

Abstract
Muscle regeneration provides a paradigm by which to study how extrinsic signals coordinate gene expression in somatic stem cells (satellite cells) by directing the genome distribution of chromatin-modifying complexes. Understanding the signal-dependent control of the epigenetic events underlying the transition of muscle stem cells through sequential regeneration stages holds the promise to reveal new targets for selective interventions toward repairing diseased muscles. This review describes the latest findings on how regeneration cues are integrated at the chromatin level to build the transcription network that regulates progression of endogenous muscle progenitors throughout the myogenic program. In particular, we describe how specific epigenetic signatures can confer responsiveness to extrinsic cues on discrete regions of the muscle stem cell genome.
Self-renewal in adult organisms and genome reprogramming in ‘muscle stem cells’ by regeneration cues
Tissue and organ progenitors are referred to as adult ‘somatic stem cells’ (SSCs) because of their functional similarities to embryonic stem cells (ESCs), including the capacity for long-term self-renewal and the potential to commit to multiple lineages. However, key differences exist between ESCs, which are totipotent and can adopt virtually all lineages, and SSCs, which are located within differentiated tissues and organs, have restricted ‘potency’, and provide an immediate reservoir of tissue-specific progenitors [1].
Satellite cells are adult muscle stem cells (MSCs) that regenerate diseased or injured skeletal muscles and support the self-renewal of myofibers during post-natal life [2]. Recent studies have demonstrated a heterogeneous composition within the satellite cell population [3–5]. Moreover, additional populations of putative MSCs distinct from satellite cells have been described [6,7]. Because all MSCs are exposed to the same cues released in the regenerative environment [8], a key issue in regenerative medicine is to understand how intracellular cascades convert regeneration cues into the information that coordinates gene expression in different sub-populations of MSCs.
The interplay between extrinsic signals and the epigenetic profile of MSCs is an area of particular interest. The regeneration signals are broadcast at the chromatin level to generate specific epigenetic signatures which, in turn, contribute to confer responsiveness to regeneration cues on discrete loci. The reciprocal regulation between extracellular signal-activated pathways and the epigenetic events that control gene transcription in MSCs are the central themes of this review.
Reprogramming of the genome in satellite cells
Upon changes in environmental cues, stem cells extensively reprogram their genome toward a specific pattern of gene expression that establishes the tissue-specific phenotype. This process is achieved epigenetically, via the genome re-distribution of chromatin-modifying enzymes, in response to intracellular cascades activated by the extrinsic cues [9,10]. The epigenetic profile of MSCs consists of a variety of chromatin modifications that are transmitted along the transition through sequential stages of the regeneration program and establish the ‘memory’ of an active or repressive gene state. These modifications contribute to reprogram the MSC genome toward a differentiated phenotype. The recruitment of chromatin-modifying enzymes and the consequent deposition of epigenetic marks at specific loci are directed by the interplay between sequence-specific transcription factors, chromatin-associated kinases activated in response to extracellular signals, and interactions with modified histones [10].
During skeletal myogenesis, the nuclei of MSCs are sequentially reprogrammed to first adopt and then maintain the myogenic identity (proliferation stage), and eventually to differentiate into myotubes. Thus, the transition from muscle progenitors to terminally differentiated muscles entails coordinated repression and activation of specific subsets of genes. The skeletal muscle lineage is determined by the expression of transcription factors that establish myogenic identity (e.g. the paired-homeobox Pax3 and Pax7, and the muscle-specific basic helix–loop–helix [bHLH], MyoD and Myf5) and by the repression of genes associated with the acquisition of non-muscle lineages – a process termed lineage-commitment [11]. Of note, the asymmetric division of satellite cells [12,13] poses the interesting issue of how the epigenetic information is segregated into two daughter cells committed to distinct fates. One cell returns to quiescence and replenishes the pool of reserve satellite cells. Another cell enters the differentiation program. Thus, asymmetric division regulates the proportion of cells that repopulate injured muscles while maintaining the integrity of satellite cell potential to sustain repeated cycles of regeneration. Recent studies have identified molecular markers that correlate with distinct fates of MSC progenies generated by asymmetric division. Pax7 expression co-segregates with the fraction of satellite cells that do not enter the differentiation program, whereas MyoD and Myf5 are expressed in differentiation-committed MSCs [5,14]. Pax7 is the upstream activator of MyoD and Myf5 [15,16], and co-expression of Pax7 and MyoD is transiently detected in activated, proliferating satellite cells at an intermediate regenerative stage [17]. Downregulation of Pax7 coincides with the ability of MyoD and Myf5 to induce the transcription of downstream genes and promote terminal differentiation [18]. This temporal and functional hierarchy between lineage determination and differentiation genes (Box 1) suggests that strategies targeting the molecular events linking Pax7, MyoD, Myf5 and downstream muscle genes are predicted to generate ‘intermediate’ cellular phenotypes. Pharmacological generation of these phenotypes could be used in regenerative medicine. The outcomes of such strategies include the expansion of endogenous MSCs available for long-term regeneration that might counter satellite cell exhaustion and implement the efficiency of myofiber regeneration. Likewise, this knowledge could be applied to devise strategies that optimize an in vitro expansion of MSCs before their transplantation.
Box 1. Muscle cell lineage and muscle-gene transcriptional control.
The skeletal muscle lineage is specified by the expression of Pax7, Pax3, MyoD and Myf5. Pax7 is expressed in quiescent satellite cells and persists during the first stages of regeneration, when it promotes proliferation and survival and induces the expression of MyoD and Myf5. Because of the asymmetric division of satellite cells, Pax7 is likely to drive two distinct transcription networks that determine the fate of satellite cell progeny. Pax7-mediated activation of MyoD and Myf5 [15,16] specifies the population of MSCs that enter the differentiation program [5]. By contrast, in the fraction of MSCs that return to quiescence, MyoD and Myf5 loci seem refractory to Pax7-mediated activation. The different response of these loci to Pax7 might depend on the epigenetic memory, which is determined by the presence of different types of histones. On the MyoD promoter, the presence of histone H3.3 establishes the epigenetic memory conducive for transcription in differentiation-committed MSCs [74]. By contrast, the presence of the H1b isoform bound to the homeoprotein Msx1 can induce repressive chromatin on the regulatory elements of MyoD in MSCs that re-enter quiescence [75].
