

Abstract
For at least a century it has been known that multiple factors play a role in the development of complex traits, and yet the notion that there are genes “for” such traits, which traces back to Mendel, is still widespread. In this paper, we illustrate how the Mendelian model has tacitly encouraged the idea that we can explain complexity by reducing it to enumerable genes. By this approach many genes associated with simple as well as complex traits have been identified. But the genetic architecture of biological traits, or how they are made, remains largely unknown. In essence, this reflects the tension between reductionism as the current “modus operandi” of science, and the emerging knowledge of the nature of complex traits. Recent interest in systems biology as a unifying approach indicates a reawakened acceptance of the complexity of complex traits, though the temptation is to replace “gene for” thinking by comparably reductionistic “network for” concepts. Both approaches implicitly mix concepts of variants and invariants in genetics. Even the basic question is unclear: what does one need to know to “understand” the genetic basis of complex traits? New operational ideas about how to deal with biological complexity are needed.
Keywords: complex traits, gene expression, in situ hybridization, mapping, pleiotropy
“…much about development and evolution is still hidden and mysterious.
“Adam Wilkins, 2002”.(1)
Prisoners of Mendel?
Make everything as simple as possible, but not simpler.
Attributed to Albert Einstein.
Where do biological traits such as a tooth or skull come from? Such a simple question has no simple answer, and indeed recent knowledge blurs rather than sharpens our understanding, raising important issues about the question itself.
Generally speaking, biological traits are the product of both a proximate and an ultimate “history.” Proximally, this means an intragenerational developmental history of a given individual. How should we characterize this? Is there a fixed genetic architecture that specifies “the” skull or “tooth?” If so, it should be possible to define the trait in terms of a list of genes “for” the trait, and their functions. Such genetic invariants, or causal stereotypes are implicitly reflected in the standard reference to the “wild-type” trait and its presumed underlying genetic basis.
Yet, the ultimate origin of biological traits is a different kind of history, an intergenerational evolutionary history among individuals. Evolution is inherently dependent on genetic “variants,” not types, and their distributions in populations or among species.
That these proximate and ultimate histories are confounded in a tenuous platonic way is exemplified by the common practice, when interpreting transgenic experiments on inbred laboratory mice, of comparing the transgenic’s phenotype to the “wild type” in the same strain, such as C57BL. But there is nothing “wild” about inbred, entirely homozygous laboratory strains, and we know that the underlying genetic architecture of most traits being studied is not in fact invariant, because the same experiment typically has different (or no) effects when done on different inbred strains. So what do we actually mean by “wild type,” and where is it and how is it to be found? Clearly, we face serious conceptual issues in both practice and theory with respect to complex developmental traits and their evolution.(2)
Gregor Mendel’s experiments with peas gave us our first glimpses into the genetic control of morphological traits. He intentionally restricted his experiments to discrete traits whose variation segregated predictably in offspring, showing that inheritance involved particulate causal agents “for” those kinds of traits. It is no accident that some of the first traits to be explained by modern genetics were in experimental organisms for which mutations in a single gene have high penetrance, thus providing the prediction of a discretely definable phenotype with a sound theoretical, even if probabilistic basis. This experimental design confounded transmission with cause, or variation with mechanism, and had the conceptually constraining effect of suggesting that the trait itself was transmitted. That legacy is still with us, as even cases of clearly complex traits, like breast cancer, diabetes, or epilepsy are sometimes said to be “Mendelian.” A zygote receives genes, but not cancer, diabetes, or epilepsy.
For nearly a century after Mendel’s rediscovery, genes were necessarily treated as causal elements of unknown nature that were “for” specified traits. Despite knowing that genes do not act alone, it was usually necessary in practice to treat them one at a time, and even today the typical approach is to look for simple genetic explanations by identifying genetic contributions one gene at a time (e.g., Refs(3,4–7)). Often these genes are discovered through studies of the association between natural or experimental variation and its effects. Yet there is distrust of statistical associations, and an understandable demand that they be confirmed by functional studies to identify the underlying mechanism. But since neither approach is independent of the other, the concept of an underlying causal structure is less clear than it might seem.
Even “simple” or “Mendelian” traits, when intensely studied as is the case for human diseases, are much more complex than first realized (e.g., Refs(8,9)). There are countless instances and few exceptions. To illustrate, approximately 100 alleles are associated with Tay Sachs Disease (www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=272800) and over 1500 alleles at the CFTR locus are currently listed in the Cystic Fibrosis Mutation Database (www.genet.sickkids.on.ca/). Known genotypes usually do not accurately predict phenotypes, or vice versa. Instead, it is more typical that identified genotypes manifest quantitative rather than qualitative “Mendelian” (recessive or dominant) phenogenetic relationships.
Even for known alleles in single genes, the causal situation can be ambiguous. Hemochromatosis in Europeans is quite strongly associated with a gene variant that is found at high frequency in the unaffected population, where it has little or no penetrance.(10,11) The lifetime risk of breast cancer associated with the known highest-penetrance alleles in BRCA1, the poster child for cancer genes, has been estimated to be between 46 and 80% or more,(12–14) a difference presumably largely due to secular trends in lifestyle risk factors. The ground is even shakier beneath heavily studied traits like obesity, blood pressure, asthma, heart disease, schizophrenia, autism, or intelligence because etiology is complex, the trait can prove to be difficult to measure or even to define, and we remain ignorant of the usually inestimable or unidentified environmental influences.
Although some say we are entering the post-genomic era, the predominant study designs are just like Mendel’s of the 1860s: select the simplest case—the frankly pathological or clear extremes of the distribution of normal variation (such as by knockout experiments), enumerate the genetic variation responsible, and extrapolate from this tail of the phenogenetic distribution to the whole distribution. The tail is wagging the causal dog, so to speak, when even the tail is complex (and usually moving!). However, similar statements apply to attempts to map the genetic basis of normal variation in traits like stature, limb, or craniofacial morphology, and the story is essentially the same in plants, animals, and even unicellular organisms.
