

Abstract
Controlling cell fate is essential for embryonic development, tissue regeneration, and the prevention of human disease. With each cell in the human body sharing a common genome, achieving the appropriate spectrum of stem cells and their differentiated lineages requires the selective activation of developmental signaling pathways, the expression of specific target genes, and the maintenance of these cellular states through epigenetic mechanisms. Small molecules that target these regulatory processes are therefore valuable tools for probing and manipulating the molecular mechanisms by which stem cells self-renew, differentiate, and arise from somatic cell reprogramming. Pharmacological modulators of cell fate could also help remediate human diseases caused by dysregulated cell proliferation or differentiation, heralding a new era in molecular therapeutics.
Keywords: Differentiation: The process by which unspecialized cells acquire specific functions, allowing the generation of complex tissues and organs. Differentiation is frequently controlled by cell signaling pathways and maintained through epigenetic mechanisms.; Ectoderm: The outer germ layer that gives rise to skin, the nervous system, and sensory organs.; Endoderm: The inner germ layer that gives rise to respiratory and digestive organs.; Embryonic stem cells: Pluripotent cells derived from embryos that can be propagated in culture.; Feeder cells: Cells co-cultured with pluripotent cells to prevent their differentiation. Feeder cells are typically mouse or human embryonic fibroblasts.; Induced pluripotent stem cells: Pluripotent cells obtained through the reprogramming of differentiated cells. Induced pluripotent stem cells are functionally similar to embryonic stem cells.; Mesoderm: The middle germ layer that gives rise to muscle, bone, connective tissues, and blood cells.; Multipotent cells: Cells that can give rise to more than one cell type of the body.; Pluripotent cells: Cells that can give rise to all differentiated cell types of the body but not extraembryonic tissues.; Totipotent cells: Cells that give rise to all differentiated cell types of the body and extraembryonic tissues such as the placenta.
The human body is composed of over 400 distinct cell types, each originating from a totipotent zygote (1, panel a) (1). During fetal development, cell differentiation arises through a succession of specification events, including the establishment of a pluripotent inner cell mass, its conversion into three multipotent germ layers (ectoderm, mesoderm, and endoderm), and the creation of increasingly specialized cells in a stepwise manner (1, panel b). These cell fate choices must be coordinated in both space and time to achieve a functional body plan, and many of the molecular reactions and interactions that regulate cell specification and tissue patterning have now been identified through large-scale mutagenesis screens in model organisms. In particular, a suite of intercellular signaling mechanisms, commonly referred to as developmental pathways, are used iteratively and combinatorially to coordinate cell fate and function during embryogenesis, as well as to control postnatal tissue homeostasis. These developmental pathways include Hedgehog (Hh), Wnt, transforming growth factor-β (TGFβ), fibroblast growth factor (FGF), and Notch signaling, and the molecular mechanisms and physiological roles of each signaling process are largely conserved across the animal kingdom (2–6). Cell fate choices actuated through these pathways are influenced and reinforced by epigenetic processes such as chromatin remodeling, thereby achieving the stereotypic cellular diversity required for organismal function amidst genomic uniformity.
Figure 1.

Progenitor cell self-renewal and differentiation contribute to tissue patterning and tumorigenesis. a) Examples of specialized cell types associated with different human tissues and organs. b) Schematic representation of the self-renewal and differentiation of progenitor cells, in both normal and oncogenic contexts. Depicted derivatives of the primary germ layers are illustrative rather than inclusive.
Small molecules that can control cell fate have garnered considerable interest in recent years, as the critical roles of stem cell self-renewal and differentiation in human physiology have become better understood. A number of these compounds target specific developmental pathways, modulating the biogenesis and/or secretion of the extracellular ligands that initiate each pathway (e.g., the Hh, Wnt, TGFβ, and FGF families of secreted factors), the activity of their cell-surface receptors, or signaling events involving downstream effectors and transcription factors. Other fate-modulating chemicals target factors that dictate the responsiveness of cells to external cues, such as chromatin remodelling enzymes and cell cycle regulators. Using these molecular probes, biologists have gained important insights into the biochemical mechanisms that determine cell fate choice, both in cell culture and in whole organisms.
Small molecules that target developmental pathways and other cell specification mechanisms also have the potential to transform how we treat certain diseases and disorders. Ontogeny and oncogenesis are mechanistically linked, as inappropriate cell fate choice can promote the onset and/or maintenance of several tumors, perhaps by engendering stem cell-like cancer cells (1, panel b) (2,3,5–7). Compounds that inhibit these processes could therefore be more efficacious than conventional chemotherapies, which indiscriminately target proliferating cells and commonly produce severe side effects. There is even the exciting prospect that pharmacological reagents might be able to enhance or create de novo regenerative activities that involve pluripotent or multipotent cells, thereby remediating traumatic injuries or degenerative diseases.
In this review we summarize recent progress toward developing small-molecule regulators of cell fate (1). Compounds that modulate the Hh, Wnt, TGFβ, FGF, or Notch pathways will first be described, since these intercellular signaling mechanisms are primary mediators of tissue patterning and regeneration. We will then survey how phenotype-based screens have been used to identify other compounds that promote stem cell self-renewal, the differentiation of specific cell types, or the reprogramming of somatic cells into pluripotent populations. The examples described herein are not intended to be comprehensive, and readers are encouraged to examine previous reviews of fate-modulating compounds (8,9). Rather, these selected studies illustrate the biological concepts, experimental approaches, and therapeutic possibilities that are associated with the discovery of small-molecule regulators of cell fate. We conclude our review with a discussion of the potential of this emerging field, its current limitations, and future challenges for the chemical biology community.
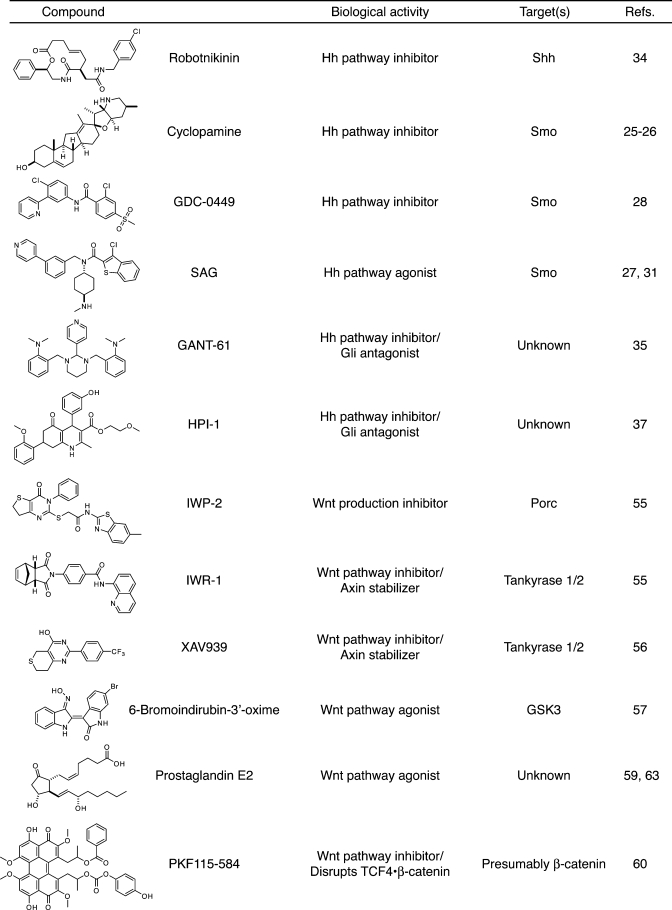
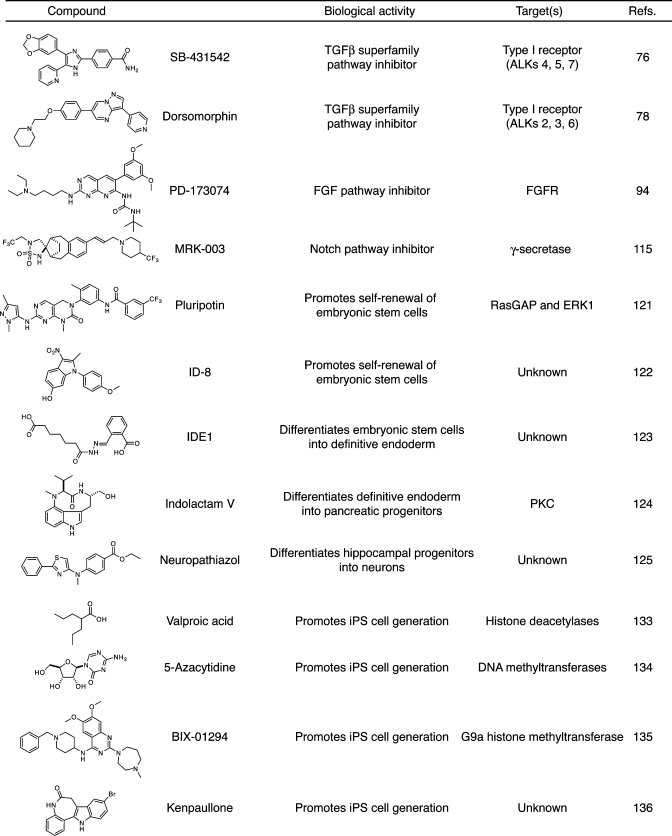
Table 1. Selected small-molecule modulators of cell fate.
 |
 |
Hh Pathway Modulators
Hh signaling is perhaps the developmental pathway that has been most extensively targeted by small molecules to date. Hh pathway activation promotes the self-renewal of certain progenitor populations such as hair follicle stem cells and cerebellar granule neuron precursors (10,11). Hh signaling also regulates neural cell fate along the dorsal-ventral axis of the developing spinal cord (12), anterior-posterior digit identity (13), and retinal cell diversification in the nascent eye (14). In each of these systems, palmitoyl- and cholesteryl-modified Hh ligands (in mammals, Sonic (Shh), Indian (Ihh), and Desert (Dhh) Hedgehog) are released in a spatially restricted manner by Hh-producing cells, creating a gradient of Hh protein (2). Hh biogenesis and secretion are specifically regulated by the transmembrane proteins Hh acyltransferase (Hhat) and Dispatched (Disp), and responsive cells then express specific target genes in a Hh concentration-dependent manner, generating an organized array of discrete cell types (2, panel a).