The bHLH muscle-specific transcriptional activators – MyoD, Myf5, myogenin and MRF4 – initiate and perpetuate the differentiation program in collaboration with the ubiquitously expressed E2A gene products (E12, E47 and HEB) and MEF2 proteins [22,23]. An additional level of control is provided by transcriptional co-activators and co-repressors, which modify the chromatin structure by catalysing post-transcriptional modifications of histone tails, including acetylation, methylation, phosphorylation and ubiquitylation. The dynamic exchange of these transcriptional co-regulators imparts to the chromatin the epigenetic profile that is either repressive (heterochromatin) or permissive (euchromatin) for gene expression. In undifferentiated myoblasts, the unscheduled activation of the differentiation program is precluded by the presence of histone deacetylases (HDACs), which prevent local hyperacetylation (Figure 1a). Class I HDACs preferentially associate with MyoD, whereas class II HDACs are dedicated repressors of MEF2 [11,27]. At least two additional events are involved in the formation of the heterochromatin on promoters of muscle genes in myoblasts: (i) Suv39h1-mediated dimethylation of H3–K9, which mediates the interaction with the chromodomain-containing heterochromatin protein 1 (HP1) [28]; and (ii) Polycomb-mediated trimethylation of H3–K27 [55] (Figure 1a). These epigenetic marks are erased by differentiation-induced events that are still unknown but that presumably involve specific demethylases and histone exchange. At the onset of differentiation, the chromatin recruitment of the acetyltransferases p300/CBP, PCAF, the arginine-methyltransferases CARM1 and PRMT5, the ATPase-dependent SWI/SNF chromatin-remodeling complexes and the MLL/TrxG-associated lysine methyl-transferases [9–11,23] endows the myogenic transcriptosome with the enzymatic activities necessary to initiate the transcription of target genes (Figure 1b). The double PHD finger protein DPF3 associates with the SWI/SNF complex and binds methylated and acetylated lysines of histones with a proposed function for the methylation ‘reader’ in remodeling chromatin on promoters of muscle genes [76]. Recent work illustrated a switching of the core promoter recognition complex that confers specificity on muscle gene transcription. The prototypic core promoter recognition complex, TFIID, which initiates the pre-initiation complex (PIC) on target sequences, is present in myoblasts but is replaced by the TAF3–TRF3 complex in myotubes [77].
An essential prerequisite to pursue such an exciting opportunity relates to our ability to fill the gap in our knowledge that currently exists between the biological information on MSCs and the epigenetic regulation of gene expression in these cells.
Transcriptional networks that orchestrate adult skeletal myogenesis
Genome-wide approaches indicate that the progression of muscle progenitors through sequential stages of skeletal myogenesis is underpinned by epigenetic changes that permit a coordinated expression of specific subsets of genes [19,20]. A logical extension of these data predicts that distinct epigenetic signatures can discriminate discrete phenotypic stages during muscle regeneration.
Muscle-specific transcription factors and chromatin-modifying enzymes form the transcriptional network that establishes a feed-forward circuit, which drives the genome reprogramming toward terminal differentiation [21–23].
When ectopically introduced into several somatic cells (such as fibroblasts, keratinocytes and other non-muscle cell types), MyoD reprograms the genome toward the expression of muscle-specific genes, a process referred to as ‘myogenic conversion’ [24]. The potential of MyoD to induce the myogenic lineage depends on its ability to penetrate and remodel the chromatin at previously silent loci [25] and to activate the transcription of muscle genes [26]. This ability is conferred by the heterodimerization with E2A gene products, by functional interaction with myocyte enhancer binding factor 2 (MEF2) proteins and by the recruitment of chromatin-modifying enzymes, such as acetyltransferases, methyltransferases and complexes implicated in chromatin modifications [9,22,23]. Displacement of transcriptional co-repressors of muscle bHLH and MEF2 proteins is also necessary to activate muscle-gene transcription [27,28] (Figure 1 and Box 1).
Figure 1.
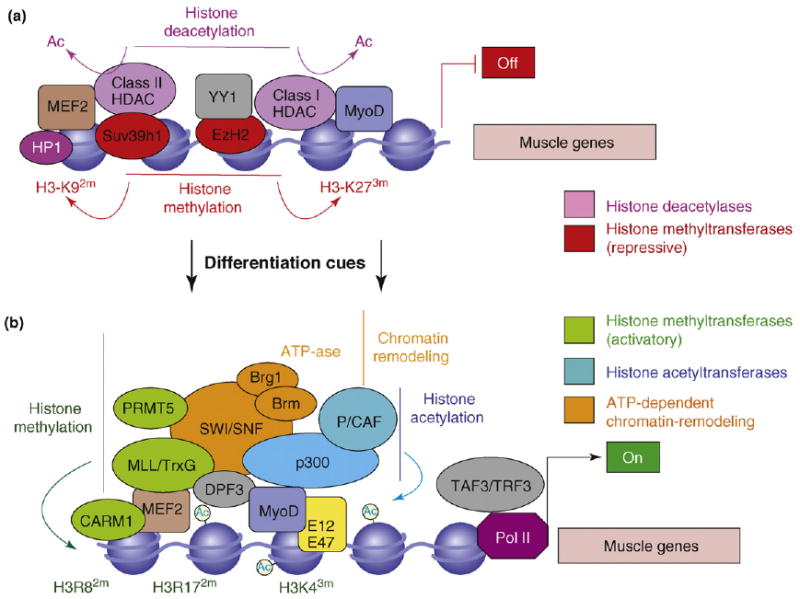
Chromatin re-configuration at muscle loci during myogenic differentiaiton. (a) In myoblasts, the regulatory regions of muscle genes are occupied by CMCs, such as class I and II histone deacetylases (HDACs; pink), the Polycomb repressive complex (PRC)-associated H3–K27 methyltransferase EzH2, and the H3–K9 methyltransferase Suv39h1 (red). These enzymes catalyse histone modifications (deacetylation, H3–K9 dimethylation and H3–K27 trimethylation) that establish a chromatin conformation repressive for transcription (heterochromatin). Transient interactions of MyoD homodimers and MEF2 proteins presumably direct these enzymes to the chromatin on the regulatory regions of muscle genes. (b) The expression of muscle genes is induced by differentiation cues via chromatin reconfiguration into a conformation permissive for transcription. This process entails the sequential erasure of the chromatin repressive marks (by HDAC displacement, histone demethylases and histone exchange mechanism), which are replaced by modifications that correlate with gene expression (H3–K4 trimethylation, H3–R8 dimethylation and H3–17 dimethylation; green). These modifications are catalysed by the interdependent activity of a variety of chromatin-modifying enzymes recruited by muscle bHLH–E12/47 heterodimers and by MEF2 proteins. Chromatin-modifying enzymes detected in the muscle transcriptosome include the acetyltransferases p300 and PCAF (blue and turquoise), the histone methyltransferases PRMT5 and CARM1, MLL (green), and the SWI/SNF chromatin remodeling complex (orange). The initiation of transcription at muscle loci is also promoted by the recruitment of TATA-associated factor 3 (TAF3) and TBP-related factors 3 (TRF3), the levels of which increase in differentiating muscle cells.
