

Abstract

Pd(II)-catalyzed enantioselective C-H olefination of diphenylacetic acid substrates has been achieved through the use of mono-protected chiral amino acid ligands. The absolute configuration of the resulting olefinated products is consistent with that of a proposed C-H insertion intermediate.
Despite substantial progress in developing various Pd-catalyzed C–heteroatom and C-C bond forming reactions via C-H activation,1 achieving enantioselectivity in these reactions through a stereoselective Pd insertion step remains a significant challenge.2–9 In our ongoing studies to design and evaluate new ligands to effect asymmetric C-H cleavage, two major problems have become apparent. First, the simultaneous binding of both the substrate and the chiral ligand to the Pd(II) center is often difficult to achieve. Second, even if such complexes are assembled, the ligand often strongly inhibits C-H activation, either because it induces an unwanted conformational change or adversely affects the electronic properties of the Pd(II) center.
We have recently found that mono-protected amino acid ligands and 2-benzylpyridine substrates coordinate with Pd(II) in a one-to-one ratio with high fidelity.3 Importantly, the resulting chiral Pd(II) complexes were found to induce asymmetric C-H cleavage with high enantioselectivity (up to 95% ee). Of critical importance for the viability of this process is the precise match between the binding ability of the pyridine substrate and the chiral ligand. This observation, however, calls into question whether this chiral ligand scaffold is broadly applicable to synthetically useful substrates, including those that contain weakly coordinating functional groups. Herein, we report an enantioselective C-H olefination reaction of α,α-diphenylacetic acids using mono-protected amino acids as chiral ligands. This new development represents an encouraging step towards the realization of synthetically useful Pd-catalyzed enantioselective C-H activation reactions.
We previously reported that both inorganic and organic cations dramatically accelerate carboxyl-directed C-H activation reactions.10 Our current hypothesis, based on the structure of a C-H insertion intermediate,10b is that the σ-chelation of the carbonyl oxygen of the carboxylate salt with Pd(II) is responsible for the facile C-H cleavage promoted by the complex-induced proximity effect. Following this hypothesis, we anticipated that a chiral carbon–Pd intermediate B could be formed in analogy to intermediate A, which is formed following enantioselective C-H activation using a pyridyl directing group. Subsequently, we envisioned this intermediate undergoing olefination to give the corresponding chiral product (Figure 1). The proposed boat conformations of A and B are based on a crystal structure of a similar 1,4-cyclohexadiene-like cyclopalladated compound.3
Figure 1.

To test this hypothesis, we began by establishing reaction conditions for a Pd(II)-catalyzed olefination reaction of α,α-diphenylacetic acid 1a using Boc-L-isoleucine (Boc-Ile-OH) L1 as a chiral ligand. Following a procedure developed for the racemic olefination of phenylacetic acid substrates,11 the olefination reaction of 1a in the presence of L1 gave the desired product in 46% yield, accompanied by substantial amounts of the decarboxylation byproduct. Nonetheless, the high enantioselectivity (95% ee) observed was encouraging (Table 1, entry 1). Through extensive screening, we found that by using the preformed sodium salt of 1a as the starting material and KHCO3 as the base, the yield could be improved to 73%, with 97% ee (entry 3). Surprisingly, the unique combination of the sodium salt of 1a and KHCO3 was crucial for the success of the reaction. Other alternatives decreased both the enantioselectivity and yield (entries 4–14). We then screened an array of mono-protected α-amino acids (Table 2). Boc-Ile-OH was the optimal chiral ligand, with Boc-Tyr(t-Bu)-OH giving similar enantioselectivity (96% ee) but significantly lower yield (45%).
Table 1.
Effect of Inorganic Cations and Basesa