A reflection of a persisting conceptual imprisonment by Mendelian thinking is the way gene nomenclature reinforces ideas about what genes are “for.” At the beginning of the 20th century, genes could only be identified by phenotypes with which they were associated, such as the Drosophila genes eyeless or hunchback (e.g., see Ref.(15)). During the mid-century expansion of genetics research, when genes were understood by their specification of proteins or enzymes, they could be named accordingly (e.g., alcohol dehydrogenase, phenylalanine hydroxylase, globin, albumin, apolipoprotein E). But in the gene-mapping arena of the past 25 years, genes began again to be named after effects through which they were discovered, as if they were for those effects. Examples include BRCA1 (breast cancer 1), PSEN1 (presenilin 1), or even BMP (bone morphogenetic protein) and MHC (major histocompatibility complex).
These names may not even recognize normal function or account for the reality that many or most gene products have multiple functions,(16,17) even though the widespread nature of pleiotropy was realized long ago.(18,19) And experience has rapidly been showing that we face a deeper problem, because terminology alone can place significant constraints on thought as well as research, and it is hard to say what a gene is “for” if we do not even know what a gene “is.”
What is a gene? A brief conceptual history
Hence, the talk of “genes for any particular character” ought to be omitted… So, as to the classical cases of peas, it is not correct to speak of the gene – or genes – for “yellow” in their cotyledons or for their “wrinkles,” yellow color and wrinkled shape being only reactions of factors that may have many other effects in the pea-plants.
Wilhelm Johannsen (1911)(20)
It is worth retracing how we got to today’s conceptual world. There is a large recent literature on the changing concepts of the gene and what genes are or what they do (e.g., Refs(21,22–25)). These are important issues that are not always discussed by biologists, who often feel they can move ahead in a de facto way without worrying about philosophy or terminology. In fact, reflecting the growing knowledge of genomic functions, modern definitions of “gene” are becoming epistemologically vacuous (e.g., Ref.(26)).
The Danish botanist Wilhelm Johannsen coined the word “gene” in 1909.(27) He acknowledged the presumed particulate nature of inheritance, but carefully neither ventured a definition nor hypothesized about mechanism. His purpose was to make a distinction between the “genetics” revealed by the recent rediscovery of Mendel’s work on variation, and Lamarck’s and Darwin’s groping mechanistic explanation of inheritance as, essentially, of miniature replicates of parental traits (a view going back at least to Hippocrates).
But even with just a metaphoric understanding of the nature of genes, their effects could be tracked with experiments that compared parent to offspring—again using variation to get at the tacitly assumed invariant underlying architecture. T. H. Morgan at Columbia University began experiments with mutant fruit flies in the 1910s to systematically catalog the Drosophila genome by locating on chromosomes the causal elements to which he attributed traits. He could do that by careful selective breeding, relying on recombination among discrete phenotypes that, like Mendel’s, were due to highly penetrant alleles—and treated traits as if they themselves were inherited. He was well aware that genes could affect more than one trait, that a trait could be influenced by more than one gene and that traits varied more than discretely.(28) But this did not disturb his research agenda because he was focused on phenotypic effects and genomic relationships rather than genetic mechanism.
By the 1920s, a Morgan student, H. J. Muller, induced mutations in Drosophila with X-rays, showing definitively that genes are localized, changeable causal molecules(29): mutation disrupted the normal function of the gene “for” the trait. By 1941, geneticist George Beadle and biochemist Edward Tatum had irradiated the bread mold Neurospora crassa,(30) and crossed irradiated with non-irradiated molds. Some could not reproduce without the addition of a specific amino acid because irradiation had mutated a gene involved in production of an enzyme that synthesized that amino acid. They concluded from this that each gene somehow produces one enzyme.
Watson and Crick cemented this view into a specific picture of genes as the causal units of life. Genes were not miniature models of traits, but instead their sequence specified the information to produce proteins of which traits are made. This and the working out of many experimental details eventually led to the “Central Dogma of Biology,” the one-way flow of information from DNA via RNA intermediates to protein, and in light of Beadle and Tatum’s work, the familiar “one gene → one protein.” This became the universal and fundamental causal underpinning of modern molecular biology and reinforced thinking of genes as being “for” specific traits (one gene → one trait).
This view of genes as discrete coding and primary functional units was highly operationalizable and enabled a torrent of discovery of the basis of countless genetic traits in plants and animals. The search was aided, only about a generation ago, by the discovery of polymorphic markers spaced across the genome of plant or animal species under study. This enabled gene mapping, searches for genome regions, and then genes, that were “for” almost any Mendelian (simply segregating) trait one would want to study. Remarkably, these genes could be found without any knowledge of the biological basis of the trait under study!
However, the torrent also brought with it a flood of discoveries beyond genes as they had been known. In fact, genes are much more than fixed protein-coding elements. We have amassed tremendous knowledge, which has forced a reconsideration of the question of what genes are.(26,31) Not only might coding regions be transcribed in both directions in different ways, but they are variously spliced in ways that may be context specific, so that a classical “gene” can code for multiple proteins used in multiple pathways.(17) There is a plethora of DNA-encoded functions other than protein coding, including non-transcribed sequence used in DNA–protein binding or epigenetic modification related to chromosome packaging, replication and gene regulation. A substantial fraction of genomes are transcribed into non-coding but functional RNAs of many types that affect gene translation and many other functions, only some of which are known(e.g., see Refs(26,31–37)). Not least of these are RNAs that interfere with protein coding (by blocking mRNA translation), and hence serve as “anti-genes.” Post-transcriptional regulatory mechanisms also play a significant role in genetic function.(38,39)
These facts are important for reasons both biological and practical. Instead of simplifying, each new discovery adds to the complexity of mechanisms by which genomes contribute to the development, maintenance, and evolution of biological traits. Candidate chromosomal regions identified by mapping can no longer be searched only for classical protein-coding genes. Instead, few if any parts of the region can be dismissed out-of-hand as non-functional: there is a lot less in the DNA junkyard than we had thought. It is usually a daunting search through wilderness to find the actual causal site (if there is but one!) in candidate regions, each of which can easily exceed 1–2 Mb in length.
Dissecting complex traits – Into what?
Everywhere, the substantive gene is a “will-o’-the-wisp.” There are merely quantitative modifiers of a developing growth pattern.