The sequential interactions between muscle-specific bHLH and MEF2 factors and co-repressors or co-activators have been determined in vitro using established muscle cell lines, which are typically synchronized by changing the culture conditions. This experimental setting creates an artificial boundary between proliferation and differentiation that might not entirely reflect the in vivo conditions. Consequently, it is difficult to transfer the information obtained from genome-wide studies performed in cultured myoblasts, which fairly recapitulate myogenic differentiation, to the heterogeneous population of MSCs that participate in muscle regeneration.
Signal-dependent assembly of the muscle transcriptosome
Changes in chromatin structure and gene transcription in tissue progenitors are typically induced by external cues, such as those released in the stem cell microenvironment – the stem cell niche [29]. Likewise, muscle regeneration is accompanied by the local release of cues that govern satellite cell transition from quiescence to terminal differentiation [11,30].
The complex role of environmental signals in the regulation of gene expression during myogenesis is well illustrated by the impact of inflammatory cues, which can either block or promote the myogenic program. An example of such a biological complexity is provided by the effect of tumor necrosis factor α (TNFα). Circulating levels of TNFα increase in conditions associated with muscle wasting, such as chronic diseases, cancer cachexia and aging [31]. In these conditions, TNFα inhibits skeletal myogenesis via activation of downstream nuclear factor (NF)-κB and Jun N-terminal kinase (JNK) signaling, as reported by in vitro studies [32,33]. Conversely, the TNFα that is released within the regenerative environment promotes muscle differentiation, by activating the p38 pathway in cooperation with other stimuli [34] (Figure 2). The final effect of inflammation on skeletal myogenesis also depends on the combination of pathways activated in response to other locally released substances, such as insulin growth factor 1 (IGF1), hepatocyte growth factor (HGF) and transforming growth factor β (TGFβ) family proteins [34].
Figure 2.
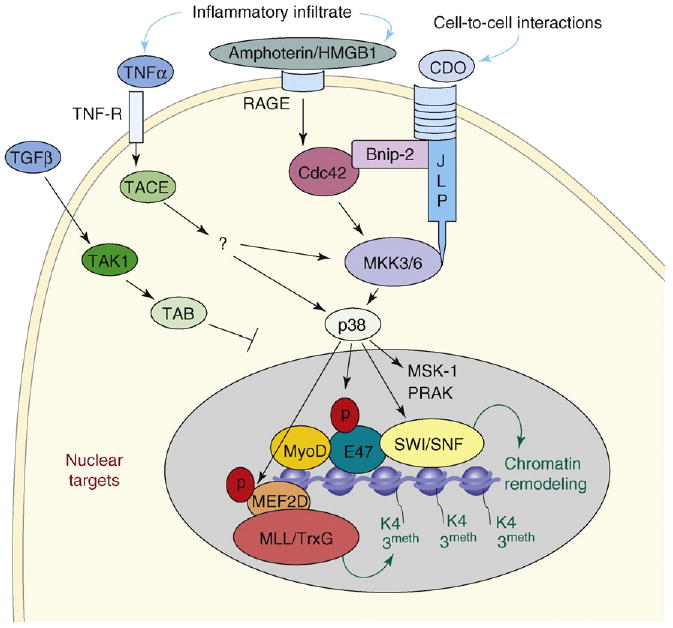
p38 signaling to the chromatin in muscle progenitor cells. Extracellular cues released in the regenerative environment, such as TNFα, Amphoterin or HMGB1, and the engagement of the cell-surface receptor CDO [51], are transduced in MSCs by combinations of intracellular signaling that culminate with the activation of p38 kinases [73]. It seems that p38α has a major role in the control of muscle-gene transcription [49]. Important determinants of p38 activity include the combination of external cues, the magnitude of activation, the intracellular distribution of p38 isoforms and the substrate availability. The nuclear translocation and chromatin localization of p38 is controlled by the TGF-β signaling via transforming-growth-factor-β-activated protein kinase (TAK), the transforming-growth-factor-β-activated 7 protein-kinase-1-binding protein (TAB) and possibly by other intracellular cascades. Potential effectors of the p38 pathway are downstream kinases, such as mitogen and stress activated protein kinase-1 (MSK-1) and p38-regulated/activated protein kinase (PRAK). In differentiating muscle progenitors, p38 kinases (and possibly other downstream kinases) are detected on the chromatin of target genes, where they control key events leading to the assembly of productive muscle transcriptosome [36]. In particular, the simultaneous recruitment of the Ash2L-containing methyltransferase complex (MLL/TrxG) via p38-mediated phosphorylation of MEF2D [53], and the SWI/SNF CMCs via p38-mediated phosphorylation of BAF60 [36] promote local H3–K4 trimethylation and chromatin remodeling – two crucial events for activation of transcription. It is possible that the formation of the MyoD–E47 homodimer – which is stimulated by p38-depndent phosphorylation of E47 [50] – contributes to the recruitment of MLL/TrxG and SWI/SNF to muscle-gene promoters.