Figure 2.

Hh and Wnt signaling pathways. Signaling proteins associated with Hh (a) and Wnt (b) pathway regulation are shown. Components or processes that currently can be targeted by small molecules to achieve selective pathway control are labeled with green (agonist) and red (antagonist) hexagons. Direct small molecule−protein interactions are depicted when known.
Reception of the Hh signal involves several conserved signaling proteins, including the twelve-pass transmembrane receptor Patched1 (Ptch1), the G protein-coupled receptor (GPCR)-like protein Smoothened (Smo), the nucleocytoplasmic factor Suppressor of Fused (Sufu), and the Gli family of transcription factors (Gli1, Gli2, and Gli3) (2). In the absence of Hh ligand, Ptch1 inhibits the activity of Smo, permitting the sequential phosphorylation of Gli2 and Gli3 by protein kinase A (PKA), glycogen synthase kinase-3 (GSK3), and casein kinase 1 (CK1). These phosphorylation events create docking sites for ubiquitination machinery, leading to proteolytic processing of the Gli proteins into N-terminal transcriptional repressors or their complete degradation. Hh ligands directly inhibit Ptch1 and therefore activate Smo, promoting the stabilization of full-length Gli2 and Gli3 and their conversion into transcriptional activators. Smo activation appears to also shield the Gli proteins from the repressive effects of Sufu, which binds to Gli factors and inhibits their activity through multiple mechanisms. Hh target genes include Ptch1 and Gli1, creating negative and positive feedback loops, respectively.
Consistent with the multiple patterning roles of Hh signaling, dysregulation of any of these events can have severe physiological consequences. Loss-of-function mutations in SHH, DHH, and GLI2 and putative gain-of-function mutations in PTCH1 have been associated with birth defects such as holoprosencephaly and gonadal dysgenesis (15–18), and genetic lesions at the GLI3 locus can cause polydactyly (19). In addition, aberrant activation of the Hh pathway is associated with the onset and/or progression of several cancers such as basal cell carcinoma (20), medulloblastoma (21,22), and pancreatic adenocarcinoma (23,24). These oncogenic events can result from inappropriate autocrine or paracrine Hh signaling, as well as pathway-activating mutations in PTCH1, SMO, or SUFU.
In principle, small molecules could target any of these signaling events, from Hh ligand biogenesis to Gli-dependent transcription. In practice, however, nearly all known small-molecule modulators of the Hh pathway target Smo, perhaps reflecting the pharmacological sensitivity of GPCR family members and related proteins. The first Hh pathway inhibitor to be discovered was the steroid alkaloid cyclopamine, a plant-derived teratogen associated with an outbreak of cyclopic lambs in Idaho during the 1950s (25). Genetic and biochemical analyses conducted nearly half a century later established that cyclopamine directly inhibits Smo (26), and subsequent high-throughput screens of synthetic chemical libraries have identified numerous other Smo inhibitors (27–30), as well as Smo agonists such as SAG and purmorphamine (27,31–33). Compounds that target other Hh signaling proteins have been more elusive. One notable exception is the 12-membered macrocycle robotnikinin, which was discovered through a small-molecule microarray screen for compounds that can bind recombinant Shh (34). Robotnikinin inhibits Hh pathway activation induced by Shh ligand, but it has no inhibitory effect on the Hh target gene expression in Ptch1−/− fibroblasts or cells activated with purmorphamine. These results indicate that robotnikinin may prevent Shh from binding to Ptch1, although the molecular details of this mechanism have not yet been established.
Until recently, the standard method for blocking Hh pathway activity downstream of Smo was to treat Hh-responsive cells with forskolin, which activates adenylate cyclase, increases cAMP levels, and promotes the inhibition of Gli function by PKA. The pleiotropic effects of forskolin, however, have prompted a search for mechanistically distinct Hh pathway inhibitors that act downstream of Smo, and a few compounds that are epistatic to endogenous or overexpressed Gli activators have now been reported. For example, the synthetic molecules GANT-58 and GANT-61 were identified in a screen for compounds that can block Hh pathway activity induced by the overexpression of Gli1 in HEK-293T cells, and natural product inhibitors of Gli-mediated transcription were found in a similar Gli1 overexpression assay (35,36). Four additional Gli antagonists (HPIs 1–4) were recently discovered in a cell-based screen for SAG repressors (37). Although the precise mechanisms of these Hh pathway inhibitors remain unknown, it is unlikely that they directly target the Gli family of transcription factors. Rather, these compounds probably interact with endogenous factors that control Gli function and therefore could be valuable probes of these regulatory mechanisms.
Several of these Hh pathway modulators have been applied toward the regulation of cell fate in vitro and in vivo. The SAG class of Smo agonists has been used to differentiate motor neurons from mouse embryonic stem cells (ESCs) and to expand neuronal precursor cells in animal models of spinal cord injury (38,39). SAG-related compounds have also successfully promoted hair growth in mouse skin (40), and purmorphamine was originally found in a screen for osteogenic compounds (33). Smo antagonists have been equally effective in murine models of medulloblastoma (41), pancreatic adenocarcinoma (23,24,42), and other tumors, and several pharmaceutical companies are developing these compounds as targeted chemotherapies. The Genentech/Curis inhibitor, GDC-0449, has demonstrated efficacy against metastatic basal cell carcinoma in phase I clinical trials (28), and lead compounds from Infinity Pharmaceuticals and Bristol Meyers Squibb/Exelixis are now in phase I trials as well. It will be interesting to determine whether downstream Hh pathway inhibitors are comparably active against murine tumor models, especially since Smo mutations that convey GDC-0449 resistance have been observed in mouse and human medulloblastomas upon drug treatment (43).
Wnt Pathway Modulators
Like the Hh pathway, Wnt signaling regulates stem cell self-renewal and differentiation in diverse physiological contexts, including progenitor cell maintenance within the intestinal crypt (44), hematopoiesis in the bone marrow (45), and the differentiation of melanocytes from neural crest precursors (46). Aberrant Wnt pathway activation is also associated with tumorigenesis, such as the onset and maintenance of colorectal cancers (47–49), hepatocellular carcinomas (50), and melanomas (51). The Hh and Wnt pathways even share interesting parallels in their signaling architecture (3). For example, the secretion of active Wnt protein similarly requires two transmembrane proteins, in this case the acyltransferase Porcupine (Porc), which modifies the ligand with palmitoleic acid, and the seven-pass transmembrane protein Wntless (Wls). Wnt ligands are also palmitoylated, the Frizzled (Fzd) family of Wnt receptors are the closest known homologues of Smo, and Wnt target gene expression is regulated by a phosphorylation- and proteolysis-dependent balance of transcriptional activators and repressors (2, panel b).
In addition to 19 Wnt and 10 Fzd family members, canonical Wnt signaling in mammals involves low-density-lipoprotein receptor-related proteins LRP5 and LRP6, the multifunctional protein adenomatosis polyposis coli (APC), the scaffolding proteins Axin1 and Dishevelled (Dvl1, Dvl2, and Dvl3), the T-cell factor/lymphoid enhancer-binding factor family of transcription factors (TCF1, TCF3, TCF4, and LEF1), and the transcriptional coactivator β-catenin (Ctnnb1) (3). TCF/LEF family members generally repress Wnt target gene expression, but they are converted into transcriptional activators when bound to β-catenin. Wnt signaling therefore controls pathway activity by modulating β-catenin levels, and in the absence of Wnt ligands, a cytosolic complex composed of Axin1, APC, CK1, and GSK3 sequentially phosphorylates β-catenin, thereby promoting its ubiquitination and proteosomal degradation. The binding of Wnts to Fzd and LRP5/6 leads to the recruitment of Dvl proteins, LRP5/6 phosphorylation, and consequently Axin1 binding. Since localization of Axin1 to the cell surface disrupts the cytosolic β-catenin degradation complex, β-catenin accumulates in the nuclei of Wnt-stimulated cells and associates with TCF/LEF proteins to activate Wnt target gene expression. Wnt-inducible genes include Axin2, which is functionally equivalent to Axin1 and establishes a negative feedback loop. Loss-of-function mutations in any of these genes can cause a variety of developmental abnormalities. For example, inactivating lesions in LRP5 and FZD4 are linked to familial exudative vitreoretinopathy (52,53), and loss of WNT7A function produces limb malformations and joint dysplasia (54). In addition, Wnt pathway-dependent tumors frequently harbor mutations that cause a loss of APC, AXIN1, or AXIN2 function or render CTNNB1 insensitive to ubiquitin-mediated destruction (47–50).
Given the parallels between Hh and Wnt signaling, one might expect that chemical modulators would readily target Fzd function. Yet this has not been observed, possibly because of the structural diversity of Fzd homologues and their combinatorial expression in various cell types. Molecules that target other aspects of Wnt signal transduction, however, have been discovered through cell-based assays and whole-organism screens, enabling the pharmacological control of both Wnt signal production and its reception. By screening a chemical library against murine fibroblasts that overexpress Wnt3a and harbor a TCF/LEF-responsive reporter, a family of benzothiazole derivatives called IWPs (inhibitors of Wnt production) were identified as Porc antagonists (55). The same study led to the discovery of two structural families of IWRs (inhibitors of Wnt response) that stabilize Axin proteins, leading to their cytosolic accumulation and increased β-catenin degradation. Subsequent investigations have shown that one IWR ligand, IWR-1, and a structurally unrelated Wnt pathway antagonist, XAV939, can inhibit poly-ADP-ribosylating enzymes tankyrase 1 and 2, which PARsylate Axin proteins and promote their degradation (56). These observations indicate that the IWRs and XAV939 inhibit Wnt pathway activation at least in part by blocking cellular factors that induce Axin proteolysis.
Conversely, β-catenin destruction can be blocked by small molecules in order to activate the Wnt pathway. The kinases that prime β-catenin for degradation are possible targets for this purpose, and several selective inhibitors of GSK3 have been reported to increase Wnt target gene expression in vitro and in vivo(57,58). β-Catenin is also stabilized by prostaglandin PGE2, which apparently acts through cAMP/PKA signaling (59). Finally, small molecules that prevent the formation of β-catenin-TCF/LEF complexes represent an alternative means of controlling Wnt pathway activity. One advantage of this strategy is that such compounds would be functionally epistatic to essentially all oncogenic mutations that abrogate β-catenin degradation, including those in CTNNB1 itself. Toward this goal, several thousand natural products were screened for their ability to disrupt β-catenin-TCF4 binding in vitro, resulting in the identification of PKF115-584 and other perylene diones as Wnt signaling antagonists that are effective in both cultured cells and embryos (60).