Sewall Wright (1934)(18)
In the early 1900s it seemed that there were two kinds of traits, qualitatively and quantitatively varying, and there was some doubt as to whether they could have similar kinds of genetic control. But if biological theory was at all correct, complex traits must also have a genetic basis. Indeed, various investigators, most notably Fisher in 1918,(40) showed that quantitative traits could result from the joint contributions of large numbers of “Mendelian” loci—that came to be known as “polygenes”—that had individually miniscule effects. This has wide applicability because most complex traits can be viewed as having underlying quantitative developmental or genetic bases.
For most of the 20th century, polygenic control could only be treated as an undifferentiated aggregate of causal effects on variation: their identity and mechanism were not knowable by available methods. However, molecular and population genetic evidence showed that the elements of polygenic control are not as internally homogeneous in effect or population frequency as often modeled or presented in textbooks, and when mapping markers became available, it was natural to ask whether these genes could also be identified by mapping their heterogeneous effects. Indeed, it became possible to search statistically for chromosomal regions that may contain variation in specific sequence elements whose contribution was sufficiently strong to be detectable using genome-wide mapping methods. A statistically implicated candidate chromosome region is known as a “quantitative trait locus,” or QTL; these fueled a burgeoning gene-mapping industry. Since then, this approach has found genes affecting traits of every type and complexity in humans and other species of any kind.
An early notable success in complex trait mapping was the identification of BRCA1 that affects human breast cancer risk.(41) However, despite tremendous success, this did not allow anything even approximating the enumeration of the entire set of genes “for” any complex trait (including breast cancer), and the problem of dissecting the genetic architecture of such traits is far from solved. Indeed, it might be appropriate to refer to QTLs as Quixotic Trait Loci,(42) because despite unquestioned successes, QTLs identified in one study are frequently not replicated or their strength of effect steadily diminishes (sometimes to zero) as additional studies are done. In fact, mapping does not detect genetic architecture per se, but relies on variation that exists in one’s particular chosen sample, providing only incomplete, sample-dependent glimpses of the trait’s genetic basis.
A fundamental aspect of polygenic aggregate control is “genotypic equivalence,” i.e., similar trait values arise from different genotypes, or for environmental reasons, and individual genotypes can be associated with a spectrum of phenotypes. Findings vary among traits, families, populations, strains within species, or among species. To date, even with many studies often of very large samples, the overall bulk of the genetic control of such traits, as measured by heritability or familial aggregation, remains unexplained by the detected QTLs (e.g., Refs(43–46)). At some point, perhaps already approaching, attempts to map infinitesimal polygenic effects through natural variation will reach epistemic limits.
In essence, the same burgeoning technology that has provided so much new knowledge about genes and how they work has also opened a Pandora’s box of complexity. We can either take on faith that life is not as complicated as it now seems, and trust that continued application of standard approaches will show this, or we can acknowledge that life is more complex than current methods account for, and rather than continue to ask “What are the genes for X?” ask instead “What makes X?”
Some assembly required. Batteries not included
We find, in experience, that we cannot safely infer from the appearance of the character what gene is producing it.
T.H. Morgan (1917).(28)
Despite growing awareness of the various complexities, genetic approaches to understand how traits are made generally still start with a version of a gene-for type of question: “What are the genes for my trait?” This is difficult to avoid because we do not yet know how to investigate traits as emergent structures. Dissatisfaction with statistical association and one-gene experimental approaches has heightened the drive to find better ways to demonstrate the presumed genetic architecture directly.
One important approach in the search for functional structure is to identify the genes that are expressed in tissues relevant to a trait under study. Expression is surely at least an indirect reflection of mechanism, and if one has a strongly Darwinian view of life, useless traits, including functionless gene expression, would be wasteful of vital resources and purged by natural selection. There are many ways to document expression, but they all yield what are basically new kinds of gene catalogs, in the end not much different from pre-Darwinian beetle collections. These catalogs may help identify aspects of genetic architecture, but without an explanation for which genes are crucial for which traits at which time or in what way, or of knowledge of which beetles are missing from the collection, they are usually not by themselves any more useful than a list of materials used to build a model airplane without a manual. Thus, like association studies, expression-array studies are suspect. In contrast to cell-specific arrays, whole-embryo in situ hybridization (ISH) studies are more demanding to do on a genome-wide scale, but can be more illuminating because they can show expression in essentially every tissue at cell-specific resolution in an intact embryo of a tested stage.
Expression studies have strongly confirmed the classical realization that pleiotropy is widespread. Each cell expresses hundreds or thousands of genes, and most genes are expressed in many different types of cells or in varying cellular contexts. Complex traits are spatio-temporal collectives of multiple interacting gene products, changing over time (from conception onward) in any target organ or tissue, and sculpted by increasingly well-documented phenomena including largely unknown environmental influences.
Expression studies do not tell us about gene function, gene-gene interaction, or gene–environment interaction and, since most expression studies to date have considered only classical protein-coding genes (by documenting their mRNA), other relevant processes involving transcribed RNA, such as RNA interference, remain largely undocumented. But even limited as they are, expression studies provide an important reminder that most genes, being highly pleiotropic, are not “for” anything in particular. It is worth describing a few examples to make this point clearly.
Figure 1 shows expression of the complement factor H (CFH) gene in a mouse at embryonic day 14.5. Canonically, CFH codes for a protein secreted into the bloodstream that is involved in the alternative complement pathway of the immune system. Several CFH alleles have been associated with human age-related macular degeneration, a retinal disease leading to loss of vision. Viewing CFH as a gene “for” immune function is thought to explain its role in macular degeneration, which appears to have inflammatory antecedents. However, CFH transcripts are found in many tissues, which suggests this gene plays roles in embryogenesis beyond immune function, that are not yet characterized—or that our notion of “immune” needs revision.
Figure 1.

Transverse section of ISH of E14.5 mouse showing expression of CFH, dark purple stain, not restricted to circulating blood or immune system cells. From GenePaint: http://zoom.gwdg.de/fif=030/EH00002983_00009B.fpx&obj=UV,1.0&wid=501&page=&page=genepaint.html.
An olfactory receptor gene, Olfr66, is expressed in the olfactory epithelium as expected, but also in brain, vertebrae, and other tissues. Amelogenin, named for its role in forming tight calcium matrices in tooth enamel, the hardest substance in the body, is also expressed in parts of the brain, as well as developing bone throughout the body. This same point can be made by looking at the expression of almost any gene.