Chromatin-associated kinases
How are signal-activated intracellular cascades integrated at the chromatin level and converted into epigenetic modifications? Direct phosphorylation of sequence-specific transcription factors is one known regulatory mechanism of gene transcription. The discovery that extracellular signal-activated kinases can also phosphorylate histones and chromatin-associated proteins [35] has established a direct biochemical link between intracellular pathways and epigenetic modifications. Chromatin-immunoprecipitation (ChIP) studies have permitted the detection of kinases on the chromatin of target genes [36–39]. These chromatin-associated kinases are predicted to regulate the enzymatic activity of chromatin-modifying complexes (CMCs) via direct phosphorylation of some of their individual components [40,41].
The p38 signaling is triggered in satellite cells by regeneration cues (Figure 2). The active p38α kinase can be detected in activated satellite cells [42] and has an essential role in muscle regeneration [43]. ChIP analyses showed that p38α was associated with the chromatin at regulatory elements of muscle genes in coincidence with the recruitment of the SWI/SNF complex [36]. Likewise, in yeast, the p38 functional homolog, Hog1 kinase, regulates the response to osmotic stress by favoring the recruitment of SWI/SNF, RNA polymerase II and other components of the general transcription machinery to target promoters [44–46].
Although the precise mechanism accounting for the kinase-dependent regulation of chromatin conformation is currently unknown, some potential targets of chromatin-associated kinases have been identified in muscle progenitors. p38 kinases α and β phosphorylate the BRG1- or BRM-associated factor 60c (BAF60) subunits of the SWI/SNF complex in vitro [36]. BAF60-a, -b and -c are structurally related variants that mediate protein–protein interactions between SWI/SNF and sequence-specific transcriptional activators [47]. BAF60c (encoded by Smarcd3 gene) is induced in the developing heart and somites during early mouse embryogenesis, and BAF60c knockout embryos show impaired cardiac and skeletal myogenesis [48], indicating an essential role for BAF60c in myogenic commitment. Future studies should define the impact of p38-mediated phosphorylation of BAF60 subunits on SWI/SNF recruitment to muscle-gene promoters.
Other direct targets of p38 α and β kinases are the HLH E2A gene product, E47, and MEF2 proteins [49]. p38-mediated phosphorylation of E47 favors the heterodimerization between E47 and muscle bHLH proteins [50]. CDO (for CAM-related or -downregulated by oncogenes) is a member of the Ig–fibronectin type III repeat subfamily of transmembrane proteins. Activation of p38 by CDO (for CAM-related or -downregulated by oncogenes) – a member of the Ig–fibronectin type III repeat subfamily of transmembrane proteins (Figure 2) – leads to the formation of MyoD–E47 heterodimers, which productively bind the E-box sequences on the regulatory regions of muscle genes [51,52]. It would be interesting to establish whether or not p38-mediated formation of MyoD–E47 heterodimers and SWI/SNF recruitment on MyoD-responsive promoters are two related events. MEF2 phosphorylation by p38 kinases was described by many groups [49]. However, only recently this event has been linked to specific epigenetic modifications. Studies from the Dilworth laboratory showed that phosphorylation of MEF2D by p38 α and β kinases mediates the recruitment of the Ash2L-containing mixed-lineage leukemia (MLL) methyltransferase complex (functionally related to Trithorax group [TrxG]) to the chromatin of muscle genes, thereby promoting H3–K4 trimethylation (H3K4me3), an epigenetic event that promotes transcription [53]. Interestingly, the catalytic subunits of SWI/SNF and TrxG are functionally linked [54]. These data suggest a model of p38-activated chromatin signaling to broadcast functionally related chromatin-remodeling complexes (SWI/SNF and MLL/TrxG) via distinct biochemical events (e.g. direct phosphorylation of BAF60 and MEF2D).
Signaling convergence to the chromatin on muscle-gene regulatory sequences
The finding that a p38 blockade in myoblasts still enables the recruitment of muscle transcription factors and acetyltransferases, and the consequent hyperacetylation on muscle-gene promoters [36,39], suggests that a pathway distinct from p38 signaling regulates acetyltransferase recruitment.
Recent studies have revealed that p38 and IGF1– protein kinase B(AKT) pathways are two parallel cascades that converge to the chromatin on regulatory elements of muscle genes [39]. IGF1-activated AKT1 and AKT2 kinases phosphorylate the C-terminal region of the acetyltransferase p300, thereby promoting the association with MyoD. The consequent local hyperacetylation at muscle loci enables the chromatin-remodeling activity of the p38-recruited SWI/SNF complex [39]. These data reveal the functional interdependence between the p38 pathway and IGF1 signaling during muscle differentiation.
Chromatin re-configuration in differentiation-committed myoblasts
Recruitment of transcriptional co-activators to muscle genes must be preceded by or take place simultaneously to the displacement of co-repressor enzymes and the deletion of pre-existing epigenetic modifications generated by these enzymes (Figure 1 and Box 1). However, the signaling that resets the chromatin of myoblasts toward the formation of a productive transcriptosome is partially known. The interplay between histone deacetylases (HDACs) and repressive histone methyltransferases contributes to the formation of the heterochromatin at repressed loci. In undifferentiated myoblasts, HDACs prevent the acetylation of H3–K9 on promoters of muscle genes and this condition facilitates H3–K92me and the formation of heterochromatin. Displacement of class II HDACs by calcium/calmodulin-dependent kinase (CaMK) and of class I HDACs by cell-cycle-related events are necessary for local hyperacetylation [27].
A recent report contributed to extend our knowledge of the mechanism that regulates muscle-gene repression in myogenic precursors [55]. In myoblasts, the chromatin at muscle genes adopts a conformation repressive for transcription, owing to the recruitment of the Polycomb group (PcG) methyltransferase Ezh2, which silences transcription by catalysing trimethylation of H3 Lys27 (H3K273me). Ezh2 is recruited to the muscle regulatory regions via interaction with yin yang 1 (YY1). Further association with HDAC1 forms a repressive complex. At the onset of differentiation, Ezh2 and HDAC1 proteins are downregulated and YY1 is replaced with serum response factor (SRF), thus enabling the binding of MyoD and the recruitment of the positive co-activators, to form a transcription-competent complex [55]. Future studies should elucidate the signaling that controls PcG function in MSCs.