As expected from the roles of Wnt signaling in stem cell self-renewal, differentiation, and oncogenesis, Wnt pathway-modulating compounds are potent regulators of cell fate. GSK3 inhibitors such as 6-bromoindirubin-3′-oxime (BIO) and 603281-31-8 have been used to induce both the self-renewal of ESCs and their differentiation to osteoblasts and dopaminergic neurons (58,61,62), divergent outcomes that perhaps reflect the participation of GSK3 in pathways other than Wnt signaling and differences between the ground states of various ESC lines. PGE2-induced Wnt target gene expression regulates the self-renewal of hematopoietic stem cells, and PGE2 and Wnt act synergistically to promote liver regeneration (63). Pharmacological blockade of Wnt target gene expression can also perturb stem cell-dependent processes, as illustrated by the inhibitory effects of IWR-1 on gastrointestinal tract renewal and caudal fin regeneration in zebrafish (55). Similarly, PKF115-584 and XAV939 have been shown to block the proliferation of multiple myeloma cells with constitutively active β-catenin and APC-deficient colorectal cancer cells, respectively (56,64).
TGFβ Superfamily Pathway Modulators
The TGFβ superfamily of secreted polypeptides represents an even larger collection of extracellular factors that regulate embryonic patterning and tissue homeostasis. There are more than 30 TGFβ family members in mammals, which can be classified further into the TGFβ, bone morphogenetic protein (BMP), Activin/Nodal, and growth differentiation factor (GDF) subgroups (4). These intercellular signaling molecules play diverse roles in cell fate choice, exemplified by the Nodal-dependent induction of mesoderm and endoderm during early embryogenesis and the osteogenic properties of BMP proteins (65,66). The relationship between cancer and TGFβ superfamily members is similarly complex. Members of the TGFβ superfamily appear to primarily promote cellular senescence, differentiation, and apoptosis, and accordingly, TGFβ and BMP signaling can actually suppress tumorigenesis. Inactivating mutations in these pathways have been found in colorectal tumors (67–69), pancreatic carcinomas (70), and head and neck cancers (69). However, TGFβ signaling also regulates immune responses and cell migration, and in certain contexts TGFβ pathway activation can protect tumors from the immune system and promote their metastasis.
All members of the TGFβ superfamily are secreted as proteolytically processed homodimers and signal through a heterotetrameric receptor complex composed of two types of serine/threonine kinases (3, panel a) (4). In mammals there are seven type I receptors (ALKs 1–7) and five type II receptors (ACVR2A, ACVR2B, TGFBR2, BMPR2, and AMHR2), and different receptor combinations are used to recognize specific ligands and achieve diverse signaling outputs. Ligand binding promotes phosphorylation of the type I receptors by their constitutively active type II counterparts, leading to the recruitment and phosphorylation of intracellular signaling proteins called Smads. These receptor-regulated Smads (also known as R-Smads 1, 2, 3, 5, and 8) form homo- and heteromeric complexes with the cofactor Smad4 and then accumulate in the nucleus to modulate target gene transcription. Specific TGFβ superfamily ligands, receptors, and Smads frequently function with preferred signaling partners, grossly partitioning into a group composed of BMP/GDF factors, ALKs 2, 3, and 6, and R-Smads 1, 5, and 8 and another group composed of TGFβ/Activin/Nodal ligands, ALKs 4, 5, and 7, and R-Smads 2 and 3. However, exceptions to these general trends exist. R-Smad function is also modulated by inhibitory Smad factors (Smads 6 and 7), providing yet another layer of pathway regulation.
Figure 3.

TGFβ superfamily and FGF signaling pathways. Signaling proteins associated with TGFβ (a) and FGF (b) pathway regulation are shown. Components or processes that currently can be targeted by small molecules to achieve selective pathway control are labeled with green (agonist) and red (antagonist) hexagons. Direct small molecule–protein interactions are depicted when known.
Consistent with this large ensemble of TGFβ superfamily signaling proteins, nearly every organ system requires one or more of these genes for proper patterning and function, and their genetic disruption has been linked to several human disorders. For example, loss-of-function GDF5 mutations can cause limb shortening and digit loss (71), and inactivating lesions in ALK1 and BMPR2 are associated with vascular diseases such as hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension, respectively (72,73). Excessive TGFβ signaling can be physiologically detrimental as well, and persistent activation of this pathway often leads to fibrosis of the kidney, liver, heart, lungs, and other organs (73). The diversification of TGFβ receptor signaling outputs through various Smad complexes also provides a mechanistic basis for the opposing roles of TGFβ pathway activation in oncogenesis. Loss-of-function mutations in the receptors ALK3, ALK5, or TGFBR2 or the cytosolic factor SMAD4 are frequently observed in tumors, and the complete loss of pathway activation resulting from these mutations appears to facilitate the progression of premalignant lesions (67–70). Certain cancers such as breast carcinomas and gliomas, however, exhibit increased TGFβ pathway activity (74,75). In these cases, the tumor suppressor arm of TGFβ signaling may be selectively inhibited, allowing other TGFβ pathway-dependent outputs to enhance tumor growth and invasiveness.
To ameliorate these conditions, several screens to identify small molecules that target TGFβ superfamily pathways have been conducted. All known inhibitors of these signaling mechanisms target type I receptor kinases, and their receptor specificities generally segregate according to the two subgroups described above. For example, cell-free kinase inhibitor screens have identified a number of compounds that selectively inactivate receptors in the TGFβ/Activin/Nodal ligand subgroup (ALKs 4, 5 and 7), such as SB-431542 and SD-208 (76,77). In contrast, a zebrafish screen for small molecules that perturb dorsoventral axis formation led to the discovery of dorsomorphin, which preferentially targets type I receptors activated by BMP/GDF ligands (ALKs 2, 3, and 6) (78). These TGFβ superfamily antagonists can both modulate cell fate decisions and suppress tumor progression. The BMP signaling antagonist dorsomorphin induces cardiomyocyte differentiation in mouse ESCs (79), and the TGFβ pathway blocker SD-208 promotes hematopoiesis and alleviates anemia in a mouse model of myelodysplastic syndrome, a disease caused by the constitutive activation of ALK5 in bone marrow precursors (80). SD-208 also inhibits the growth and metastasis of gliomas transplanted intercranially into mice, in part by enhancing the immunogenicity of these cancers (77). Other type I receptor antagonists have been used successfully to suppress fibriotic responses to tissue injury in animal models (81).
FGF Pathway Modulators
As with the TGFβ superfamily, FGF signaling involves a diverse range of extracellular ligand and signaling outputs. There are at least 22 members of the FGF ligand superfamily, 18 of which interact with FGF receptors (FGFRs 1−4 and their splice variants) (5). Moreover, FGF signaling activates several downstream signaling modules, including the phospholipase C-γ (PLCγ), phosphatidylinositol-3-kinase (PI3K), and mitogen-activated protein kinase (MAPK) pathways. This molecular complexity is mirrored by the manifold roles of FGF signaling in vertebrate physiology, ranging from mesoderm induction during gastrulation to control of hair follicle growth in postnatal life (82,83). Dysregulation of FGF pathway state can lead to skeletal disorders (84,85), hypogonadism (86), as well as the onset or progression of multiple myeloma, cervical carcinomas, and other human cancers (87,88).
FGF pathway activation is initiated by the FGF-dependent homodimerization of FGFRs, leading to trans-autophosphorylation by their intracellular tyrosine kinase domains (3, panel b). This process increases FGFR kinase activity and creates docking sites for downstream effectors that contain Src Homology 2 (SH2) or phosphotyrosine-binding (PTB) domains, enabling the activated FGFRs to trigger several intracellular signaling pathways. In the case of MAPK signaling, FGFR substrate 2 (FRS2) is sequestered by the activated FGFR homodimer and phosphorylated, enabling it to recruit the growth factor receptor-bound 2 (Grb2) adaptor protein and guanine nucleotide exchange factor Son of sevenless (Sos) to the plasma membrane. Membrane-localized Sos then promotes Ras-dependent activation of Raf kinase, triggering the MEK/ERK kinase cascade and culminating in the actuation of MAPK-responsive transcription factors. MAPK pathway target genes include promoters of G1-phase cell cycle entry and progression, such as cyclin D1, and prolonged MAPK signaling is a potent inducer of cell differentiation. In addition to these signaling mechanisms, FGF-dependent MAPK pathway activation is negatively regulated by cytoplasmic factors, such as the Sprouty (Spry) family of inhibitors that target both Grb2 and Raf and dual-specificity phosphatases that inactivate ERK proteins (Dusp6, Dusp7, and Dusp9). Activated FGFRs can similarly modulate PLCγ and PI3K signaling by recruiting and phosphorylating other effector molecules in FGF-responsive cells.
Due to the many prominent roles of FGF signaling in tissue patterning and homeostasis, it is not surprising that genetic lesions affecting this pathway have been associated with a large number of human disorders. FGF ligands are primarily susceptible to loss-of-function mutations. For example, inactivating mutations in FGF3 can cause congenital deafness and structural abnormalities of the outer ear and teeth (89), while loss of FGF8 signaling is associated with salivary and tear gland defects (90). In contrast, FGFRs can be rendered inoperative or constitutively active depending on the structural modification. Loss-of-function mutations in FGFR1 have been linked to Kallmann syndrome, which is characterized by hypogonadism, defective olfaction, and in some cases cleft lip or palate (86). Germline mutations in FGFR1, FGFR2 and FGFR3 can also lead to their ligand-independent homodimerization and activation, causing a broad spectrum of skeletal abnormalities such acrocephaly, polysyndactyly, craniosynostosis and dwarfism (84,85,91). Analogous somatic lesions have been found to promote multiple myeloma, endometrial carcinoma, and bladder cancer, providing yet another example of the molecular kinship between ontogeny and oncogenesis (87,88,92).
