc in the presence of calcium to assess background binding. Transfected cells were plated in seven replicate wells and treated as required.
RNA isolation and semi-quantitative RT-PCR
RNA was isolated by Trizol extraction according to the manufacturer’s instructions (Invitrogen). Complementary DNA was obtained with Superscript III reverse transcriptase using random hexamers (Invitrogen). The primer sets for quantitative PCR are given in supplementary material Table S1. Amplifications were run in a 7500 Fast real-time PCR system (Applied Biosystems/Invitrogen). Each value was calculated using the comparative Ct method (Livak and Schmittgen, 2001) and normalized to Gapdh or Actb (β-actin) internal controls. All samples were run in at least triplicate, and each experiment was performed from five separate cell/RNA preparations (n=5). For RNA isolation from mouse intestinal tissue, areas of normal non-involved tissue and adenoma were identified using a dissecting microscope and removed. Tissue samples of 3–5 mm2 were incubated in 10× volume of RNAlater solution (Ambion) at 4°C prior to Trizol extraction. All samples were run in at least quadruplicate. For accuracy of comparison, all primer sets were designed to amplify across exon boundaries, confirmed to produce a single product, and to have amplification efficiencies roughly equal to that of the reference set (data not shown).
KRIT1 ELISA
ELISA high binding plates (Fisher, Pittsburgh, PA) were coated with 5.0 μg/ml of mouse anti-KRIT1 15B2 in 0.05 M NaHCO3, pH 9.5, for 1 hour at 37°C. Plates were then blocked with 150 μl of blocking buffer (1% BSA in PBS, heat-inactivated). After washing with wash buffer (PBS with 0.2% Tween-20), the diluted sample (in 1% BSA in PBS with 0.2% Tween-20) was added to the appropriate wells and incubated at 37°C for 1 hour. Plates were washed again, and primary antibody (rabbit anti-KRIT1 6832, 1:1000 in 1% BSA in PBS with 0.2% Tween-20) was then added. After 1 hour at 37°C, the secondary antibody (goat anti-rabbit HRP conjugate, Invitrogen, 1:1000) was added and incubated for a further 30 minutes. The signal was developed using enhanced chemiluminescence (ECL) solution (Amersham Biosciences, UK), and measured using a Wallac Victor2 multilabel plate reader. Non-coated and secondary-only wells were used to assess background binding. All samples and controls were run in triplicate. The amount of KRIT1 in the cell lysate was estimated by comparison to a standard curve of recombinant GST-KRIT1 F123, which also served as a positive control.
Mouse models
KRIT1 hemizygous (Krit1+/−) mice were obtained from Dr Dean Li (University of Utah) and maintained on a clean C57BL/6 background; PCR-based genotyping was performed as described in the initial characterization of this model (Whitehead et al., 2004). ApcMin/+ mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and genotyped using the recommended primer sequences and protocol. Mice were bred and maintained under standard conditions in the University of California, San Diego animal facility that is accredited by the American Association for Accreditation of Laboratory Animal Care. All animal protocols received prior approval by the institutional review board.
Krit1+/− and Krit1+/+ littermates on an ApcMin/+ background were maintained on normal mice chow until sacrifice at 90 or 120 days of age (adenoma study) or until moribund (survival study). Krit1+/−mice were born at the expected Mendelian ratio on the ApcMin/+ background (data not shown). For analysis of adenoma formation, age- and sex-matched littermate mice were euthanized, and the intestinal tract from the duodenum to the rectum was removed. Intestinal segments were rinsed with saline and fixed in 10% formalin solution at 4°C overnight. Segments were then stained with Methylene Blue (0.13% in 75% ethanol) for 30 minutes and then rinsed with 75% ethanol for 15 minutes. Adenoma formation was quantified using a dissecting microscope (2× magnification) by an investigator who was blind to the mouse genotype. Imaging was performed using an eyepiece-mounted digital camera (Canon Powershot A520) and processed using Adobe Photoshop. Following analysis of whole mounts, representative sections of each segment, including adenomatous and adjacent normal tissue, were taken for paraffin embedding and sectioning.