| Entry | M | Base | % Yieldb | %eec |
|---|---|---|---|---|
| 1 | H | KHCO3d | 46 | 95 |
| 2 | Na | - | 51 | 36 |
| 3 | Na | KHCO3 | 73e | 97 |
| 4 | NH4 | KHCO3 | - | - |
| 5 | K | KHCO3 | 49 | 84 |
| 6 | Cs | KCH2O3 | - | - |
| 7 | Na | K2CO3 | 25 | 87 |
| 8 | Na | NaHCO3 | 56 | 89 |
| 9 | Na | Na2CO3 | 61 | 91 |
| 10 | Na | Cs2CO3 | - | - |
| 11 | Na | K2HPO4 | 37 | 83 |
| 12 | Na | Li2CO3 | 44 | 85 |
| 13 | Na | NaOTsf | 57 | 79 |
| 14 | K | NaHCO3 | 53 | 91 |
0.5 mmol 1a, 5 mol% Pd(OAc)2, 10 mol% L1, 5 mol% BQ, 0.5 equiv. Base, 1 atm O2 in 3 mL tert-amyl alcohol at 90 °C for 48 h;
The yield was determined by 1H NMR using CH2Br2 as a calibrated internal standard.
ee was determined by chiral HPLC;
2 equiv. KHCO3;
Isolated yield;
1 equiv. NaOTs.
Table 2.
Evaluation of Amino Acidsa
| Entry | Ligand | % Yield | % ee |
|---|---|---|---|
| 1 | Boc-Ala-OH | 46 | 54 |
| 2 | Boc-Abu-OH | 51 | 67 |
| 3 | Boc-Nva-OH | 63 | 61 |
| 4 | Boc-Nle-OH | 59 | 81 |
| 5 | Boc-Val-OH | 39 | 93 |
| 6 | Boc-Ser(Bzl)-OH | 61 | 91 |
| 7 | Boc-Phe-OH | 25 | 93 |
| 8 | Boc-Thr(t-Bu)-OH | 50 | 86 |
| 9 | Boc-Tyr(t-Bu)-OH | 45 | 96 |
| 10 | Boc-Tle-OH | 43 | 94 |
| 11 | Boc-lle-OH•0.5H2O | 73 | 97 |
| 12 | Boc-Leu-OH | 60 | 86 |
| 13 | Formyl-Leu-OH | 44 | 79 |
| 14 | PG1-Leu-OH | 57 | 84 |
| 15 | PG2-Leu-OH | 44 | 69 |
| 16 | PG3-Leu-OH | 37 | 65 |

The reaction conditions are identical to those described in Table 1.
We next proceeded to establish the scope of the styrene coupling partner. para- and meta-Alkyl substituted styrenes gave high enantioselectivity (92–97% ee, Table 3, entries 2, 3 and 7) while ortho-methyl substituted styrene gave only 81% ee (entry 4). para-Chlorostyrene afforded both high enantioselectivity (96% ee) and reactivity (74% yield); however, para-fluorostyrene gave both decreased yield and enantioselectivity (entry 6).
Table 3.
Enantioselective C-H Activation/Olefination Using Substituted Styrenes as the Coupling Partnersa

| Entry | 2 | R | R1 | R2 | % Yieldb | % eec(Config) |
|---|---|---|---|---|---|---|
| 1 | 2a | Me | H | H | 73 | 97 |
| 2 | 2b | Me | H | p-Me | 71 | 97 |
| 3 | 2c | Me | H | m-Me | 63 | 92 |
| 4 | 2d | Me | H | o-Me | 51 | 80 |
| 5 | 2e | Me | H | p-CI | 74 | 96 (R)d |
| 6 | 2f | Me | H | p-F | 51 | 89 |
| 7 | 2g | Me | H | p-t-Bu | 51 | 95 |
| 8 | 2h | Me | p-Me | H | 63 | 90e |
| 9 | 2i | Me | m-Me | H | 58 | 92 |
| 10 | 2j | Me | 3,4-Dimethyl | H | 63 | 82e |
| 11 | 2k | Me | p-t-Bu | H | 45 | 88e |
| 12 | 2l | Me | p-OPiv | H | 51 | 95 |
| 13 | 2m | Me | p-CI | H | 35 | 87 |
| 14 | 2n | Me | 3-chloro-4-methoxy | H | 47 | 90 |
| 15 | 2o | Me | 3-methyl-4-methoxy | H | 40 | 75 |
| 16 | 2p | Me | 4-methoxy-3-trifluoromethyl | H | 39 | 89 |
| 17 | 2q | Et | H | H | 61 | 72 |
| 18 | 2r | Pr | H | H | 52 | 76e |
| 19 | 2s | H | H | H | 69 | 58f |
The reaction conditions are identical to those described in Table 1;
Isolated yield;
ee was determined by chiral HPLC;
The absolute configuration was determined by analysis of the X-ray crystal structure;
Boc-Tyr(t-Bu)-OH was used as ligand;
Racemization occurred during the reaction.
Different carboxylic acid substrates were also subjected to this reaction protocol. Alkyl-substituted sodium carboxylates 1h–1k were converted to the corresponding products with good to high enantioselectivity (entries 8–11). Boc-Tyr(t-Bu)-OH was found to be better chiral ligand for sodium carboxylates 1h, 1j and 1k. The reaction was also found to tolerate substrates containing electron-donating groups (p-OPiv, 1l, entry 12) and moderately electron-withdrawing groups (p-Cl, 1m, entry 13), although olefination of 1m gave 2m in only 35% yield. 3,4-Disubstituted substrates were also olefinated effectively giving moderate to high levels of enantioselectivity (entries 14–16). Reactions of sodium 2,2-diphenylbutanoate 1q and sodium 2,2-diphenylpentanoate 1r with styrene gave lower enantioselectivity (entries 17–18). Unfortunately, the reaction of α-hydrogen-containing 1s only gave 58% ee, and it was found that 2s was partially racemized under the reaction conditions (entry 19). Notably, the absolute configuration of the olefination product 2e was determined to be R by X-ray crystallographic analysis (Figure 2), which was consistent with the proposed intermediate B (Figure 1).
Figure 2.