Since FGF signaling can lead to the activation of multiple downstream effectors, each of which can participate in other signaling pathways, efforts to specifically control the FGF pathway have necessarily focused on the most upstream components. FGFR tyrosine kinase activity is the most obvious pharmacological target, and relatively specific FGFR inhibitors such as SU5402 and PD173074 have been developed (93,94). These antagonists can counteract dysregulated FGF signaling in cultured cells and animal models, demonstrating preclinical success in models of multiple myeloma, small-cell lung cancer, and endometrial carcinoma (92,95,96). Alternatively, small molecules can be used to selectively target the MAPK, PLCγ, or PI3K arms of FGF signaling, such as the Raf inhibitor BAY-43-9006 and MEK antagonist PD0325901 (97,98). The Dusp6 inhibitor (E)-2-benzylidene-3-(cyclohexylamino)-2,3-dihydro-1H-inden-1-one (BCI) has also been used to enhance FGF target gene expression and promote cardiac cell lineages in zebrafish embryos (99).
Notch Pathway Modulators
While many aspects of tissue patterning can be achieved through distinct cellular responses to secreted factors, certain cellular architectures rely upon signaling processes that are limited to immediately adjacent cells. The Notch pathway is one mechanism by which neighboring cells interact (6), and it is used to promote the self-renewal of hematopoietic and neuronal progenitor cells in specific niches (100,101). Notch signaling is also frequently associated with lateral inhibition, in which Notch pathway activation in one cell population suppresses pathway activity in adjacent cells. This process can generate structurally complex tissues by simultanously limiting the number of progenitor cells that adopt a particular cell fate and ensuring that their neighboring cells are functionally distinct. For example, Notch signaling is used to produce arrays of sensory hair cells and supporting cells in the inner ear (102), to establish segmentation boundaries during somitogenesis (103), and to determine arterial versus venous cell fates within the cardiovascular system (104).
Activation of the mammalian Notch pathway is initiated by the direct binding of two families of type I transmembrane proteins: Notch receptors (Notch 1–4) that are expressed in one cell population and members of the Delta-like (Dll1, Dll3, and Dll4) and Jagged (Jag1 and Jag2) families that are displayed on adjacent cells (4) (6). Notch receptors are heterodimeric proteins, resulting from the furin-mediated cleavage of a single polypeptide precursor, and their ligand specificity is regulated in part through glycosylation. Notch proteins undergo a conformational change upon binding to Delta-like/Jagged family members, and this structural shift exposes an extracellular region in Notch proximal to the transmembrane domain, promoting its proteolysis by tumor necrosis factor-α-converting enzyme (TACE). The remaining membrane-bound Notch fragment is then endocytosed and cleaved within its transmembrane domain by the multiprotein complex γ-secretase. This third and final proteolytic event releases the Notch intercellular domain (NICD) from the plasma membrane, allowing it to enter the nucleus and bind the transcription factor CSL (CBF1/Suppressor of Hairless/Lag1). CSL normally inhibits Notch target gene expression by recruiting co-repressors and histone deacetylases (HDACs) to specific promoter elements, and displacement of these proteins by NICD promotes the association of CSL with transcriptional coactivators such as the Mastermind-like gene family (MAML1, MAML2, and MAML3). Notch target genes include members of the Hes/Hey family of basic helix–loop–helix transcriptional repressors, which play critical roles in stem cell biology.
Figure 4.
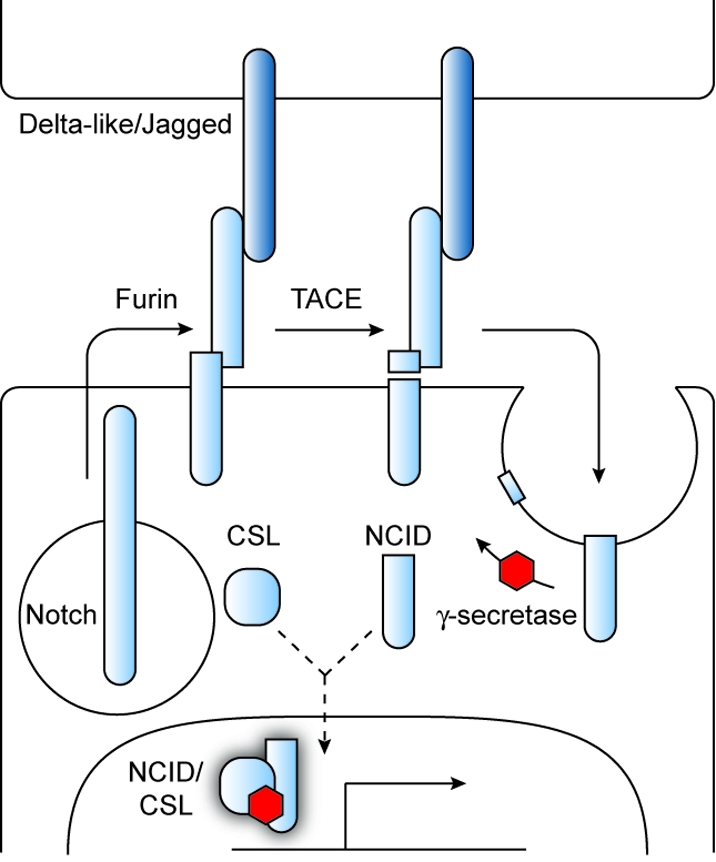
Notch signaling pathway. Signaling proteins associated with Notch signaling are shown. Components that currently can be inhibited by synthetic molecules to achieve selective pathway control are labeled with red hexagons.
Perturbations of these Notch pathway components can lead to aberrant cell fates associated with human disorders and diseases. For example, inactivation of NOTCH2 and JAG1 can give rise to Alagille syndrome (105–107), which is characterized by mispatterning of the liver, heart, eye, and skeleton, and NOTCH3 mutations have been linked to CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) (108), a vascular disorder that primarily affects cerebral arteries and causes cognitive and mood dysfunction. Deformities associated with loss of Delta-like ligand function have been observed as well, illustrated by the vertebrae segmentation defects associated with DLL3 mutant alleles (109). In analogy to the Hh, Wnt, and FGF pathways, Notch signaling can also be co-opted postnatally to promote the self-renewal in disease states. Co-expression of JAG1 and NOTCH1 at high levels correlates with poor clinical outcomes for breast cancer (110), and the majority of T cell acute lymphoblastic leukemia/lymphoma (T-ALL) cases are caused by gain-of-function mutations in NOTCH1(111).
Small molecules that modulate Notch pathway state could therefore have significant therapeutic value. However, identifying such compounds has been a challenge. While Notch pathway activation requires multiple cellular processes such as receptor proteolysis, endocytosis, and nuclear transport, these pharmacologically targetable signaling mechanisms are not necessarily unique to the Notch pathway. Furthermore, Notch receptor/ligand and NICD/CSL complexes may be difficult to target with small molecules. With one exception, all synthetic Notch pathway modulators known to date inhibit γ-secretase, which is perhaps best known for its cleavage of amyloid precursor protein (APP). One of these γ-secretase antagonists, N-[N-(3,5-difluorophenacetyl-l-alanyl)]-S-phenylglycine tert-butyl ester (DAPT) (112), has been used to promote neuronal regeneration in an animal model of retinal injury (113), and another, MDL-28170, can induce hair cell formation in the cochleas of neonate mice (114). Small-molecule inhibitors of γ-secretase can also inhibit the progression of Notch pathway-dependent tumors by preventing the proliferation of their progenitor cells. For example, the compound MRK-003 decreases Notch target gene expression in T-ALL cells, leading to their exit from the cell cycle and programmed cell death (115). Whether the effects of these compounds on other γ-secretase substrates will be clinically deleterious remains to be determined, but the development of small molecules that selectively block NICD production may be possible since Notch and APP are processed in distinct cellular compartments (116).
The only mechanistically distinct Notch pathway inhibitor known to date is not a small molecule but rather a “stapled” α-helical MAML1-derived peptide (SAHM1) (117). This synthetic, cell-permeable peptide contains two non-natural alkenyl amino acids that are conjugated by ruthenium-catalyzed olefin metathesis, and it can bind to the NICD/CSL heterodimer and prevent its association with MAML1 to form a transcriptionally active complex. Consistent with this mode of action, NOTCH1-dependent T-ALL cells treated with SAHM1 exhibit a set of downregulated transcripts that is enriched for γ-secretase inhibitor-sensitive genes. SAHM1 can also inhibit T-ALL cell proliferation in vitro and block the progression of T-ALL cell-dependent leukemia in mouse models.
Small-Molecule Modulators of Stem Cell Self-Renewal
The Hh, Wnt, TGFβ, FGF, and Notch pathways therefore constitute a core set of cell fate regulators, and small molecules that selectively modulate these signaling processes are valuable research and clinical tools. However, these pathways alone cannot account for the cellular diversity that is observed in metazoans. Cell fate choice is regulated by a complex network of genetic and epigenetic mechanisms, many of which have not yet been identified, and our understanding of how developmental pathways interact with these pluripotency and differentiation factors is still incomplete. To uncover and exploit these regulatory events, several laboratories have used phenotypic assays to identify chemical modulators of these cellular processes, including known bioactive reagents and synthetic compounds selected for their structural diversity.
Since one challenge in stem cell biology has been the long-term culture of pluripotent cell lines, technologies that promote stem cell self-renewal have been of particular interest. Conventional protocols utilize protein factors that promote rapid G1-phase transit during cell-cycle progression and inhibit differentiation, as it is generally believed that cells are most sensitive to differentiation-inducing stimuli during early G1 phase. For instance, mouse ESCs can be maintained in a pluripotent state by culturing them in the presence of two extrinsic factors: LIF (leukemia inhibitory factor), a cytokine that binds to the gp130 receptor and activates the pro-self-renewal STAT3 (signal transducers and activators of transcription) pathway and the pro-differentiation MAPK pathway, and BMP4 protein, which induces the expression of Id (inhibitor of differentiation) proteins and attenuates the activity of MAPK-responsive transcription factors (118). Thus, the combined actions of LIF and BMP4 selectively promote mouse ESC self-renewal. Human ESCs, however, are unresponsive to LIF and are generally cultured in the presence of either mouse embryonic fibroblast (MEF) feeder cells, MEF-conditioned medium, or more recently in defined medium containing recombinant TGFβ and FGF growth factors (119). While these mouse and human ESC culture conditions are generally effective for research applications, the use of exogenous biological factors restricts the practical scale at which these cells can be cultured and introduces batch-dependent differences resulting from inconsistent growth factor production.