We can further illustrate this with several examples of ongoing work from our own laboratories. These take the logic the other way: rather than finding unexpected sites of expression of known genes, we want to find genes in chromosomal regions implicated by mapping that have expected sites of expression.
Primate cranial evolution
In collaboration with a number of people at other institutions, our group is studying the development and evolution of the primate skull by looking at variation among primate lineages (http://www.hominid.psu.edu). A skull is a vital, functional, evolutionarily important trait, and its development, as that of all complex traits, involves the interaction of many pleiotropic genes and modifying factors, most with small effects, interacting with environmental influences. The somewhat daunting challenge is to go beyond the usual “gene for” approach to construct an integrated picture of the development of the skull and then try to determine how that integration might be choreographed and directed. But even preliminary mapping results suggest this is not going to be easy.
For example, data from many resources makes it clear that a majority of genes in every chromosomal region are expressed in the developing head. Indeed, genes in tentative candidate regions found by mouse and baboon mapping are no more frequently expressed in the head than genes in random chromosomal regions. Nor do the mapped candidate regions highly overlap between test species, and within species multiple apparently unrelated aspects of craniofacial variation sometimes map to the same region (unpublished work in progress).
Variation in the mouse dentition
One might object that this is a straw man because the skull is not meaningfully viewed as a single trait, but the problem is not ameliorated by concentrating only on substructures. Consider the humble tooth. Tooth development has been intensely studied with some of its developmental genetic mechanisms at least partially understood (http://bite-it.helsinki.fi), mainly through work on severe effects such as from experimental knockouts. However, evolution usually works on smaller, non-pathological variation, and we have identified a simple naturally occurring dental cusp variant that we are mapping using a recombinant inbred (RI) platform (see Fig. 2).
Figure 2.
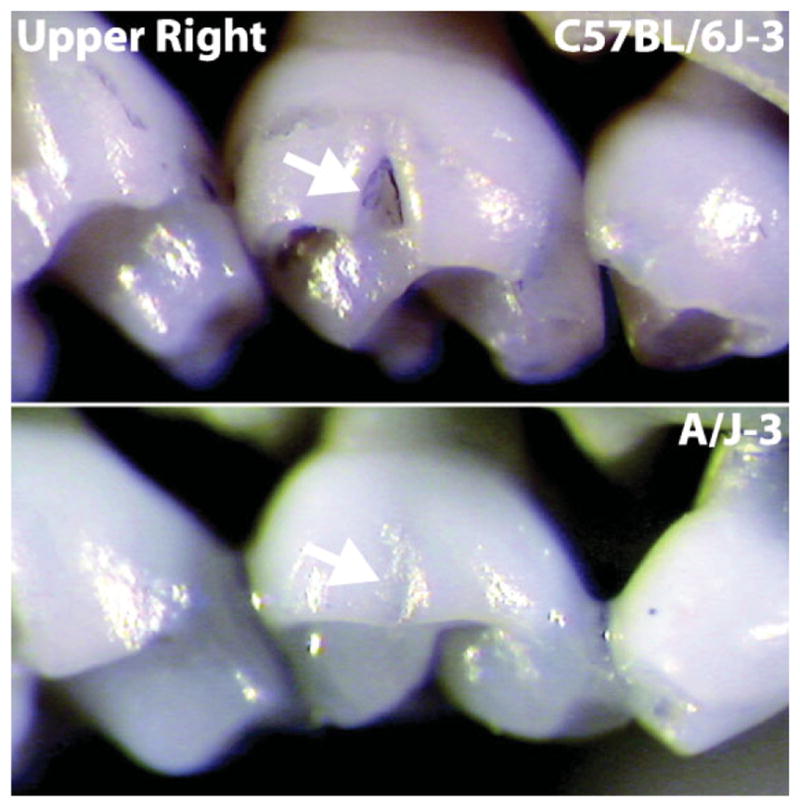
Tooth trait variation between two inbred lines of mice.
Because the variant is a discrete trait that breeds true in the RI cross, in a sense we are following Mendel’s model ourselves by identifying a trait that should be tractable in an inbred laboratory organism, but that may not represent the full range of variation in outbred, wild populations.
Mapping identified three chromosome regions statistically associated with this trait variant. The regions span several megabases, and they include up to several hundred genes (the number of functional non-coding regions is at present unknowable). As for all such studies, the question is how to prioritize genes for further study. Choice of candidates typically relies on prior knowledge of the function or expression locus of genes in a given region. One must be mindful of the vagaries of gene ontology resources, and that previous research might have concentrated solely on a gene’s role in an anomaly or in regard to some entirely different trait. To avoid sources of bias, we chose to consider each gene in the mapped intervals to be a potential candidate, amassing information from the easily retrievable resources available on the internet.
Our first step was an extensive literature search, which suggested about 30 candidate genes based on previous description in tooth development, gene family relationships, or expressed sequence tags. Through direct sequence comparison of the genome sequences of the two parental mouse strains, we were able to quickly reduce this to a handful of possibly plausible genes with inter-strain differences either altering the amino acid sequence of the encoded protein or in transcription factor binding sites predicted by various criteria that potentially could affect gene expression.
Prior to the availability of major high-throughput gene expression resources (e.g., microarray databases, the Gene Expression Omnibus (GEO) at NCBI (http://www.ncbi.nlm.nih.gov/geo/), the Gene Expression Database (GXD) at Jackson Labs (http://www.informatics.jax.org/expression.shtml), or GenePaint developed by the Department of Molecular Embryology at the Max-Planck Institute of Experimental Endocrinology, Hannover, Germany (http://www.gene-paint.org)) we would have stopped here, focusing further study on these few candidate genes. But the new resources made it possible to search each gene in the identified chromosomal regions, looking for expression in the tooth primordia. Illustrative of the point is the GenePaint database (www.GenePaint.org), which provides whole-embryo ISH results for about 18 000 genes studied to date. While not all (protein-coding) genes are yet in the database, and ISH results for most have been done only at mid-gestational day E14.5, mainly in a single mouse strain (NMRI), this is the most complete database currently available that allows expression of so many genes to be visualized, at the cell level, in all tissues. Even considering the limits of this particular resource, the evidence is sobering.