The involvement of histone methylation in the dynamic regulation of muscle-gene expression implicates a role for histone demethylases in the control of myogenesis. Proteins containing the Jumonji (Jmj) domain catalyse histone demethylation and are involved in the switching of the epigenetic profile of discrete genes in response to environmental changes [56–59]. This regulation includes the removal of both epigenetic marks of repression (H3–K273me) and activation (H3–K43me), which are typically located at transcription start sites. Recent studies have revealed the existence of a cross-talk between histone methyltransferases and demethylases. H3–K27 demethylases associate with the TrxG to generate the epigenetic profile of active promoters (H3–K43me) [59], and H3–K4 demethylases, such as RBP2, associate with PcG to promote selective H3-K273me enrichment on repressed promoters [60]. Thus, coordinate activity of PcG and TrxG methyltransferases and Jmj demethylases are predicted to generate the epigenetic profile that restricts the expression of target genes to specific stages of cellular differentiation. Importantly, coexistence of H3–K43me and H3–K273me is found on silent promoters in stem cells and is considered to be a ‘bivalent mark’ that maintains these promoters in a ‘poised’ state accessible for the activation of lineage-specific genes [61,62]. When extended to muscle regeneration, this knowledge leads to the speculation that, in endogenous MSCs, the bivalent status defines the promoters of signal-responsive genes. This concept could provide the epigenetic basis for the selective gene responsiveness to regeneration cues in MSCs and might inspire the design of epigenetic drugs that manipulate the expression of specific genes in MSCs.
Processivity, redundancy, cooperation and variability: shaping epigenetic signatures
One conceptual challenge in the field of skeletal myogenesis concerns the mechanism that ensures the temporal sequence of expression of muscle genes (e.g. early and late genes) in response to one initial event. This concept applies well to MyoD-dependent myogenic conversion [24]. Approximately 10 years ago, the prevailing model was that MyoD–E12/47 heterodimers could directly activate the transcription of all muscle genes containing E-box sequences on the regulatory sequences. This model has recently been revised, based upon discoveries that revealed the complexity of the epigenetic regulation of muscle-gene transcription. A seminal finding was the demonstration of the existence of discrete sub-programs that coordinate the temporal expression of muscle genes in fibroblasts converted by MyoD [26]. An integrated analysis of genome-wide ChIP and expression profiling in muscle cells revealed a stepwise progression of skeletal myogenesis through a variety of events that amplify the initial induction by preexisting bHLH factors, MyoD and Myf5 [25]. Several favorable conditions must enable transcriptional activation by MyoD and Myf5, which are otherwise inactive in myoblasts. These conditions include interactions with transcription factors that bind DNA sequences in proximity to E-box motifs, within the promoters of early muscle genes. For example, MyoD interacts with an adjacent protein complex containing the homeodomain protein Pbx/Meis, which is constitutively bound at the chromatin of the myogenin promoter [63,64]. These interactions might facilitate the binding of MyoD to non-canonical E-box sequences and ‘poise’ the chromatin of target genes for rapid induction upon differentiation cues. MyoD acetylation [65] is another important regulatory signal for the temporal pattern of gene expression in cultured myoblasts [66] and during muscle regeneration [67]. Other proteins that collaborate with MyoD in the activation of early muscle-gene transcription are MEF2 and Six1 [68], which are directly induced by MyoD and can activate other muscle genes through cooperative interactions.
Activation of myogenin is an early crucial event in the differentiation program because the absence of myogenin precludes muscle development [69]. Thus, MyoD-mediated activation of myogenin illustrates an example of ‘processive’ activation within muscle bHLH proteins that propagates the myogenic program. Despite the structural and functional similarities among muscle bHLH proteins, the degree of ‘redundancy’ with regard to their ability to activate target genes is still not completely understood. Recent studies showed that MyoD and myogenin occupy the chromatin of overlapping genes, but have distinct functions [20]. Chromatin recruitment of MyoD at late genes initiates local epigenetic modifications that facilitate the recruitment of myogenin, which in turn is required for transcriptional activation of these genes in collaboration with MEF2D [70], an example of co-operation between transcriptional activators. Additional targets of MyoD seem to contribute to the activation of late muscle genes [19,20]. This implies that MyoD downstream genes confer on MyoD the competence to induce the transcription of genes that would otherwise be refractory to activation. One candidate mechanism by which MyoD downstream genes participate in the activation of muscle genes entails the recruitment of transcriptional co-activators, either via direct interaction with bHLH and/or MEF2 proteins or by the occupancy of adjacent chromatin domains. Thus, newly synthesized proteins might generate the ‘epigenetic variability’ that permits discrimination of different muscle genes, despite the redundant presence of E-box and MEF2 sites on their promoters. This model postulates that additional transcription factors enable bHLH and MEF2 factors to activate target genes by promoter recruitment of CMCs with complementary enzymatic activities that impart the epigenetic modifications conducive for transcription. Examples of different modalities of muscle-gene activation are illustrated in Figure 3.
Figure 3.