Small molecules that permit the propagation of ESCs in the absence of feeder cells, serum, or extrinsic protein factors could provide a solution to this problem, facilitating the use of pluripotent cells for medical applications. One approach has been to recapitulate the anti-MAPK pathway activity of BMP4 with the MEK blocker PD0325901, while using the GSK3 inhibitor CHIR99021 to release cyclins and other proliferative factors from GSK3-mediated suppression (120). Using this cocktail of chemical antagonists, mouse ESCs can be sustained without exogenous growth factors or cytokines. Other research groups have conducted chemical screens to identify new compounds that promote ESC self-renewal, assessing pluripotency by the expression of alkaline phosphatase, Nanog, Oct4, Sox2, and other genes associated with the undifferentiated state. One such compound is the dihydropyrimidine pluripotin, and mouse ESCs cultured in the presence of pluripotin rather than protein factors can be used to generate derivatives of the three primary germ layers in chimeric mice, as well as germline tissues (121). Affinity chromatography studies revealed that pluripotin binds directly to Ras GTPase-activating protein and ERK1, and it has been proposed that this small molecule activates the phosphatidylinositol-3-kinase (PI3K) arm of Ras signaling to promote mouse ESC cell cycle progression while inhibiting ESC differentiation induced by Ras-dependent MAPK pathway activation. It has also been reported that mouse ESCs cultured in serum-free medium supplemented with an indole derivative called ID-8 maintain their pluripotency, albeit through an unknown mechanism of action (122). Whether these chemical regimens are equally effective at maintaining human ESC pluripotency has not yet been established, and it is possible that their cellular targets are not functionally conserved across species. However, the ability of TGFβ and FGF proteins to sustain human ESCs in defined medium (119) provides at least a starting point from which small-molecule inducers of human ESC self-renewal could be developed.
Small-Molecule Modulators of Cell Differentiation
Just as small molecules can direct ESC regulatory mechanisms to sustain pluripotency, certain chemicals have been found to promote the conversion of stem cells into specialized derivatives. For example, mouse and human ESCs have been chemically differentiated into pancreatic progenitor cells through a two-step procedure (123). First, a screen for small molecules that promote ESC differentiation into definitive endoderm revealed two alkyl hydrazone derivatives, IDE1 and IDE2 (123). Cells treated with these synthetic compounds express a variety of endodermal markers and populate the developing gut tube when injected into mouse embryos. In fact, the IDE compounds are more effective endoderm-inducing agents than commonly used protein factors such as Activin A or Nodal. The precise mechanism of these small molecules remains unclear, but they both induce Smad2 phosphorylation and Nodal transcription. IDE1 and IDE2 therefore appear to pharmacologically recapitulate signaling mechanisms normally initiated by members of the TGFβ ligand superfamily.
A second screen for small molecules that promote the conversion of definitive endoderm into pancreatic progenitors was concurrently undertaken, using expression of the pancreatic marker Pdx1 as an indicator of differentiation efficiency (124). This study focused on compounds with known cellular targets, and the protein kinase C activator indolactam V was found to increase the fraction of Pdx1-expressing cells by over 100-fold compared to definitive endoderm treated with a vehicle control. The indolactam V-induced pancreatic progenitor cells are functionally indistinguishable from those obtained through growth factor treatments; they express several pancreatic lineage markers and give rise to insulin-producing endocrine cells when transplanted into mice. Sequential treatment of ESCs by IDE1/IDE2 and indolactam V can therefore yield pancreatic progenitors without a need for exogenous growth factors (123), achieving an overall conversion efficiency greater than that currently achieved with protein factor-based protocols.
Chemical library screens have revealed compounds that facilitate the differentiation of ectodermal and mesodermal cell types as well. The aminothiazole derivative neuropathiazol has been reported to induce the neuronal differentiation of hippocampal progenitor cells (125), while sulfonylhydrazones can convert cultured mouse embryonic carcinomas into cardiovascular progenitor cells (126). The mechanisms by which these compounds promote lineage-restricted populations have not been determined, and the functional competence of these differentiated cells awaits further study. Preliminary studies of sulfonylhydrazone-induced progenitor cells are promising, however, as they can generate spontaneously beating cardiomyocytes in cell culture and contribute to the repair of cryoinjured hearts in rat models (126).
Small-Molecule Modulators of Cell Reprogramming
One of the most transformative scientific breakthroughs in this decade has been the discovery that transient overexpression of a few transcription factors can reprogram differentiated cells into pluripotent populations, commonly referred to as induced pluripotent stem (iPS) cells. This was first achieved by expressing the transcription factors Oct4, Sox2, Klf4, and c-Myc in MEFs using retroviruses, and the resulting iPS cells are functionally identical to mouse ESCs (127). The technology has subsequently been applied to human cells (128), and iPS cells could significantly advance the development of cell-based therapies for human disorders and diseases.
Fully realizing the biomedical potential of iPS cells, however, will require addressing certain challenges. First, somatic cell reprogramming is very inefficient and slow, with typically 0.01% of virally infected fibroblasts giving rise to ESC-like colonies after two to three weeks. Second, the integration of these vectors into the host genome can have deleterious effects. Third, although expression of the exogenous reprogramming factors is eventually silenced during iPS cell generation, there is a significant risk of tumorigenesis if these exogenous genes are inadvertently reactivated. In particular, c-Myc and Klf4 are known oncogenes, and a high incidence of cancer has been observed in chimeric mice derived from germline-competent iPS cells (129).
While several genetic methods have been employed to bypass these issues (130−132), the possibility of using small molecules to increase the efficiency of iPS cell generation or to functionally replace one or more reprogramming factors has been explored by a number of laboratories. Compounds that promote chromatin remodeling have been of particular interest because of the critical roles of DNA and histone modifications in establishing and maintaining differentiated cell fates. For example, the HDAC inhibitor valproic acid has been reported to increase four-factor reprogramming efficiency by nearly 400-fold and iPS cell clones can even be obtained from valproic acid-treated human fibroblasts transduced with only Oct4 and Sox2 (133). Recent studies further suggest that blocking the methylation of DNA or histones can similarly facilitate cell dedifferentiation. When applied at the appropriate time during somatic cell reprogramming, the DNA methyltransferase inhibitor 5-azacytidine can improve the efficiency of iPS cell generation by approximately 100-fold (134). Similarly, using the G9a histone methyltransferase inhibitor BIX-01294 and the DNA methyltransferase inhibitor N-phthalyl-tryptophan (also known as RG108) in combination has been reported to promote the dedifferentiation of mouse embryonic fibroblasts transduced with only Oct4 and Klf4 (135).
More recently, compounds that target upstream signaling pathways have been pursued as an alternative strategy for optimizing iPS cell induction. In a large-scale, high-throughput screen for small molecules that can functionally replace Klf4, it was determined that the benzazepinone kenpaullone can work with Oct4, Sox2, and c-Myc to convert murine embryonic fibroblasts into iPS cells, albeit at a lower efficiency than that observed with the complete set of reprogramming factors (136). The mechanism by which this compound promotes cell dedifferentiation, however, is unknown and will likely be difficult to decipher. Kenpaullone can inhibit GSK3 and several cyclin-dependent kinases, and short-hairpin RNA-mediated silencing of these kinases, either individually or in combination, does not increase the expression of ESC markers. These observations suggest kenpaullone may act through a novel and perhaps complex mechanism.
Approaches that specifically target developmental signaling pathways have been successful as well. For example, TGFβ pathway antagonists such as SB-431542 can increase the efficiency of four-factor reprogramming of murine fibroblasts by 30-fold and permit their conversion into iPS cells using three-factor protocols that lack either Sox2 or c-Myc (137). TGFβ signaling blockade can also reduce the time required for efficient cell reprogramming from weeks to days. While inhibiting TGFβ pathway activation alone does not improve the efficiency of human iPS cell induction (137), it has been reported that using SB-431542 and the MEK inhibitor PD0325901 in combination can improve the efficiency and rate of this process by 100- and 4-fold, respectively (138). Presumably these compounds activate proliferative and/or inhibit pro-differentiation signaling mechanisms, but the precise mechanisms by which they facilitate somatic cell reprogramming are unclear. Moreover, since TGFβ and FGF signaling has been found to promote human ESC self-renewal (119), the duration of SB-431542 and PD0325901 treatment may influence the utility of this approach.
Concluding Remarks
The demand for small molecules that can control cell fate continues to grow. In part, this need arises from the dynamic and combinatorial manner in which signaling pathways control cell pluripotency and differentiation state, mechanisms that can be difficult to recapitulate with current genetic technologies but can be approximated through the application of one or more chemical reagents at specific times, durations, and doses. Chemical modulators of cell fate similarly offer certain advantages over biologic approaches for the treatment of human diseases and disorders. Small molecules can readily interact with intracellular signaling molecules that might be inaccessible to protein-based therapies, and the ability of compounds to simultaneously target homologous proteins can be beneficial in certain therapeutic contexts. From a practical point of view, there also are well-established procedures for producing, testing, and administering pharmaceutical-grade compounds, while safe practice guidelines for virus- and cell-based therapies and other biologic approaches are still under development.
Yet despite this potential, the spectrum of compounds that can be used to study and therapeutically manipulate cell fate choice is still narrow. For example, the Hh, Wnt, TGFβ, FGF, and Notch pathways are each regulated by an ensemble of signaling proteins, but in most cases only one or two of these factors have known small-molecule modulators. Smo is the most common pharmacological target within the Hh pathway, Wnt pathway modulators predominantly abrogate tankyrase and GSK3 activities, all known inhibitors of the TGFβ superfamily block type I receptor kinase activity, and all but one of the reported Notch pathway antagonists target the γ-secretase protein complex. This lack of mechanistic breadth limits the range of biological systems that can be pharmacologically controlled, particularly with respect to disease states caused by mutations in specific developmental pathway components. Moreover, tankyrase, GSK3, and γ-secretase functions are not restricted to Wnt or Notch pathway regulation, and inhibitors of these enzymatic activities will likely perturb multiple signaling processes in vivo. Whether this targeting bias is primarily due to the inherent small-molecule sensitivity of individual signaling proteins versus the limited structural diversity of current chemical libraries is not clear. It is likely, however, that expanding our repertoire of chemical probes will require both new assays of protein function and compound collections with greater molecular complexity. Small molecules that selectively inhibit Hh protein biogenesis or Ptch1 activity, regulate Fzd receptor function, activate TGFβ signaling pathways, actuate FGFRs, or abrogate NICD/CSL function, to name a few examples, would be especially desirable.