Of the genes in the three chromosome regions identified by mapping that are in GenePaint, more than half are expressed in developing teeth (Table 1) and must be considered as candidates. These included many genes misleadingly named for a particular function or expression domain such as Hap1 (huntingtin-associated protein, Fig. 3A) and Tex2 (testis expressed gene 2, Fig. 3B), genes that we would have dismissed if we were relying only on reports in the literature of what these genes do.
Table 1.
Chromosomal regions with suggestive LOD scores identified by mapping tooth trait, total genes in region, number with tooth expression found in GenePaint, and number with tooth expression reported in the literature
| Chromosome | Peak LOD | ~1 LOD interval (Mb) | Total genes | Genes in tooth (literature) | Genes in GenePaint | GenePaint genes expressed in teeth |
|---|---|---|---|---|---|---|
| 11 | 2.24 | 92–110 | 351 | 17 | 242 | 136 |
| 13 | 2.24 | 30–50 | 113 | 7 | 84 | 43 |
| 19 | 3.01 | 26–32 | 42 | 8 | 26 | 18 |
Figure 3.

A: Hap1: huntingtin-associated protein 1, inset shows expression in developing molar (http://zoom.gwdg.de/fif=033/EG00001730_00019B.fpx&obj=UV,1.0&wid=501&page=genepaint.html); B: Tex2: testis-expressed gene 2, showing molar and incisor regions (insets) (http://zoom.gwdg.de/fif=029/EH00002754_00008B.fpx&obj=UV,1.0&wid=501&page=genepaint.html).
How do we make sense of all this information? The potential targets for experimental intervention become more daunting the more we know. Expression data only reveal protein-coding genes, not regulatory elements of various kinds or transcribed non-coding RNA genes (most of which are not yet known). Even assuming that the mapping signals are not false positives, just in a cross between two inbred strains there are around 1000–2000 SNPs (single nucleotide polymorphisms) per Mb. Which can be dismissed as irrelevant? These same issues will be true for any trait. An obvious solution is to increase sample sizes so as to greatly narrow the candidate mapped region, which is clearly important to do but a potentially illusory economy—the other genes in the genome that are known to have potentially relevant expression may not affect variation in our particular sample, but cannot be dismissed as unrelated to the underlying genetic architecture.
Down syndrome craniofacial phenotype
The problem is just as difficult if we consider only rare or abnormal phenotypes, which might on first thought seem to have simpler origins. Down syndrome (DS), caused by triplication of the genes on Chromosome 21 (Chr 21), is characterized by over 80 clinical features,(47) all of which occur in the non-DS population(48). Only a subset of the 80 clinical phenotypes occur in all individuals born with DS (e.g., cognitive impairment, characteristic craniofacial appearance, and histopathology of Alzheimer’s disease), and there is a great degree of variability in extent and/or severity of each of these.
One of the main issues confronting DS researchers concerns how dosage imbalance of Chr 21 genes disrupts development to produce DS phenotypic features.(49) The craniofacial appearance is an immediately recognizable, invariant DS phenotype, although craniofacial structures are not grossly malformed. Until recently the dominant theory in DS research was that dosage imbalance of the genes in the “DS critical region” (DSCR; see OMIM entry 605298) was responsible for many DS phenotypes including the typical craniofacial appearance, but the failure of trisomy of the DSCR gene orthologs to cause the craniofacial phenotype in a chromosomally engineered mouse demonstrated that the genetics underlying these traits is far more complex than hoped.(50)
Genes on Chr 21 do not cause anomalous conditions when expressed at appropriate levels, yet certainly contribute to DS phenotypes when overexpressed. Using a specifically designed high-content Chr 21 expression microarray, Aït Yahya-Graison et al.(51) were able to sort Chr 21 genes into four classes: those expressed proportional to the gene dosage effect, those that are compensated, those that are amplified, and those that show high interindividual variability in their effect. The authors propose that the genes that are overexpressed are those most likely to account for the DS phenotypes but it remains unclear which genes or combination of genes on Chr 21 (and elsewhere) underlie which trait(s).
For a gene to cause craniofacial dysmorphology in DS, it would at least need to be expressed in tissues of the head during normal development. Using available gene expression resources, we attempted to narrow our search for those genes that may be responsible for craniofacial dysmorphogenesis in DS by determining which genes on Chr 21 are expressed in some tissue of the head during normal development, using mouse orthologs of Chr 21 genes. Of the nearly 140 genes on Chr 21 for which expression data are available, only 20 were not expressed in some potentially relevant craniofacial tissue, even just at day E14.5, Thus, at least 120 genes are potential players in the making of the DS head. And again this is only a consideration of classical coding genes, without any search for variation in regulatory or non-coding RNA sequence in the trisomic regions.
Indeed, one need not rely on our specific case studies, nor restrict attention to candidate regions identified by statistical gene mapping. Readers can see the issues for themselves by identifying some organ or structure, and picking a (say) 1–5 Mb random region of the genome, then looking at GenePaint or other expression data to see how many genes in that region have potentially relevant expression. In our experience, the results are not comforting.
Systems, networks, hierarchical interactions
In both science and engineering, the study of “systems” is an increasingly popular activity. Its popularity is more a response to a pressing need for synthesizing and analyzing complexity than it is to any large development of a body of knowledge and technique for dealing with complexity. If this popularity is to be more than a fad, necessity will have to mother invention and provide substance to go with the name.
Herbert Simon (1962)(52)
Most genes are pleiotropic, and for few if any do we know all their functions. Pleiotropy may be widespread because new traits evolve by re-use of existing genetic pathways by relatively easy change of expression context that can be brought about by mutation in a gene’s flanking regulatory sequences. However, genes do not act alone. In given contexts, sets of genes (to date, this means classical genes) are coordinately expressed in ontogenetic paths for which control hierarchies can be identified (e.g., Ref.(53)), a fact that has long been known.(19,28,54) These are often referred to as networks. The genes or even the networks are typically used in tissues of unrelated embryological origins, and the development of eukaryotic organs typically involves the interaction of multiple cell-fate lineages.