Absolute Configuration of Olefination Product 2e
Acrylates were also found to be efficient coupling partners under these conditions, affording 99% ee. However, a mixture of the desired olefination product and the corresponding conjugated addition product was obtained (Scheme 1). The use of sodium carboxylate salt also improved the yield.
Scheme 1.

Enantioselective C-H Activation/Olefination Using Acrylates as the Coupling Partnersa,b
a The reaction conditions are identical to those described in Table 1; b The ratio of products and dr were determined by 1H NMR.
Finally, these olefinated products could be readily converted to aldehydes or lactones by simple chemical transformations with complete retention of stereochemistry (Scheme 2).
Scheme 2.
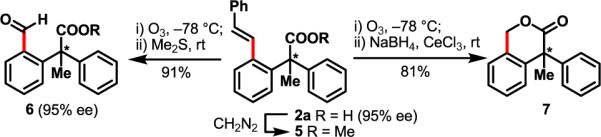
Derivatization of the Olefination Products
In summary, we have demonstrated that mono-protected α-amino acids are effective chiral ligands for Pd(II)-catalyzed enantioselective C-H activation reactions of carboxylic acid substrates. Expansion of this asymmetric technology to enantioselective sp3 C-H functionalization is underway.
Supplementary Material
Acknowledgement
We gratefully acknowledge The Scripps Research Institute, the National Institutes of Health (NIGMS, 1 R01 GM084019-01A1), Amgen and Lilly for financial support.
Footnotes
Supporting Information Available: X-ray diffraction analysis for 2e, experimental procedure and characterization of all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For recent reviews see: Dick AR, Sanford MS. Tetrahedron. 2006;62:2439.. Daugulis O, Zaitsev VG, Shabashov D, Pham Q-N, Lazareva A. Synlett. 2006:3382.. Yu J-Q, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k.. Campeau L-C, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35.. Rubin M, Rubina M, Gevorgyan V. Chem. Rev. 2007;107:3117. doi: 10.1021/cr050988l.
- (2).For a review on stereoselective C-H functionalization, see: Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Chem. Soc. Rev. 2009;38:3242. doi: 10.1039/b816707a.
- (3).Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Angew. Chem., Int. Ed. 2008;47:4761. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- (4).For a Ru-catalyzed atropselective alkylation, see: Kakiuchi F, Le Gendre P, Yamada A, Ohtaki H, Murai S. Tetrahedron: Asymmetry. 2000;11:2647.
- (5).For a Pd(0)-catalyzed enantioselective arylation see: Albicker MR, Cramer N. Angew. Chem., Int. Ed. 2009;48:9139. doi: 10.1002/anie.200905060.
- (6).For enantioselective hydroarylation and hydroacylations see: Bosnich B, Wang X. Organometallics. 1994;13:4131. Mikami K, Hatano M, Terada M. Chem. Lett. 1999:55.. Thalji RK, Ellman JA, Bergman RG. J. Am. Chem. Soc. 2004;126:7192. doi: 10.1021/ja0394986.. Coulter MM, Dornan PK, Dong VM. J. Am. Chem. Soc. 2009;131:6932. doi: 10.1021/ja901915u.. Shibata Yu, Tanaka K. J. Am. Chem. Soc. 2009;131:12552. doi: 10.1021/ja905908z.
- (7).For enantioselective carbenoid and nitrenoid insertions, see: Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485.. Doyle MP. J. Org. Chem. 2006;71:9253. doi: 10.1021/jo061411m.. Du Bois J, Zalatan DN. J. Am. Chem. Soc. 2008;130:9220. doi: 10.1021/ja8031955.. Milczek E, Boudet N, Blakey S. Angew. Chem., Int. Ed. 2008;47:6825. doi: 10.1002/anie.200801445.
- (8).For enantioselective Heck and cross-coupling reactions see: Sato Y, Sodeoka M, Shibasaki M. J. Org. Chem. 1989;54:4738.. Carpenter NE, Kucera DJ, Overman LE. J. Org. Chem. 1989;54:5846.. Arp FO, Fu GC. J. Am. Chem. Soc. 2005;127:10482. doi: 10.1021/ja053751f.
- (9).For enantioselective oxidation of alcohols see: Ferreira EM, Stoltz BM. J. Am. Chem. Soc. 2001;123:7725. doi: 10.1021/ja015791z.. Jensen DR, Pugsley JS, Sigman MS. J. Am. Chem. Soc. 2001;123:7475. doi: 10.1021/ja015827n.
- (10).(a) Mei T-S, Giri R, Maugel N, Yu J-Q. Angew. Chem., Int. Ed. 2008;47:5215. doi: 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]; (b) Giri R, Yu J-Q. J. Am. Chem. Soc. 2008;130:14082. doi: 10.1021/ja8063827. [DOI] [PubMed] [Google Scholar]
- (11).Wang D-H, Engle KM, Shi B-F, Yu J-Q. Science. DOI: 10.1126/science.1182512. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.