The development of small molecules that can modulate cell self-renewal, differentiation, and reprogramming through other cellular targets is also in a nascent stage. Although a few of these compounds have known biological activities, the mechanisms by which they influence cellular states are not well understood. Indeed, the ability of these small molecules to perturb multiple signaling proteins may be an essential aspect of their cellular phenotypes. Valproic acid, BIX-01294, 5-azacytidine, N-phthalyl-tryptophan, and other chemical reagents that alter chromatin structure affect the expression of hundreds if not thousands of genes, and it is possible that pluripotin, IDE1/IDE2, indolactam V, and kenpaullone also act through complex mechanisms. The wide-ranging effects of these compounds on signaling pathways and gene transcription make it challenging to decipher how they influence stem cell biology. It is also uncertain how broadly these compounds can be applied to induce or maintain specific cell fates.
Reliably and efficiently controlling cell fate with chemical reagents will likely require a more deliberate approach, using compounds that rival endogenous factors with respect to potency and selectivity. While phenotypic screens will continue to play an important role in these efforts, identifying new compounds that can modulate cell pluripotency and differentiation in certain contexts is only a starting point. Establishing their mechanisms of action with biochemical and genetic rigor and thoroughly investigating their applicability to other biological systems is essential for maximizing the impact of these small molecules at the laboratory bench and in the clinic. In addition, new strategies for the discovery of cell fate regulators, including more targeted approaches, will undoubtedly arise as we gain new insights into stem cell biology. For example, future compounds could target cellular mechanisms that link developmental signaling pathways to pluripotency factor networks and differentiation programs, regulate cellular and genomic responsiveness to differentiation-inducing signals, or alter cell cycle dynamics to favor pluripotent or committed states. Chemical biologists therefore have an opportunity to make unique contributions to our understanding of how pluripotent cells self-renew and differentiate, as well as our ability to control these processes for therapeutic purposes. Through these efforts, we may someday have pharmacological agents that help remediate vision or hearing loss, spinal cord injuries, cardiac ischemic damage, neurodegenerative diseases, and malignant tumors. Realizing these possibilities is an exciting prospect, a fate that the chemistry community should not leave entirely to chance.
Acknowledgments
We thank C. Cho, M. Wernig, and J. Mich for helpful discussions and acknowledge financial support from the National Institutes of Health Director’s Pioneer Award (DP1 OD003792) and the NIH/NCI (R01 CA136574).
Funding Statement
National Institutes of Health, United States
References
- Vickaryous M. K.; Hall B. K. (2006) Human cell type diversity, evolution, development, and classification with special reference to cells derived from the neural crest. Biol. Rev. Cambridge Philos. Soc. 81, 425–455. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Hui C.-c. (2008) Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald B. T.; Tamai K.; He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. Y.; Hill C. S. (2009) Tgf-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 16, 329–343. [DOI] [PubMed] [Google Scholar]
- Beenken A.; Mohammadi M. (2009) The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discovery 8, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolós V.; Grego-Bessa J.; de la Pompa J. L. (2007) Notch signaling in Development and cancer. Endocr. Rev. 28, 339–363. [DOI] [PubMed] [Google Scholar]
- Massagué J. (2008) TGFβ in cancer. Cell 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schugar R. C.; Robbins P. D.; Deasy B. M. (2008) Small molecules in stem cell self-renewal and differentiation. Gene Ther. 15, 126–135. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Shi Y.; Ding S. (2008) A chemical approach to stem-cell biology and regenerative medicine. Nature 453, 338–344. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R. J.; Scott M. P. (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22, 103–114. [DOI] [PubMed] [Google Scholar]
- Levy V.; Lindon C.; Harfe B. D.; Morgan B. A. (2005) Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell 9, 855–861. [DOI] [PubMed] [Google Scholar]
- Roelink H.; Augsburger A.; Heemskerk J.; Korzh V.; Norlin S.; Ruiz i Altaba A.; Tanabe Y.; Placzek M.; Edlund T.; Jessell T. M.; et al. (1994) Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell 76, 761–775. [DOI] [PubMed] [Google Scholar]
- Litingtung Y.; Dahn R. D.; Li Y.; Fallon J. F.; Chiang C. (2002) Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 418, 979–983. [DOI] [PubMed] [Google Scholar]
- Neumann C. J.; Nuesslein-Volhard C. (2000) Patterning of the zebrafish retina by a wave of sonic hedgehog activity. Science 289, 2137–2139. [DOI] [PubMed] [Google Scholar]
- Canto P.; Soderlund D.; Reyes E.; Mendez J. P. (2004) Mutations in the desert hedgehog (DHH) gene in patients with 46, XY complete pure gonadal dysgenesis. J. Clin. Endocrinol. Metab. 89, 4480–4483. [DOI] [PubMed] [Google Scholar]
- Belloni E.; Muenke M.; Roessler E.; Traverso G.; Siegel-Bartelt J.; Frumkin A.; Mitchell H. F.; Donis-Keller H.; Helms C.; Hing A. V.; Heng H. H.; Koop B.; Martindale D.; Rommens J. M.; Tsui L. C.; Scherer S. W. (1996) Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 14, 353–356. [DOI] [PubMed] [Google Scholar]
- Roessler E.; Du Y. Z.; Mullor J. L.; Casas E.; Allen W. P.; Gillessen-Kaesbach G.; Roeder E. R.; Ming J. E.; Ruiz i Altaba A.; Muenke M. (2003) Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. U.S.A. 100, 13424–13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming J. E.; Kaupas M. E.; Roessler E.; Brunner H. G.; Golabi M.; Tekin M.; Stratton R. F.; Sujansky E.; Bale S. J.; Muenke M. (2002) Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum. Genet. 110, 297–301. [DOI] [PubMed] [Google Scholar]
- Radhakrishna U.; Wild A.; Grzeschik K. H.; Antonarakis S. E. (1997) Mutation in GLI3 in postaxial polydactyly type A. Nat. Genet. 17, 269–271. [DOI] [PubMed] [Google Scholar]
- Xie J.; Murone M.; Luoh S. M.; Ryan A.; Gu Q.; Zhang C.; Bonifas J. M.; Lam C. W.; Hynes M.; Goddard A.; Rosenthal A.; Epstein E. H. Jr.; de Sauvage F. J. (1998) Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92. [DOI] [PubMed] [Google Scholar]
- Johnson R. L.; Rothman A. L.; Xie J.; Goodrich L. V.; Bare J. W.; Bonifas J. M.; Quinn A. G.; Myers R. M.; Cox D. R.; Epstein E. H. Jr.; Scott M. P. (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272, 1668–1671. [DOI] [PubMed] [Google Scholar]
- Taylor M. D.; Liu L.; Raffel C.; Hui C. C.; Mainprize T. G.; Zhang X.; Agatep R.; Chiappa S.; Gao L.; Lowrance A.; Hao A.; Goldstein A. M.; Stavrou T.; Scherer S. W.; Dura W. T.; Wainwright B.; Squire J. A.; Rutka J. T.; Hogg D. (2002) Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 31, 306–310. [DOI] [PubMed] [Google Scholar]
- Berman D. M.; Karhadkar S. S.; Maitra A.; Montes De Oca R.; Gerstenblith M. R.; Briggs K.; Parker A. R.; Shimada Y.; Eshleman J. R.; Watkins D. N.; Beachy P. A. (2003) Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425, 846–851. [DOI] [PubMed] [Google Scholar]
- Thayer S. P.; di Magliano M. P.; Heiser P. W.; Nielsen C. M.; Roberts D. J.; Lauwers G. Y.; Qi Y. P.; Gysin S.; Fernández-del Castillo C.; Yajnik V.; Antoniu B.; McMahon M.; Warshaw A. L.; Hebrok M. (2003) Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns W.; Thacker E. J.; James L. F.; Huffman W. T. (1959) A congenital cyclopiantype malformation in lambs. J. Am. Vet. Med. Assoc. 134, 180–183. [PubMed] [Google Scholar]
- Chen J. K.; Taipale J.; Cooper M. K.; Beachy P. A. (2002) Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 16, 2743–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. K.; Taipale J.; Young K. E.; Maiti T.; Beachy P. A. (2002) Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. U.S.A. 99, 14071–14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff D.; Lorusso P.; Rudin C.; Reddy J.; Yauch R.; Tibes R.; Weiss G.; Borad M.; Hann C.; Brahmer J.; Mackey H.; Lum B.; Darbonne W.; Marsters J.; de Sauvage F.; Low J. (2009) Inhibition of the Hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. . [DOI] [PubMed] [Google Scholar]
- Tremblay M. R.; Lescarbeau A.; Grogan M. J.; Tan E.; Lin G.; Austad B. C.; Yu L. C.; Behnke M. L.; Nair S. J.; Hagel M.; White K.; Conley J.; Manna J. D.; Alvarez-Diez T. M.; Hoyt J.; Woodward C. N.; Sydor J. R.; Pink M.; MacDougall J.; Campbell M. J.; Cushing J.; Ferguson J.; Curtis M. S.; McGovern K.; Read M. A.; Palombella V. J.; Adams J.; Castro A. C. (2009) Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 52, 4400–4418. [DOI] [PubMed] [Google Scholar]