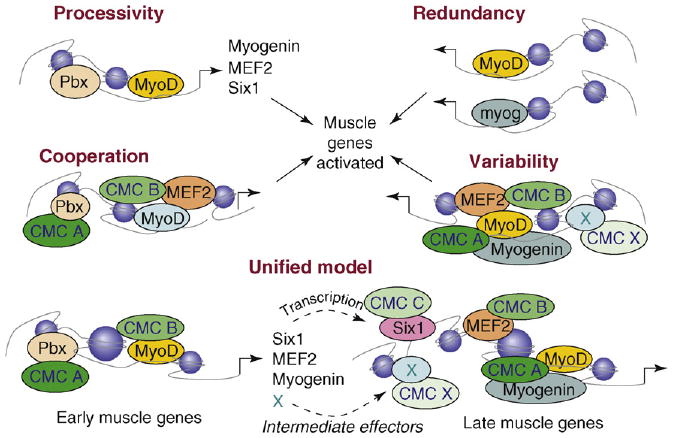
Mechanisms of activation of muscle-gene transcription. Muscle-gene transcription can be activated by different mechanisms. Cross-activation between muscle bHLH proteins, such as MyoD and myogenin, is an example of processivity by which myoblasts expand the number of master regulators of differentiation. They can, in turn, activate the same target genes because of their structural and functional redundancy or synergize to activate late genes in a model of cooperation that might also require other direct targets of MyoD, such as MEF2 and Six1. Genome-wide studies indicate that different subsets of genes are expressed according to a temporal regulation owing to the intervention of additional factors that are often induced by muscle regulatory factors at earlier stages. Although the identity of these factors remains to be established (indicated by X), they can provide the variability that permits the activation of promoters containing similar motifs (Ebox and MEF2) at different times. A key element in the cooperation model is provided by the recruitment of different CMCs, possibly with complementary enzymatic activities. In this regard, the recruitment of additional transcription factors in the vicinity of Ebox and MEFs sites offers an obvious advantage. These different models of transcriptional activation are not mutually exclusive and can all fit into one unified general model that enables coordination of the temporal pattern of gene expression.
An additional level of control is provided by intracellular pathways, which are elicited in response to MyoD activation. MyoD-dependent expression of surface receptors, signaling components (kinases or phosphatases) and scaffold proteins can re-shape the intracellular signaling network to prepare stage-specific signal responsiveness. The same mechanism can confer on myoblasts an autonomous control of the myogenic program. One notable example is provided by the persistent induction of the p38 pathway by MyoD [21,71], which extends the initial activation of p38 kinases by regeneration cues to late stages of skeletal myogenesis. It is likely that the interplay between signaling pathways and epigenetic changes induced during myogenic differentiation selects the repertoire of active loci in the genome of MSCs.
Concluding remarks
The application of genome-wide technologies to complex systems such as muscle regeneration promises to elucidate the regulatory mechanism underlying signal-dependent distribution of epigenetic marks in the genome of muscle progenitors. This technology has revealed the existence of particular epigenetic modifications that regulate gene expression in stem cells [61,62] and might dictate the domains of the MSC genome that respond to extrinsic signals. In this respect, the elucidation of the functional inter-play between histone methyltransferases and demethylases on promoters of genes such as Pax7, MyoD and Myf5, which regulate MSC proliferation and differentiation, seems to be of key importance (Box 2). Likewise, modulation of Pax7-mediated activation of downstream genes can be used to control muscle regeneration and, in particular, the proportion of cells committed to differentiation versus those that replenish the pool of quiescent, reserve MSCs.
Box 2. Chromatin targets of epigenetic drugs for potential therapeutic interventions.
The elucidation of the epigenetic events that regulate gene transcription in MSCs might reveal the target for pharmacological manipulation of muscle regeneration. In this regard, several speculations can be formulated on the potential application of drugs that interfere with key events in the control of gene transcription at different stages of MSC progression to differentiated muscles. Pax7 occupies a nodal position in MSC biology because its expression specifies the satellite cell identity [78]. Thus, pharmacological induction of endogenous Pax7 has the potential to convert ESCs or SSCs into satellite cells. Understanding the mechanism that restricts Pax7 expression to satellite cells will help to devise such a pharmacological approach. Likewise, molecular insight into the control of Pax7 expression in satellite cells is awaited to devise strategies that control the proportion of cells that regenerate muscles or restore the reserve pool.
Recent studies shed light on the mechanism by which Pax7 activates downstream genes in satellite cells. Pax7-mediated activation of Myf5 is mediated by the association with the Wdr5– Ash2L–MLL2 histone methyltransferase complex (TrxG), which directs H3–K43me on the chromatin of Myf5 locus [15]. Activation of MyoD in satellite cells requires cooperation between Pax7 and FoxO3 [16]. The transcriptional activation of Myf5 and MyoD commits satellite cells to the differentiation program and is therefore a candidate target for interventions that implement the ability of satellite cells to regenerate muscles.
Current knowledge of the molecular mechanism that regulates muscle-gene transcription has already inspired pharmacological interventions to boost muscle regeneration. Histone acetyltransferases and deacetylases regulate the acetylation status of target genes and are a target of epigenetic drugs. Inhibition of histone deacetylase in MSCs by drugs currently used in clinical practice (deacetylase inhibitors) implements muscle regeneration and counters the progression of muscular dystrophy in dystrophic mice [79].
Another relevant issue relates to the promoter selectivity of CMCs and the epigenetic regulation of individual genes. Recent works are revealing a different composition of complexes associated with promoters of early versus late muscle genes. For instance, the PcG catalytic subunit EzH2 is found on the regulatory regions of late but not early muscle genes [55]. The arginine methyltransferases PRMT5 and CARM1 also show a specific distribution to the promoters of early and late muscle genes, respectively [72]. This evidence suggests that different epigenetic signatures contribute to coordinate the temporal expression of different subsets of genes. Future investigation should determine the precise composition of CMCs at individual promoters, and whether changes in composition can provide the surface variability to adapt to dynamic protein– protein interactions and to respond to signaling activated by external cues.
The temporal relationship between promoter occupancy by transcription factors, epigenetic changes and signal-responsiveness should also be clarified. The discovery that the p38 and IGF1–AKT cascades direct two distinct epigenetic events on the chromatin of muscle genes – the recruitment of SWI/SNF and acetyltransferases, respectively – suggests that pharmacological dissociation of these pathways could generate interesting cellular phenotypes. Indeed, blockade of p38α and p38β kinases in satellite cells exposed to IGF1 resulted in the expansion of satellite cells in which the chromatin at muscle promoters is hyperacetylated but not remodeled [39]. This ‘poised’ conformation can be readily converted into a productive state by restoring p38 signaling, which rapidly promotes muscle-gene expression. This evidence suggests that intermittent blockade of the p38 pathway can generate intermediate cellular phenotypes that are exploitable to increase the efficiency of muscle regeneration.
Acknowledgments
P.L.P. is the Associate Telethon Scientist of the Dulbecco Telethon Institute and the recipient of R01 AR053779 from the National Institute of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMSD).