Statistical analysis
Statistical analysis was performed using PRISM software (version 4.0, GraphPad Software, La Jolla, CA). Statistical testing included one-way ANOVA or Student’s t-test, where appropriate; the test used is indicated in each figure legend. Pairwise comparison of conditions was performed following ANOVA using the Bonferroni post-hoc test as indicated.
Supplementary Material
Acknowledgments
This work was supported by NIH Grants HL078784, AR27214 and HL31950 to M.H.G. and an American Heart Association Scientist Development Grant to A.J.G. Deposited in PMC for release after 12 months.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
A.J.G. and M.H.G. conceived and designed the experiments and co-wrote the paper. All experiments were performed and all data analyzed by A.J.G.
SUPPLEMENTARY INFORMATION
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.003293/-/DC1.
REFERENCES
- Aberle H, Schwartz H, Kemler R. (1996). Cadherin-catenin complex: protein interactions and their implications for cadherin function. J Cell Biochem. 61, 514–523 [DOI] [PubMed] [Google Scholar]
- Akers AL, Johnson E, Steinberg GK, Zabramski JM, Marchuk DA. (2008). Biallelic somatic and germline mutations in cerebral cavernous malformations (CCM): evidence for a two-hit mechanism of CCM pathogenesis. Hum Mol Genet. 18, 191–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW. (2008). p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood 112, 2770–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachrati CZ, Downes CS, Rasko I. (1999). Chemical reverse transformation of CHO-K1 cells induces changes in expression of a candidate tumour suppressor and of a gene not previously characterised as transformation related. Eur J Cell Biol. 78, 561–566 [DOI] [PubMed] [Google Scholar]
- Basirico R, Pirrotta R, Fabbiano F, Mirto S, Cascio L, Pagano M, Cammarata G, Magrin S, Santoro A. (2003). Submicroscopic deletions at 7q region are associated with recurrent chromosome abnormalities in acute leukemia. Haematologica 88, 429–437 [PubMed] [Google Scholar]
- Beckers CM, Garcia-Vallejo JJ, van Hinsbergh VW, van Nieuw Amerongen GP. (2008). Nuclear targeting of {beta}-catenin and p120ctn during thrombin-induced endothelial barrier dysfunction. Cardiovasc Res. 79, 679–688 [DOI] [PubMed] [Google Scholar]
- Berman JR, Kenyon C. (2006). Germ-cell loss extends C. elegans life span through regulation of DAF-16 by kri-1 and lipophilic-hormone signaling. Cell 124, 1055–1068 [DOI] [PubMed] [Google Scholar]
- Boulday G, Blecon A, Petit N, Chareyre F, Garcia LA, Niwa-Kawakita M, Giovannini M, Tournier-Lasserve E. (2009). Tissue-specific conditional CCM2 knockout mice establish the essential role of endothelial CCM2 in angiogenesis: implications for human cerebral cavernous malformations. Dis Model Mech. 2, 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burney TL, Rockove S, Eiseman JL, Jacobs SC, Kyprianou N. (1994). Partial growth suppression of human prostate cancer cells by the Krev-1 suppressor gene. Prostate 25, 177–188 [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Lampugnani M, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, et al. (1999). Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98, 147–157 [DOI] [PubMed] [Google Scholar]
- Chou F, Hill J, Hsieh J, Pouyssegur J, Brunet A, Glading A, Uberall F, Ramos J, Werner M, Ginsberg M. (2003). PEA-15 binding to ERK1/2 MAPKs is required for its modulation of integrin activation. J Biol Chem. 278, 52587–52597 [DOI] [PubMed] [Google Scholar]
- Clatterbuck R, Eberhart C, Crain B, Rigamonti D. (2001). Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J Neurol Neurosurg Psychiatr. 71, 188–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- Cullere X, Shaw S, Andersson L, Hirahashi J, Luscinskas F, Mayadas T. (2005). Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105, 1950–1955 [DOI] [PubMed] [Google Scholar]