- Miller-Moslin K.; Peukert S.; Jain R. K.; McEwan M. A.; Karki R.; Llamas L.; Yusuff N.; He F.; Li Y.; Sun Y.; Dai M.; Perez L.; Michael W.; Sheng T.; Lei H.; Zhang R.; Williams J.; Bourret A.; Ramamurthy A.; Yuan J.; Guo R.; Matsumoto M.; Vattay A.; Maniara W.; Amaral A.; Dorsch M.; Kelleher J. F., 3rd (2009) 1-Amino-4-benzylphthalazines as orally bioavailable smoothened antagonists with antitumor activity. J. Med. Chem. 52, 3954–3968. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M.; Zhang X. M.; Bottega S.; Guicherit O.; Wichterle H.; Dudek H.; Bumcrot D.; Wang F. Y.; Jones S.; Shulok J.; Rubin L. L.; Porter J. A. (2002) Small-molecule modulators of Hedgehog signaling: identification and characterization of Smoothened agonists and antagonists. J. Biol. 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S.; Chen J. K. (2006) Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2, 29–30. [DOI] [PubMed] [Google Scholar]
- Wu X.; Ding S.; Ding Q.; Gray N. S.; Schultz P. G. (2002) A small molecule with osteogenesis-inducing activity in multipotent mesenchymal progenitor cells. J. Am. Chem. Soc. 124, 14520–14521. [DOI] [PubMed] [Google Scholar]
- Stanton B. Z.; Peng L. F.; Maloof N.; Nakai K.; Wang X.; Duffner J. L.; Taveras K. M.; Hyman J. M.; Lee S. W.; Koehler A. N.; Chen J. K.; Fox J. L.; Mandinova A.; Schreiber S. L. (2009) A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 5, 154–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth M.; Bergström A.; Shimokawa T.; Toftgård R. (2007) Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. U.S.A. 104, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya T.; Arai M. A.; Koyano T.; Kowithayakorn T.; Ishibashi M. (2008) Naturally occurring small-molecule inhibitors of hedgehog/GLI-mediated transcription. ChemBioChem 9, 1082–1092. [DOI] [PubMed] [Google Scholar]
- Hyman J. M.; Firestone A. J.; Heine V. M.; Zhao Y.; Ocasio C. A.; Han K.; Sun M.; Rack P. G.; Sinha S.; Wu J. J.; Solow-Cordero D. E.; Jiang J.; Rowitch D. H.; Chen J. K. (2009) Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. U.S.A. 106, 14132–14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bambakidis N. C.; Horn E. M.; Nakaji P.; Theodore N.; Bless E.; Dellovade T.; Ma C.; Wang X.; Preul M. C.; Coons S. W.; Spetzler R. F.; Sonntag V. K. (2009) Endogenous stem cell proliferation induced by intravenous hedgehog agonist administration after contusion in the adult rat spinal cord. J. Neurosurg. Spine 10, 171–176. [DOI] [PubMed] [Google Scholar]
- Soundararajan P.; Miles G. B.; Rubin L. L.; Brownstone R. M.; Rafuse V. F. (2006) Motoneurons derived from embryonic stem cells express transcription factors and develop phenotypes characteristic of medial motor column neurons. J. Neurosci. 26, 3256–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladini R. D.; Saleh J.; Qian C.; Xu G.-X.; Rubin L. L. (2005) Modulation of hair growth with small molecule agonists of the hedgehog signaling pathway. J. Invest. Dermatol. 125, 638–646. [DOI] [PubMed] [Google Scholar]
- Berman D. M.; Karhadkar S. S.; Hallahan A. R.; Pritchard J. I.; Eberhart C. G.; Watkins D. N.; Chen J. K.; Cooper M. K.; Taipale J.; Olson J. M.; Beachy P. A. (2002) Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 297, 1559–1561. [DOI] [PubMed] [Google Scholar]
- Yauch R. L.; Gould S. E.; Scales S. J.; Tang T.; Tian H.; Ahn C. P.; Marshall D.; Fu L.; Januario T.; Kallop D.; Nannini-Pepe M.; Kotkow K.; Marsters J. C.; Rubin L. L.; de Sauvage F. J. (2008) A paracrine requirement for hedgehog signalling in cancer. Nature 455, 406–410. [DOI] [PubMed] [Google Scholar]
- Yauch R.; Dijkgraaf G.; Alicke B.; Januario T.; Ahn C.; Holcomb T.; Pujara K.; Stinson J.; Callahan C.; Tang T.; Bazan J.; Kan Z.; Seshagiri S.; Hann C.; Gould S.; Low J.; Rudin C.; de Sauvage F. (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326, 572–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M.; Sancho E.; Verweij C.; de Lau W.; Oving I.; Hurlstone A.; van der Horn K.; Batlle E.; Coudreuse D.; Haramis A. P.; Tjon-Pon-Fong M.; Moerer P.; van den Born M.; Soete G.; Pals S.; Eilers M.; Medema R.; Clevers H. (2002) The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111, 241–250. [DOI] [PubMed] [Google Scholar]
- Austin T. W.; Solar G. P.; Ziegler F. C.; Liem L.; Matthews W. (1997) A role for the Wnt gene family in hematopoiesis: expansion of multilineage progenitor cells. Blood 89, 3624–3635. [PubMed] [Google Scholar]
- Dunn K. J.; Williams B. O.; Li Y.; Pavan W. J. (2000) Neural crest-directed gene transfer demonstrates Wnt1 role in melanocyte expansion and differentiation during mouse development. Proc. Natl. Acad. Sci. U.S.A. 97, 10050–10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell S. M.; Zilz N.; Beazer-Barclay Y.; Bryan T. M.; Hamilton S. R.; Thibodeau S. N.; Vogelstein B.; Kinzler K. W. (1992) APC mutations occur early during colorectal tumorigenesis. Nature 359, 235–237. [DOI] [PubMed] [Google Scholar]
- Liu W.; Dong X.; Mai M.; Seelan R. S.; Taniguchi K.; Krishnadath K. K.; Halling K. C.; Cunningham J. M.; Boardman L. A.; Qian C.; Christensen E.; Schmidt S. S.; Roche P. C.; Smith D. I.; Thibodeau S. N. (2000) Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating β-catenin/TCF signalling. Nat. Genet. 26, 146–147. [DOI] [PubMed] [Google Scholar]
- Morin P. J.; Sparks A. B.; Korinek V.; Barker N.; Clevers H.; Vogelstein B.; Kinzler K. W. (1997) Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- Satoh S.; Daigo Y.; Furukawa Y.; Kato T.; Miwa N.; Nishiwaki T.; Kawasoe T.; Ishiguro H.; Fujita M.; Tokino T.; Sasaki Y.; Imaoka S.; Murata M.; Shimano T.; Yamaoka Y.; Nakamura Y. (2000) AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 24, 245–250. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B.; Robbins P.; El-Gamil M.; Albert I.; Porfiri E.; Polakis P. (1997) Stabilization of β-catenin by genetic defects in melanoma cell lines. Science 275, 1790–1792. [DOI] [PubMed] [Google Scholar]
- Jiao X.; Ventruto V.; Trese M. T.; Shastry B. S.; Hejtmancik J. F. (2004) Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am. J. Hum. Genet. 75, 878–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille J.; Macdonald M. L.; Kaykas A.; Sheldahl L. C.; Zeisler J.; Dube M. P.; Zhang L. H.; Singaraja R. R.; Guernsey D. L.; Zheng B.; Siebert L. F.; Hoskin-Mott A.; Trese M. T.; Pimstone S. N.; Shastry B. S.; Moon R. T.; Hayden M. R.; Goldberg Y. P.; Samuels M. E. (2002) Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 32, 326–330. [DOI] [PubMed] [Google Scholar]
- Woods C. G.; Stricker S.; Seemann P.; Stern R.; Cox J.; Sherridan E.; Roberts E.; Springell K.; Scott S.; Karbani G.; Sharif S. M.; Toomes C.; Bond J.; Kumar D.; Al-Gazali L.; Mundlos S. (2006) Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am. J. Hum. Genet. 79, 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Dodge M. E.; Tang W.; Lu J.; Ma Z.; Fan C.-W.; Wei S.; Hao W.; Kilgore J.; Williams N. S.; Roth M. G.; Amatruda J. F.; Chen C.; Lum L. (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.-M. A.; Mishina Y. M.; Liu S.; Cheung A.; Stegmeier F.; Michaud G. A.; Charlat O.; Wiellette E.; Zhang Y.; Wiessner S.; Hild M.; Shi X.; Wilson C. J.; Mickanin C.; Myer V.; Fazal A.; Tomlinson R.; Serluca F.; Shao W.; Cheng H.; Shultz M.; Rau C.; Schirle M.; Schlegl J.; Ghidelli S.; Fawell S.; Lu C.; Curtis D.; Kirschner M. W.; Lengauer C.; Finan P. M.; Tallarico J. A.; Bouwmeester T.; Porter J. A.; Bauer A.; Cong F. (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620. [DOI] [PubMed] [Google Scholar]
- Meijer L.; Skaltsounis A. L.; Magiatis P.; Polychronopoulos P.; Knockaert M.; Leost M.; Ryan X. P.; Vonica C. A.; Brivanlou A.; Dajani R.; Crovace C.; Tarricone C.; Musacchio A.; Roe S. M.; Pearl L.; Greengard P. (2003) GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 10, 1255–1266. [DOI] [PubMed] [Google Scholar]
- Kulkarni N. H.; Onyia J. E.; Zeng Q.; Tian X.; Liu M.; Halladay D. L.; Frolik C. A.; Engler T.; Wei T.; Kriauciunas A.; Martin T. J.; Sato M.; Bryant H. U.; Ma Y. L. (2006) Orally bioavailable GSK-3α/β dual inhibitor increases markers of cellular differentiation in vitro and bone mass in vivo. J. Bone Miner. Res. 21, 910–920. [DOI] [PubMed] [Google Scholar]
- North T. E.; Goessling W.; Walkley C. R.; Lengerke C.; Kopani K. R.; Lord A. M.; Weber G. J.; Bowman T. V.; Jang I.-H.; Grosser T.; Fitzgerald G. A.; Daley G. Q.; Orkin S. H.; Zon L. I. (2007) Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 447, 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepourcelet M.; Chen Y.-N. P.; France D. S.; Wang H.; Crews P.; Petersen F.; Bruseo C.; Wood A. W.; Shivdasani R. A. (2004) Small-molecule antagonists of the oncogenic Tcf/β-catenin protein complex. Cancer Cell 5, 91–102. [DOI] [PubMed] [Google Scholar]
- Sato N.; Meijer L.; Skaltsounis L.; Greengard P.; Brivanlou A. H. (2004) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 10, 55–63. [DOI] [PubMed] [Google Scholar]