In the face of the problems we have been discussing, there has been a movement to think differently about complex traits, not in terms of individual genes that are “for” them, but in terms of the participating networks or systems, such as signaling networks, viewed as ontogenetic units. Networks are represented by hierarchical or hub-and-spoke diagrams of the interactions of the participating genes. The idea is the seemingly correct one that complex traits are made by “relational structures” that jointly constitute the long-sought underlying genetic architecture, whose occasional allelic variation serendipitously captured in mapping samples reveals some of the genes involved.
Network thinking is suggested as a way to move from unitary “gene for” notions to more biologically realistic understanding of the production of traits in terms of generating processes. However, the same kinds of issues that apply to our understanding of individual gene function may also apply to networks. Even if the idea of an invariant causal architecture is correct, networks can involve tens or hundreds of genes, and traits involve multiple networks in differing combinations at different stages of development. Indeed, combinatorial signaling mechanisms are fundamental to development, and signals are usually produced by cells different, and distant, from the receiving cells. Thus, expression in localized tissues of interest may systematically fail to detect relevant activity taking place elsewhere in the embryo. Under these circumstances, documenting the actual function or effect of each gene, or perhaps even each interaction among genes, is daunting if it is even possible. Intensive studies have recently shown the nature of network complexity even just between two haploid genomes (in both mice and yeast), that is, even in the most reduced sample of natural variation in the presumed underlying architecture (e.g., Refs(55–57)).
With mapping as well as expression evidence in hand there are many ways to search for a gene’s functional role (e.g., see Refs(58,59) for discussions of increasing the rigor of such searches). What is new is that we can now begin to enumerate the genes and pathways underlying it. By treating networks as causal units, however, we risk simply replacing the old “gene for” concepts with a new kind of platonic entity—a “network for” concept. Neither increased specificity, new terminology, nor more complex statistical characterization implies that we will understand how the parts make the whole anytime soon. Assembly and batteries are still required, and there is good reason to think that a standard instruction manual does not exist.
What do we want to know….and how will we know when we know it?
All of the results could… be accounted for consistently by postulating multifactorial segregation, giving practically normal distributions of all arrays in association with considerable non-genetic variation, on a physiological scale…
Sewall Wright (1984)(19)
The problem of complexity has been recognized by numerous authors, in many different disciplines, over many decades, to the extent that some would say that “everybody knows” the issues. New statistical and molecular approaches are being developed and refined; systems biology, the search for gene pathways and networks, the mining of microarray data, all recognize that traits are complex and the venerable “gene for” approach cannot explain them. However, to say that these issues have already been realized is not the same as recognizing that they are important enough to change the way we do our work. We still do not have a good solution to the problem of emergent complexity and, despite some broader network approaches, single-gene thinking and study designs are still predominant (even if now often treating networks as causal units driven by their eponymous genes, as for example assigning a role to “Bmp signaling”). More explicit, fleshed-out network approaches remain technically if not conceptually overwhelming.
A fundamental part of the problem is that it is hard to know when we have answered the question—or even whether we are asking a meaningful question. Superficially, it seems perfectly sensibletoask:What is the genetic basis of the skull? But, which skull do we use to answer this? And must we know every gene that is involved? In every species with “the” trait? We could ask a more specific question instead: What is the genetic basis of “the” mouse skull? or “the” human skull? or the vertebrate skull? Again, which skull do we use, or must we use many? Need we enumerate every allele in every individual?
It is possible, or even probable, that invariants—developmental stereotypes such as “the” skull, or even “the” mouse skull—may exist only in an imaginary world. If as much of the genome is involved in biological traits as the current evidence suggests, it constitutes a large mutational target, and no two individuals may be alike, each a unique, and essentially untestable combination of factors and effects: there is no “wild type”!
We can offer but an exploratory attempt at looking at these fundamental problems differently by returning to the difference between intergenerational evolutionary and intragenerational developmental histories. Selective agricultural breeding treats traits per se, and can produce change over generations in the net or aggregate effect of genes without the need to enumerate them individually. The problem in development is that, to date, we continue to enumerate genes because we do not yet have a suitable genetic analogy to an aggregate approach within individuals. While the goal of explaining traits is different from the breeder’s goal of manipulating them, developmental biologists might be well-served by trying out the breeder’s conceptual framework.
The genetic architecture of a complex trait is the net result of a multiplicity of relational processes involving synchronized cooperation of many different genes, cells, and tissues throughout development. A skull, for example, involves more than just, say, gene expression in osteoprogenitor cells in bone primordia, their immediate environment, and their descendants that form bone. Differentiation is typically based on signaling and response, interactions among diverse cells over developmental time, some factors may be contiguous while others may be organismally distributed (e.g., circulating cells, extracellular paracrine, autocrine, endocrine signals, etc.).
We suggest this schematically in Fig. 4. Hundreds of functional genomic elements are involved, both intra- and intercellularly. For reasons we have described, they may not be enumerable and natural variation may make each individual unique. However, it could be possible in principle to intercept the flow of information, perhaps especially via signals occurring in intercellular spaces at critical points. Aggregate measures of levels of this subset of factors might reflect the net effect of the generating processes, without the futile need to enumerate all the hundreds of factors involved, nor the genotypic variants and stochastic idiosyncrasies unique to each particular instance. This is an extension of expression-based methods that identify networks by correlated responses in specific cell types, but would not require correlation, unity, or invariant genetic structures. In principle, this could help resolve a number of conceptual and definitional issues, including ideas of genetic architectural stereotypes and what constitutes sufficient knowledge. We do not say it would be easy, but some sort of aggregate approach would at least reflect the complex, dispersed, multicellular, largely signal-based nature of complex phenotypes. Focusing on extracellular signaling, while largely ignoring intracellular complexity, would concentrate attention on the triggers responsible for intracellular action, perhaps obviating the need to enumerate the responding genes and networks within cells, because the critical action is in the interaction among them.
Figure 4.