References
- 1.Singec I, et al. The leading edge of stem cell therapeutics. Annu Rev Med. 2007;58:313–328. doi: 10.1146/annurev.med.58.070605.115252. [DOI] [PubMed] [Google Scholar]
- 2.Kuang S, Rudnicki MA. The emerging biology of satellite cells and their therapeutic potential. Trends Mol Med. 2008;14:82–91. doi: 10.1016/j.molmed.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Sherwood RI, et al. Isolation of adult mouse myogenic progenitors: functional heterogeneity of cells within and engrafting skeletal muscle. Cell. 2004;119:543–554. doi: 10.1016/j.cell.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 4.Collins CA, et al. Stem cell function, self renewal, and behavioral heterogeneity of cells from the adult muscle satellite niche. Cell. 2005;122:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Kuang S, et al. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006;20:1692–1708. doi: 10.1101/gad.1419406. [DOI] [PubMed] [Google Scholar]
- 7.Péault B, et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;15:867–877. doi: 10.1038/mt.sj.6300145. [DOI] [PubMed] [Google Scholar]
- 8.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 9.de la Serna IL, et al. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat Rev Genet. 2006;7:461–473. doi: 10.1038/nrg1882. [DOI] [PubMed] [Google Scholar]
- 10.Forcales SV, Puri PL. Signaling to the chromatin during skeletal myogenesis: novel targets for pharmacological modulation of gene expression. Semin Cell Dev Biol. 2005;16:596–611. doi: 10.1016/j.semcdb.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Palacios D, Puri PL. The epigenetic network regulating muscle development and regeneration. J Cell Physiol. 2006;207:1–11. doi: 10.1002/jcp.20489. [DOI] [PubMed] [Google Scholar]
- 12.Conboy IM, Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell. 2002;3:397–409. doi: 10.1016/s1534-5807(02)00254-x. [DOI] [PubMed] [Google Scholar]
- 13.Shinin V, et al. Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol. 2006;8:677–678. doi: 10.1038/ncb1425. [DOI] [PubMed] [Google Scholar]
- 14.Zammit PS, et al. Muscle satellite cells adopt divergent fates: a mechanism for self-renewal? J Cell Biol. 2004;166:347–357. doi: 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKinnell IW, et al. Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat Cell Biol. 2008;10:77–84. doi: 10.1038/ncb1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu P, et al. Codependent activators direct myoblast-specific MyoD transcription. Dev Cell. 2008;15:534–546. doi: 10.1016/j.devcel.2008.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zammit PS, et al. Pax7 and myogenic progression in skeletal muscle satellite cells. J Cell Sci. 2006;119:1824–1832. doi: 10.1242/jcs.02908. [DOI] [PubMed] [Google Scholar]
- 18.Olguin HC, et al. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J Cell Biol. 2007;177:769–779. doi: 10.1083/jcb.200608122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blais A, et al. An initial blueprint for myogenic differentiation. Genes Dev. 2005;19:553–556. doi: 10.1101/gad.1281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao Y, et al. Global and gene-specific analyses show distinct roles for Myod and Myog at a common set of promoters. EMBO J. 2006;25:502–511. doi: 10.1038/sj.emboj.7600958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penn BH, et al. A MyoD-generated feed-forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev. 2004;18:2348–2353. doi: 10.1101/gad.1234304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puri PL, Sartorelli V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J Cell Physiol. 2000;185:155–173. doi: 10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 23.Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol. 2005;16:585–595. doi: 10.1016/j.semcdb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Weintraub H, et al. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc Natl Acad Sci U S A. 1989;86:5434–5438. doi: 10.1073/pnas.86.14.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerber AN, et al. Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev. 1997;11:436–450. doi: 10.1101/gad.11.4.436. [DOI] [PubMed] [Google Scholar]
- 26.Bergstrom DA, et al. Promoter-specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol Cell. 2002;9:587–600. doi: 10.1016/s1097-2765(02)00481-1. [DOI] [PubMed] [Google Scholar]
- 27.McKinsey TA, et al. Signaling chromatin to make muscle. Curr Opin Cell Biol. 2002;14:763–772. doi: 10.1016/s0955-0674(02)00389-7. [DOI] [PubMed] [Google Scholar]
- 28.Ait-Si-Ali S, et al. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004;23:605–615. doi: 10.1038/sj.emboj.7600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9:11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 30.Kuang S, et al. Niche regulation of muscle satellite cell self-renewal and differentiation. Cell Stem Cell. 2008;2:22–31. doi: 10.1016/j.stem.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 31.Guasconi V, Puri PL. Epigenetic drugs in the treatment of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care. 2008;11:233–241. doi: 10.1097/MCO.0b013e3282fa1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guttridge DC, et al. NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- 33.Alter J, et al. Inhibition of myoblast differentiation by tumor necrosis factor αis mediated by c-Jun N-terminal kinase 1 and leukemia inhibitory factor. J Biol Chem. 2008;283:23224–23234. doi: 10.1074/jbc.M801379200. [DOI] [PubMed] [Google Scholar]
- 34.Mozzetta C, et al. Regenerative pharmacology in the treatment of genetic diseases: The paradigm of muscular dystrophy. Int J Biochem Cell Biol. 2009;41:701–710. doi: 10.1016/j.biocel.2008.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edmunds JW, Mahadevan LC. MAP kinases as structural adaptors and enzymatic activators in transcription complexes. J Cell Sci. 2004;117:3715–3723. doi: 10.1242/jcs.01346. [DOI] [PubMed] [Google Scholar]
- 36.Simone C, et al. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet. 2004;36:738–743. doi: 10.1038/ng1378. [DOI] [PubMed] [Google Scholar]
- 37.Pokholok DK, et al. Activated signal transduction kinases frequently occupy target genes. Science. 2006;313:533–536. doi: 10.1126/science.1127677. [DOI] [PubMed] [Google Scholar]
- 38.Proft M, et al. The stress-activated Hog1 kinase is a selective transcriptional elongation factor for genes responding to osmotic stress. Mol Cell. 2006;23:241–250. doi: 10.1016/j.molcel.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 39.Serra C, et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell. 2007;28:200–213. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chow CW, Davis RJ. Proteins kinases: chromatin-associated enzymes. Cell. 2006;127:887–890. doi: 10.1016/j.cell.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 41.Edmunds JW, Mahadevan LC. Cell signaling. Protein kinases seek close encounters with active genes. Science. 2006;313:449–451. doi: 10.1126/science.1131158. [DOI] [PubMed] [Google Scholar]