- Damak S, Harnboonsong Y, George PM, Bullock DW. (1996). Expression of human Krev-1 gene in lungs of transgenic mice and subsequent reduction in multiplicity of ethyl carbamate-induced lung adenomas. Mol Carcinog. 17, 84–91 [DOI] [PubMed] [Google Scholar]
- Dejana E. (2004). Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 5, 261–270 [DOI] [PubMed] [Google Scholar]
- Denier C, Gasc J, Chapon F, Domenga V, Lescoat C, Joutel A, Tournier-Lasserve E. (2002). Krit1/cerebral cavernous malformation 1 mRNA is preferentially expressed in neurons and epithelial cells in embryo and adult. Mech Dev. 117, 363–367 [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP. (2001). The role of vascular endothelial growth factor in angiogenesis. Acta Haematol. 106, 148–156 [DOI] [PubMed] [Google Scholar]
- Fischer K, Frohling S, Scherer SW, McAllister Brown J, Scholl C, Stilgenbauer S, Tsui LC, Lichter P, Dohner H. (1997). Molecular cytogenetic delineation of deletions and translocations involving chromosome band 7q22 in myeloid leukemias. Blood 89, 2036–2041 [PubMed] [Google Scholar]
- Fuchs S, Ougolkov A, Spiegelman V, Minamoto T. (2005). Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle 4, 1522–1539 [DOI] [PubMed] [Google Scholar]
- Gavard J, Gutkind J. (2006). VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 8, 1223–1234 [DOI] [PubMed] [Google Scholar]
- Glading A, Han J, Stockton RA, Ginsberg MH. (2007). KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 179, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin A, Sullivan K, D’Amore P. (2006). Cultured endothelial cells display endogenous activation of the canonical Wnt signaling pathway and express multiple ligands, receptors, and secreted modulators of Wnt signaling. Dev Dyn. 235, 3110–3120 [DOI] [PubMed] [Google Scholar]
- Gore AV, Lampugnani MG, Dye L, Dejana E, Weinstein BM. (2008). Combinatorial interaction between CCM pathway genes precipitates hemorrhagic stroke. Dis Model Mech. 1, 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotsch U, Borges E, Bosse R, Boggemeyer E, Simon M, Mossmann H, Vestweber D. (1997). VE-cadherin antibody accelerates neutrophil recruitment in vivo. J Cell Sci. 110, 583–588 [DOI] [PubMed] [Google Scholar]
- Gottardi CJ, Wong E, Gumbiner BM. (2001). E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol. 153, 1049–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. (2007). Cell-matrix adhesion in vascular development. J. Thromb. Haemost. 5 Suppl. 1, 32–40 [DOI] [PubMed] [Google Scholar]
- Ichikawa T, Hosoki S, Suzuki H, Akakura K, Igarashi T, Furuya Y, Oshimura M, Rinker-Schaeffer CW, Nihei N, Barrett JC, et al. (2000). Mapping of metastasis suppressor genes for prostate cancer by microcell-mediated chromosome transfer. Asian J Androl. 2, 167–171 [PubMed] [Google Scholar]
- Ignatenko NA, Holubec H, Besselsen DG, Blohm-Mangone KA, Padilla-Torres JL, Nagle RB, de Alboranc IM, Guillen RJ, Gerner EW. (2006). Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol Ther. 5, 1658–1664 [DOI] [PubMed] [Google Scholar]
- Ivanov DB, Philippova MP, Tkachuk VA. (2001). Structure and functions of classical cadherins. Biochemistry (Mosc) 66, 1174–1186 [DOI] [PubMed] [Google Scholar]
- Jeanes A, Gottardi CJ, Yap AS. (2008). Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 27, 6920–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic T, Pamir MN, Kullu S, Eren F, Ozek MM, Black PM. (2000). Expression of structural proteins and angiogenic factors in cerebrovascular anomalies. Neurosurgery 46, 1179–1191; discussion 1191–1192 [DOI] [PubMed] [Google Scholar]
- Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. (1989). A ras-related gene with transformation suppressor activity. Cell 56, 77–84 [DOI] [PubMed] [Google Scholar]