A biological trait, like a skull or even just a tooth, involves interactions, typically based on signaling and response. Shown schematically, four types of cells receive and send signals to a final tissue of interest, such as the bones that form the skull. They may also send signals to each other, or to other tissues in close approximation (muscles of mastication, neural fibers, hairs) or far away (cartilage of the knee) and these other tissues may or may not enter into the interactions that ultimately form the skull. Internal signal-response gene expression is shown symbolically for each cell. Extracellular signals represented by shapes specific to the sending cells circulate to receiving cells. The inset shows the network relationships. The net result is based on the aggregate of these effects, which in principle could be accessible in extracellular space for various kinds of quantitation, to characterize the resulting trait without enumerating the genotypes and stochastic variation that may be unique to each individual. Skull drawing by Vesalius.(60)
By analogy, we can account for the functioning of a radio in terms of receiving and tuning processes, without having to know the details of its cabinet and buttons, or what station each radio is tuned to, or what stations are available in any given area—or even how to build a radio. Similarly, we can aspire to identify the relational systems that are involved in a given trait and its phylogeny, knowing that each instance will vary in ways we cannot always document, and that this is in the very nature of life and evolution. However, it is not easy to seek even that kind of answer without a more perceptive statement of the question itself.
Acknowledgments
We thank anonymous reviewers for helping us improve this perspective. We gratefully acknowledge support by grants from the US National Institutes of Health, grant R01 HD 38384 (NICHD/NCI) to RH Reeves and R01DE018500 (NIDCR) to JTR; the National Science Foundation: grants BCS 0343442, BCS 0725227 to KMW and JTR; and the Penn State Evan Pugh Professors fund to KMW.
Footnotes
We dedicate this paper to Adam Wilkins, to honor a generation of service to integrated biological thinking, as Editor of BioEssays
References
- 1.Wilkins AS. The evolution of developmental pathways. Sunderland, MA: Sinauer; 2002. [Google Scholar]
- 2.Sholtis S, Weiss KM. Phenogenetics, genotypes, phenotypes, and variation. In: Hall B, Halgrimsson B, editors. Variation. New York: Academic Press; 2005. pp. 499–523. [Google Scholar]
- 3.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatr. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 4.Kreek MJ, Nielsen DA, Butelman ER, LaForge KS. Genetic influences on impulsivity, risk taking, stress responsivity and vulnerability to drug abuse and addiction. Nat Neurosci. 2005;8:1450–1457. doi: 10.1038/nn1583. [DOI] [PubMed] [Google Scholar]
- 5.Ober C, Hoffjan S. Asthma genetics 2006: the long and winding road to gene discovery. Genes Immun. 2006;7:95–100. doi: 10.1038/sj.gene.6364284. [DOI] [PubMed] [Google Scholar]
- 6.Plomin R, McClearn GE, McGuffin P, DeFries JC. Behavioral genetics. New York: Worth; 2000. [Google Scholar]
- 7.Yang MS, Gill M. A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int J Dev Neurosci. 2007;25:69–85. doi: 10.1016/j.ijdevneu.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Scriver CR, Waters PJ. Monogenic traits are not simple: lessons from phenylketonuria. Trends Genet. 1999;15:267–272. doi: 10.1016/s0168-9525(99)01761-8. [DOI] [PubMed] [Google Scholar]
- 9.Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007;28:831–845. doi: 10.1002/humu.20526. [DOI] [PubMed] [Google Scholar]
- 10.Laine F, Jouannolle AM, Morcet J, Brigand A, Pouchard M, et al. Phenotypic expression in detected C282Y homozygous women depends on body mass index. J Hepatol. 2005;43:1055–1059. doi: 10.1016/j.jhep.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 11.Waalen J, Felitti V, Gelbart T, Ho NJ, Beutler E. Penetrance of hemochromatosis. Blood Cells Mol Dis. 2002;29:418–432. doi: 10.1006/bcmd.2002.0596. [DOI] [PubMed] [Google Scholar]
- 12.Chen S, Iversen ES, Friebel T, Finkelstein D, Weber BL, et al. Characterization of BRCA1 and BRCA2 mutations in a large United States sample. J Clin Oncol. 2006;24:863–871. doi: 10.1200/JCO.2005.03.6772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fodor FH, Weston A, Bleiweiss IJ, McCurdy LD, Walsh MM, et al. Frequency and carrier risk associated with common BRCA1 and BRCA2 mutations in Ashkenazi Jewish breast cancer patients. Am J Hum Genet. 1998;63:45–51. doi: 10.1086/301903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 15.Wilkins AS. Gene names: the approaching end of a century-long dilemma. Bioessays. 2001;23:377–378. doi: 10.1002/bies.1054. [DOI] [PubMed] [Google Scholar]
- 16.Jeffery CJ. Multifunctional proteins: examples of gene sharing. Ann Med. 2003;35:28–35. doi: 10.1080/07853890310004101. [DOI] [PubMed] [Google Scholar]
- 17.Piatigorsky J. Gene sharing and evolution: the diversity of protein functions. Cambridge, MA: Harvard University Press; 2007. [Google Scholar]
- 18.Wright S. Genetics of abnormal growth in the guinea pig Cold Spring Harbor Symposium. Quant Biol. 1934;2:137–147. [Google Scholar]
- 19.Wright S. The first Meckel oration: on the causes of morphological differences in a population of guinea pigs. Am J Med Genet. 1984;18:591–616. doi: 10.1002/ajmg.1320180408. [DOI] [PubMed] [Google Scholar]
- 20.Johannsen W. The genotype conception of heredity. Am Naturalist. 1911;45:129–159. doi: 10.1093/ije/dyu063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths PE, Stotz K. Genes in the postgenomic era. Theor Med Bioeth. 2006;27:499–521. doi: 10.1007/s11017-006-9020-y. [DOI] [PubMed] [Google Scholar]
- 22.Kay LE. A history of the genetic code. Stanford, Calif: Stanford University Press; 2000. Who wrote the book of life? [Google Scholar]
- 23.Keller EF. The century of the gene. Cambridge, MA: Harvard University Press; 2000. [Google Scholar]
- 24.Moss L NetLibrary Inc. What genes can’t do. Cambridge, MA: MIT Press; 2003. [Google Scholar]