- 42.Jones NC, et al. The p38α/β MAPK functions as a molecular switch to activate the quiescent satellite cell. J Cell Biol. 2005;169:105–116. doi: 10.1083/jcb.200408066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz-Bonilla V, et al. Efficient adult skeletal muscle regeneration in mice deficient in p38β, p38γ and p38δ MAP kinases. Cell Cycle. 2008;7:2208–2214. doi: 10.4161/cc.7.14.6273. [DOI] [PubMed] [Google Scholar]
- 44.Proft M, Struhl K. Hog1 kinase converts the Sko1-Cyc8-Tup1 repressor complex into an activator that recruits SAGA and SWI/SNF in response to osmotic stress. Mol Cell. 2002;9:1307–1317. doi: 10.1016/s1097-2765(02)00557-9. [DOI] [PubMed] [Google Scholar]
- 45.Alepuz PM, et al. Osmostress-induced transcription by Hot1 depends on a Hog1-mediated recruitment of the RNA Pol II. EMBO J. 2003;22:2433–2442. doi: 10.1093/emboj/cdg243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alepuz PM, et al. Stress-induced map kinase Hog1 is part of transcription activation complexes. Mol Cell. 2001;7:767–777. doi: 10.1016/s1097-2765(01)00221-0. [DOI] [PubMed] [Google Scholar]
- 47.Chi T. A BAF-centred view of the immune system. Nat Rev Immunol. 2004;4:965–977. doi: 10.1038/nri1501. [DOI] [PubMed] [Google Scholar]
- 48.Lickert H, et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- 49.Lluís F, et al. Regulation of skeletal muscle gene expression by p38 MAP kinases. Trends Cell Biol. 2006;16:36–44. doi: 10.1016/j.tcb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Lluís F, et al. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J. 2005;24:974–984. doi: 10.1038/sj.emboj.7600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cole F, et al. Positive regulation of myogenic bHLH factors and skeletal muscle development by the cell surface receptor CDO. Dev Cell. 2004;7:843–854. doi: 10.1016/j.devcel.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 52.Takaesu G, et al. Activation of p38α/β MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo. J Cell Biol. 2006;175:383–388. doi: 10.1083/jcb.200608031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rampalli S, et al. p38 MAPK signaling regulates recruitment of Ash2L-containing methyltransferase complexes to specific genes during differentiation. Nat Struct Mol Biol. 2007;14:1150–1156. doi: 10.1038/nsmb1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ringrose L, Paro R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet. 2004;38:413–443. doi: 10.1146/annurev.genet.38.072902.091907. [DOI] [PubMed] [Google Scholar]
- 55.Caretti G, et al. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004;18:2627–2638. doi: 10.1101/gad.1241904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsukada Y, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- 57.Klose RJ, et al. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- 58.De Santa F, et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–1094. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 59.Agger K, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 60.Pasini D, et al. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008;22:1345–1355. doi: 10.1101/gad.470008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 62.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berkes CA, et al. Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol Cell. 2004;14:465–477. doi: 10.1016/s1097-2765(04)00260-6. [DOI] [PubMed] [Google Scholar]
- 64.Heidt AB, et al. Determinants of myogenic specificity within MyoD are required for noncanonical E box binding. Mol Cell Biol. 2007;27:5910–5920. doi: 10.1128/MCB.01700-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sartorelli V, et al. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol Cell. 1999;4:725–773. doi: 10.1016/s1097-2765(00)80383-4. [DOI] [PubMed] [Google Scholar]
- 66.Di Padova M, et al. MyoD acetylation influences temporal patterns of skeletal muscle gene expression. J Biol Chem. 2007;282:37650–37659. doi: 10.1074/jbc.M707309200. [DOI] [PubMed] [Google Scholar]
- 67.Duquet A, et al. Acetylation is important for MyoD function in adult mice. EMBO Rep. 2006;7:1140–1146. doi: 10.1038/sj.embor.7400820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spitz F, et al. Expression of myogenin during embryogenesis is controlled by Six/sine oculis homeoproteins through a conserved MEF3 binding site. Proc Natl Acad Sci U S A. 1998;95:14220–14225. doi: 10.1073/pnas.95.24.14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hasty P, et al. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature. 1993;364:501–506. doi: 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- 70.Ohkawa Y, et al. Skeletal muscle specification by myogenin and Mef2D via the SWI/SNF ATPase Brg1. EMBO J. 2006;25:490–501. doi: 10.1038/sj.emboj.7600943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Puri PL, et al. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in Human Rhabdomyosarcomas. Genes Dev. 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- 72.Dacwag CS. Distinct protein arginine methyltransferases promote ATP-dependent chromatin remodeling function at different stages of skeletal muscle differentiation. Mol Cell Biol. 2009 doi: 10.1128/MCB.00742-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen SE, et al. TNF-α regulates myogenesis and muscle regeneration by activating p38 MAPK. Am J Physiol Cell Physiol. 2007;292:C1660–C1671. doi: 10.1152/ajpcell.00486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ng RK, Gurdon JB. Epigenetic memory of an active gene state depends on histone H3.3 incorporation into chromatin in the absence of transcription. Nat Cell Biol. 2008;10:102–109. doi: 10.1038/ncb1674. [DOI] [PubMed] [Google Scholar]
- 75.Lee H, et al. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science. 2004;304:1675–1678. doi: 10.1126/science.1098096. [DOI] [PubMed] [Google Scholar]
- 76.Lange M, et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 2008;22:2370–2384. doi: 10.1101/gad.471408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deato MD, et al. MyoD targets TAF3/TRF3 to activate myogenin transcription. Mol Cell. 2008;32:96–105. doi: 10.1016/j.molcel.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seale P, et al. Pax7 is required for the specification of myogenic satellite cells. Cell. 2000;102:777–786. doi: 10.1016/s0092-8674(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 79.Minetti GC, et al. Functional and morphological recovery of systrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]