- Kleaveland B, Zheng X, Liu JJ, Blum Y, Tung JJ, Zou Z, Chen M, Guo L, Lu MM, Zhou D, et al. (2009). Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat Med. 15, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongkanuntn R, Bubb VJ, Sansom OJ, Wyllie AH, Harrison DJ, Clarke AR. (1999). Dysregulated expression of beta-catenin marks early neoplastic change in Apc mutant mice, but not all lesions arising in Msh2 deficient mice. Oncogene 18, 7219–7225 [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. (1997). Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 275, 1784–1787 [DOI] [PubMed] [Google Scholar]
- Kristjansson AK, Eiriksdottir G, Ragnarsson G, Sigurdsson A, Gudmundsson J, Barkardottir RB, Jonasson JG, Egilsson V, Ingvarsson S. (1997). Loss of heterozygosity at chromosome 7q in human breast cancer: association with clinical variables. Anticancer Res. 17, 93–98 [PubMed] [Google Scholar]
- Kuphal F, Behrens J. (2006). E-cadherin modulates Wnt-dependent transcription in colorectal cancer cells but does not alter Wnt-independent gene expression in fibroblasts. Exp Cell Res. 312, 457–467 [DOI] [PubMed] [Google Scholar]
- Laberge-le Couteulx S, Jung H, Labauge P, Houtteville J, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach J, Tournier-Lasserve E. (1999). Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 23, 189–193 [DOI] [PubMed] [Google Scholar]
- Leach SD, Berger DH, Davidson BS, Curley SA, Tainsky MA. (1998). Enhanced Krev-1 expression inhibits the growth of pancreatic adenocarcinoma cells. Pancreas 16, 491–498 [DOI] [PubMed] [Google Scholar]
- Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER. (2002). Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 277, 21657–21665 [DOI] [PubMed] [Google Scholar]
- Lilien J, Balsamo J. (2005). The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 17, 459–465 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- Mably JD, Chuang LP, Serluca FC, Mohideen MA, Chen JN, Fishman MC. (2006). santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development 133, 3139–3146 [DOI] [PubMed] [Google Scholar]
- Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. (1995). ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur. J. Cancer 31A, 1061–1064 [DOI] [PubMed] [Google Scholar]
- Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. (1999). E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J Cell Sci. 112, 1237–1245 [DOI] [PubMed] [Google Scholar]
- Oyama T, Yamada Y, Hata K, Tomita H, Hirata A, Sheng H, Hara A, Aoki H, Kunisada T, Yamashita S, et al. (2008). Further upregulation of beta-catenin/Tcf transcription is involved in the development of macroscopic tumors in the colon of ApcMin/+ mice. Carcinogenesis 29, 666–672 [DOI] [PubMed] [Google Scholar]
- Plummer N, Zawistowski J, Marchuk D. (2005). Genetics of cerebral cavernous malformations. Curr Neurol Neurosci Rep. 5, 391–396 [DOI] [PubMed] [Google Scholar]
- Potter MD, Barbero S, Cheresh DA. (2005). Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 280, 31906–31912 [DOI] [PubMed] [Google Scholar]
- Price L, Hajdo-Milasinovic A, Zhao J, Zwartkruis F, Collard J, Bos J. (2004). Rap1 regulates E-cadherin-mediated cell-cell adhesion. J Biol Chem. 279, 35127–35132 [DOI] [PubMed] [Google Scholar]
- Ray R, Cabal-Manzano R, Moser AR, Waldman T, Zipper LM, Aigner A, Byers SW, Riegel AT, Wellstein A. (2003). Up-regulation of fibroblast growth factor-binding protein, by beta-catenin during colon carcinogenesis. Cancer Res. 63, 8085–8089 [PubMed] [Google Scholar]
- Revencu N, Vikkula M. (2006). Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet. 43, 716–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AB, Carnahan RH. (2004). Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol. 15, 657–663 [DOI] [PubMed] [Google Scholar]
- Rothbart D, Awad IA, Lee J, Kim J, Harbaugh R, Criscuolo GR. (1996). Expression of angiogenic factors and structural proteins in central nervous system vascular malformations. Neurosurgery 38, 915–924; discussion 924–925 [DOI] [PubMed] [Google Scholar]