- 25.Stotz K, Griffiths P. Genes: philosophical analyses put to the test. Hist Philos Life Sci. 2004;26:5–28. doi: 10.1080/03919710412331341621. [DOI] [PubMed] [Google Scholar]
- 26.Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, et al. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17:669–681. doi: 10.1101/gr.6339607. [DOI] [PubMed] [Google Scholar]
- 27.Johannsen W. The elements of heredity (Elemente der exakten Erblich-keitslehre) Jena, Sweden: Gustav Fischer; 1909. [Google Scholar]
- 28.Morgan TH. The theory of the gene. Am Naturalist. 1917;51:513–544. [Google Scholar]
- 29.Muller H. The gene as the basis of life. Proc Int Congr Plant Sci. 1929;1:897–921. [Google Scholar]
- 30.Beadle GW, Tatum EL. Genetic control of biochemical reactions in Neurospora. Proc Natl Acad Sci USA. 1941;27:499–506. doi: 10.1073/pnas.27.11.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czech B, Malone CD, Zhou R, Stark A, Schlingeheyde C, et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banerjee D, Slack F. Control of developmental timing by small temporal RNAs: a paradigm for RNA-mediated regulation of gene expression. Bioessays. 2002;24:119–129. doi: 10.1002/bies.10046. [DOI] [PubMed] [Google Scholar]
- 34.Chang TC, Mendell JT. MicroRNAs in vertebrate physiology and human disease. Annu Rev Genomics Hum Genet. 2007;8:215–239. doi: 10.1146/annurev.genom.8.080706.092351. [DOI] [PubMed] [Google Scholar]
- 35.Johnson JM, Edwards S, Shoemaker D, Schadt EE. Dark matter in the genome: evidence of widespread transcription detected by microarray tiling experiments. Trends Genet. 2005;21:93–102. doi: 10.1016/j.tig.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 36.Amaral PP, Dinger ME, Mercer TR, Mattick JS. The eukaryotic genome as an RNA machine. Science. 2008;319:1787–1789. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- 37.Pheasant M, Mattick JS. Raising the estimate of functional human sequences. Genome Res. 2007;17:1245–1253. doi: 10.1101/gr.6406307. [DOI] [PubMed] [Google Scholar]
- 38.Isalan M, Lemerle C, Michalodimitrakis K, Horn C, Beltrao P, et al. Evolvability and hierarchy in rewired bacterial gene networks. Nature. 2008;452:840–845. doi: 10.1038/nature06847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett MR, Hasty J. Systems biology: genome rewired. Nature. 2008;452:824–825. doi: 10.1038/452824a. [DOI] [PubMed] [Google Scholar]
- 40.Fisher R. The correlations between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinburgh. 1918;52:399–433. [Google Scholar]
- 41.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 42.Weiss KM. Tilting at quixotic trait loci (QTLs): an evolutionary perspective on genetic association studies. Genetics. 2008;179:1741–1756. doi: 10.1534/genetics.108.094128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buchanan AV, Weiss KM, Fullerton SM. Dissecting complex disease: the quest for the Philosopher’s Stone? Int J Epidemiol. 2006;35:562–571. doi: 10.1093/ije/dyl001. [DOI] [PubMed] [Google Scholar]
- 44.Terwilliger JD, Weiss KM. Confounding, ascertainment bias, and the blind quest for a genetic ‘fountain of youth’. Ann Med. 2003;35:532–544. doi: 10.1080/07853890310015181. [DOI] [PubMed] [Google Scholar]
- 45.Ioannides J. Genetic associations: false or true? Trends Mol Med. 2003;9:135–138. doi: 10.1016/s1471-4914(03)00030-3. [DOI] [PubMed] [Google Scholar]
- 46.Weiss KM, Clark AG. Linkage disequilibrium and the mapping of complex human traits. Trends Genet. 2002;18:19–24. doi: 10.1016/s0168-9525(01)02550-1. [DOI] [PubMed] [Google Scholar]
- 47.Cohen WI. Health care guidelines for individuals with Down syndrome: revision. Down Syndr Q. 1999;4:1–16. [Google Scholar]
- 48.Roper RJ, Reeves RH. Understanding the basis for Down syndrome phenotypes. PLoS Genet. 2006;2:e50. doi: 10.1371/journal.pgen.0020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reeves RH, Baxter LL, Richtsmeier JT. Too much of a good thing: mechanisms of gene action in Down syndrome. Trends Genet. 2001;17:83–88. doi: 10.1016/s0168-9525(00)02172-7. [DOI] [PubMed] [Google Scholar]
- 50.Olson LE, Richtsmeier JT, Leszl J, Reeves RH. A chromosome 21 critical region does not cause specific Down syndrome phenotypes. Science. 2004;306:687–690. doi: 10.1126/science.1098992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ait Yahya-Graison E, Aubert J, Dauphinot L, Rivals I, Prieur M, et al. Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am J Hum Genet. 2007;81:475–491. doi: 10.1086/520000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simon H. The architecture of complexity. Proc Am Philos Soc. 1962;106:467–482. [Google Scholar]
- 53.Visel A, Carson J, Oldekamp J, Warnecke M, Jakubcakova V, et al. Regulatory pathway analysis by high-throughput in situ hybridization. PLoS Genet. 2007;3:1867–1883. doi: 10.1371/journal.pgen.0030178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waddington CH. The strategy of the genes. London: George Allwin and Unwin Ltd; 1957. [Google Scholar]
- 55.Chen Y, Zhu J, Lum PY, Yang X, Pinto S, et al. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452:429–435. doi: 10.1038/nature06757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hunter DJ, Khoury MJ, Drazen JM. Letting the genome out of the bottle–will we get our wish? N Engl J Med. 2008;358:105–107. doi: 10.1056/NEJMp0708162. [DOI] [PubMed] [Google Scholar]
- 57.Keller MP, Choi Y, Wang P, Belt Davis D, Rabaglia ME, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18:706–716. doi: 10.1101/gr.074914.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glazier AM, Nadeau JH, Aitman TJ. Finding genes that underlie complex traits. Science. 2002;298:2345–2349. doi: 10.1126/science.1076641. [DOI] [PubMed] [Google Scholar]
- 59.Goldstein DB. Genomics and biology come together to fight HIV. PLoS Biol. 2008;6:e76. doi: 10.1371/journal.pbio.0060076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vesalius A. De Humani corporis fabrica liborum, epitome. Basel: Oporinus; 1543. (Copied from Dover reprint, edition) [Google Scholar]