- Sadot E, Simcha I, Shtutman M, Ben-Ze’ev A, Geiger B. (1998). Inhibition of beta-catenin-mediated transactivation by cadherin derivatives. Proc Natl Acad Sci USA 95, 15339–15344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee-Lin SQ, Kosofsky B, et al. (1999). Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 8, 2325–2333 [DOI] [PubMed] [Google Scholar]
- Sakoda T, Kaibuchi K, Kishi K, Kishida S, Doi K, Hoshino M, Hattori S, Takai Y. (1992). smg/rap1/Krev-1 p21s inhibit the signal pathway to the c-fos promoter/enhancer from c-Ki-ras p21 but not from c-raf-1 kinase in NIH3T3 cells. Oncogene 7, 1705–1711 [PubMed] [Google Scholar]
- Sanson B, White P, Vincent JP. (1996). Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 383, 627–630 [DOI] [PubMed] [Google Scholar]
- Sato T, Fujita N, Yamada A, Ooshio T, Okamoto R, Irie K, Takai Y. (2006). Regulation of the assembly and adhesion activity of E-cadherin by nectin and afadin for the formation of adherens junctions in Madin-Darby canine kidney cells. J Biol Chem. 281, 5288–5299 [DOI] [PubMed] [Google Scholar]
- Serebriiskii I, Estojak J, Sonoda G, Testa JR, Golemis EA. (1997). Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21–22. Oncogene 15, 1043–1049 [DOI] [PubMed] [Google Scholar]
- Skurk C, Maatz H, Rocnik E, Bialik A, Force T, Walsh K. (2005). Glycogen-Synthase Kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ Res. 96, 308–318 [DOI] [PubMed] [Google Scholar]
- Solanas G, Porta-de-la-Riva M, Agusti C, Casagolda D, Sanchez-Aguilera F, Larriba MJ, Pons F, Peiro S, Escriva M, Munoz A, et al. (2008). E-cadherin controls beta-catenin and NF-kappaB transcriptional activity in mesenchymal gene expression. J Cell Sci. 121, 2224–2234 [DOI] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, Dejana E. (2008). Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 10, 923–934 [DOI] [PubMed] [Google Scholar]
- Trovato M, Ulivieri A, Dominici R, Ruggeri RM, Vitarelli E, Benvenga S, Barresi G, Trimarchi F, Brunetti E, Vecchione A, et al. (2004). Clinico-pathological significance of cell-type-specific loss of heterozygosity on chromosome 7q21: analysis of 318 microdissected thyroid lesions. Endocr Relat Cancer 11, 365–376 [DOI] [PubMed] [Google Scholar]
- Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD, Felbor U. (2007). CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics 8, 249–256 [DOI] [PubMed] [Google Scholar]
- Voss K, Stahl S, Hogan BM, Reinders J, Schleider E, Schulte-Merker S, Felbor U. (2009). Functional analyses of human and zebrafish 18-amino acid in-frame deletion pave the way for domain mapping of the cerebral cavernous malformation 3 protein. Hum Mutat. 30, 1003–1011 [DOI] [PubMed] [Google Scholar]
- Whitehead K, Plummer N, Adams J, Marchuk D, Li D. (2004). Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development 131, 1437–1448 [DOI] [PubMed] [Google Scholar]
- Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, Mayo AH, Drakos SG, Marchuk DA, Davis GE, et al. (2009). The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 15, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter MC, Shasby SS, Ries DR, Shasby DM. (2004). Histamine selectively interrupts VE-cadherin adhesion independently of capacitive calcium entry. Am J Physiol Lung Cell Mol Physiol. 287, L816–L823 [DOI] [PubMed] [Google Scholar]
- Yamada Y, Hata K, Hirose Y, Hara A, Sugie S, Kuno T, Yoshimi N, Tanaka T, Mori H. (2002). Microadenomatous lesions involving loss of Apc heterozygosity in the colon of adult Apc(Min/+) mice. Cancer Res. 62, 6367–6370 [PubMed] [Google Scholar]
- Yekkala K, Baudino TA. (2007). Inhibition of intestinal polyposis with reduced angiogenesis in ApcMin/+ mice due to decreases in c-Myc expression. Mol Cancer Res. 5, 1296–1303 [DOI] [PubMed] [Google Scholar]
- Zawistowski J, Stalheim L, Uhlik M, Abell A, Ancrile B, Johnson G, Marchuk D. (2005). CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 14, 2521–2531 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.