- Castelo-Branco G.; Rawal N.; Arenas E. (2004) GSK-3β inhibition/β-catenin stabilization in ventral midbrain precursors increases differentiation into dopamine neurons. J. Cell Sci. 117, 5731–5737. [DOI] [PubMed] [Google Scholar]
- Goessling W.; North T. E.; Loewer S.; Lord A. M.; Lee S.; Stoick-Cooper C. L.; Weidinger G.; Puder M.; Daley G. Q.; Moon R. T.; Zon L. I. (2009) Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 136, 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhdeo K.; Mani M.; Zhang Y.; Dutta J.; Yasui H.; Rooney M. D.; Carrasco D. E.; Zheng M.; He H.; Tai Y. T.; Mitsiades C.; Anderson K. C.; Carrasco D. R. (2007) Targeting the β-catenin/TCF transcriptional complex in the treatment of multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 104, 7516–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozney J. M.; Rosen V.; Celeste A. J.; Mitsock L. M.; Whitters M. J.; Kriz R. W.; Hewick R. M.; Wang E. A. (1988) Novel regulators of bone formation: molecular clones and activities. Science 242, 1528–1534. [DOI] [PubMed] [Google Scholar]
- Jones C. M.; Kuehn M. R.; Hogan B. L.; Smith J. C.; Wright C. V. (1995) Nodal-related signals induce axial mesoderm and dorsalize mesoderm during gastrulation. Development 121, 3651–3662. [DOI] [PubMed] [Google Scholar]
- Howe J. R.; Bair J. L.; Sayed M. G.; Anderson M. E.; Mitros F. A.; Petersen G. M.; Velculescu V. E.; Traverso G.; Vogelstein B. (2001) Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 28, 184–187. [DOI] [PubMed] [Google Scholar]
- Markowitz S.; Wang J.; Myeroff L.; Parsons R.; Sun L.; Lutterbaugh J.; Fan R. S.; Zborowska E.; Kinzler K. W.; Vogelstein B.; et al. (1995) Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 268, 1336–1338. [DOI] [PubMed] [Google Scholar]
- Pasche B.; Knobloch T. J.; Bian Y.; Liu J.; Phukan S.; Rosman D.; Kaklamani V.; Baddi L.; Siddiqui F. S.; Frankel W.; Prior T. W.; Schuller D. E.; Agrawal A.; Lang J.; Dolan M. E.; Vokes E. E.; Lane W. S.; Huang C. C.; Caldes T.; Di Cristofano A.; Hampel H.; Nilsson I.; von Heijne G.; Fodde R.; Murty V. V.; de la Chapelle A.; Weghorst C. M. (2005) Somatic acquisition and signaling of TGFBR1*6A in cancer. J. Am. Med. Assoc. 294, 1634–1646. [DOI] [PubMed] [Google Scholar]
- Schutte M.; Hruban R. H.; Hedrick L.; Cho K. R.; Nadasdy G. M.; Weinstein C. L.; Bova G. S.; Isaacs W. B.; Cairns P.; Nawroz H.; Sidransky D.; Casero R. A. Jr.; Meltzer P. S.; Hahn S. A.; Kern S. E. (1996) DPC4 gene in various tumor types. Cancer Res. 56, 2527–2530. [PubMed] [Google Scholar]
- Faiyaz-Ul-Haque M.; Ahmad W.; Zaidi S. H.; Haque S.; Teebi A. S.; Ahmad M.; Cohn D. H.; Tsui L. C. (2002) Mutation in the cartilage-derived morphogenetic protein-1 (CDMP1) gene in a kindred affected with fibular hypoplasia and complex brachydactyly (DuPan syndrome). Clin. Genet. 61, 454–458. [DOI] [PubMed] [Google Scholar]
- Johnson D. W.; Berg J. N.; Baldwin M. A.; Gallione C. J.; Marondel I.; Yoon S. J.; Stenzel T. T.; Speer M.; Pericak-Vance M. A.; Diamond A.; Guttmacher A. E.; Jackson C. E.; Attisano L.; Kucherlapati R.; Porteous M. E.; Marchuk D. A. (1996) Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 13, 189–195. [DOI] [PubMed] [Google Scholar]
- Lane K. B.; Machado R. D.; Pauciulo M. W.; Thomson J. R.; Phillips J. A. 3rd; Loyd J. E.; Nichols W. C.; Trembath R. C. (2000) Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 26, 81–84. [DOI] [PubMed] [Google Scholar]
- Dalal B. I.; Keown P. A.; Greenberg A. H. (1993) Immunocytochemical localization of secreted transforming growth factor-β 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am. J. Pathol. 143, 381–389. [PMC free article] [PubMed] [Google Scholar]
- Bruna A.; Darken R. S.; Rojo F.; Ocana A.; Penuelas S.; Arias A.; Paris R.; Tortosa A.; Mora J.; Baselga J.; Seoane J. (2007) High TGFβ-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–160. [DOI] [PubMed] [Google Scholar]
- Inman G. J.; Nicolás F. J.; Callahan J. F.; Harling J. D.; Gaster L. M.; Reith A. D.; Laping N. J.; Hill C. S. (2002) SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62, 65–74. [DOI] [PubMed] [Google Scholar]
- Uhl M.; Aulwurm S.; Wischhusen J.; Weiler M.; Ma J. Y.; Almirez R.; Mangadu R.; Liu Y.-W.; Platten M.; Herrlinger U.; Murphy A.; Wong D. H.; Wick W.; Higgins L. S.; Weller M. (2004) SD-208, a novel transforming growth factor β receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 64, 7954–7961. [DOI] [PubMed] [Google Scholar]
- Yu P. B.; Hong C. C.; Sachidanandan C.; Babitt J. L.; Deng D. Y.; Hoyng S. A.; Lin H. Y.; Bloch K. D.; Peterson R. T. (2008) Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 4, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J.; Daleo M. A.; Murphy C. K.; Yu P. B.; Ho J. N.; Hu J.; Peterson R. T.; Hatzopoulos A. K.; Hong C. C. (2008) Dorsomorphin, a selective small molecule inhibitor of BMP signaling, promotes cardiomyogenesis in embryonic stem cells. PLoS ONE 3, e2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Nguyen A. N.; Sohal D.; Ying Ma J.; Pahanish P.; Gundabolu K.; Hayman J.; Chubak A.; Mo Y.; Bhagat T. D.; Das B.; Kapoun A. M.; Navas T. A.; Parmar S.; Kambhampati S.; Pellagatti A.; Braunchweig I.; Zhang Y.; Wickrema A.; Medicherla S.; Boultwood J.; Platanias L. C.; Higgins L. S.; List A. F.; Bitzer M.; Verma A. (2008) Inhibition of the TGF-β receptor I kinase promotes hematopoiesis in MDS. Blood 112, 3434–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon J.-A.; Kim H.-T.; Cho I.-S.; Sheen Y. Y.; Kim D.-K. (2006) IN-1130, a novel transforming growth factor-β type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 70, 1234–1243. [DOI] [PubMed] [Google Scholar]
- Kimelman D.; Kirschner M. (1987) Synergistic induction of mesoderm by FGF and TGF-β and the identification of an mRNA coding for FGF in the early Xenopus embryo. Cell 51, 869–877. [DOI] [PubMed] [Google Scholar]
- Hebert J. M.; Rosenquist T.; Gotz J.; Martin G. R. (1994) FGF5 as a regulator of the hair growth cycle: evidence from targeted and spontaneous mutations. Cell 78, 1017–1025. [DOI] [PubMed] [Google Scholar]
- Reardon W.; Winter R. M.; Rutland P.; Pulleyn L. J.; Jones B. M.; Malcolm S. (1994) Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat. Genet. 8, 98–103. [DOI] [PubMed] [Google Scholar]
- Shiang R.; Thompson L. M.; Zhu Y. Z.; Church D. M.; Fielder T. J.; Bocian M.; Winokur S. T.; Wasmuth J. J. (1994) Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78, 335–342. [DOI] [PubMed] [Google Scholar]
- Pitteloud N.; Acierno J. S. Jr.; Meysing A.; Eliseenkova A. V.; Ma J.; Ibrahimi O. A.; Metzger D. L.; Hayes F. J.; Dwyer A. A.; Hughes V. A.; Yialamas M.; Hall J. E.; Grant E.; Mohammadi M.; Crowley W. F. Jr. (2006) Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. U. S. A. 103, 6281–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi M.; Nardini E.; Brents L. A.; Schrock E.; Ried T.; Kuehl W. M.; Bergsagel P. L. (1997) Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 16, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D.; De Oliveira C.; Ricol D.; de Medina S.; Bourdin J.; Sastre-Garau X.; Chopin D.; Thiery J. P.; Radvanyi F. (1999) Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat. Genet. 23, 18–20. [DOI] [PubMed] [Google Scholar]
- Tekin M.; Hismi B. O.; Fitoz S.; Ozdag H.; Cengiz F. B.; Sirmaci A.; Aslan I.; Inceoglu B.; Yuksel-Konuk E. B.; Yilmaz S. T.; Yasun O.; Akar N. (2007) Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am. J. Hum. Genet. 80, 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entesarian M.; Dahlqvist J.; Shashi V.; Stanley C. S.; Falahat B.; Reardon W.; Dahl N. (2007) FGF10 missense mutations in aplasia of lacrimal and salivary glands (ALSG). Eur. J. Hum. Genet. 15, 379–382. [DOI] [PubMed] [Google Scholar]
- Muenke M.; Schell U.; Hehr A.; Robin N. H.; Losken H. W.; Schinzel A.; Pulleyn L. J.; Rutland P.; Reardon W.; Malcolm S.; et al. (1994) A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat. Genet. 8, 269–274. [DOI] [PubMed] [Google Scholar]
- Dutt A.; Salvesen H. B.; Chen T. H.; Ramos A. H.; Onofrio R. C.; Hatton C.; Nicoletti R.; Winckler W.; Grewal R.; Hanna M.; Wyhs N.; Ziaugra L.; Richter D. J.; Trovik J.; Engelsen I. B.; Stefansson I. M.; Fennell T.; Cibulskis K.; Zody M. C.; Akslen L. A.; Gabriel S.; Wong K. K.; Sellers W. R.; Meyerson M.; Greulich H. (2008) Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc. Natl. Acad. Sci. U.S.A. 105, 8713–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M.; McMahon G.; Sun L.; Tang C.; Hirth P.; Yeh B. K.; Hubbard S. R.; Schlessinger J. (1997) Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 276, 955–960. [DOI] [PubMed] [Google Scholar]
- Mohammadi M.; Froum S.; Hamby J. M.; Schroeder M. C.; Panek R. L.; Lu G. H.; Eliseenkova A. V.; Green D.; Schlessinger J.; Hubbard S. R. (1998) Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 17, 5896–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grand E. K.; Chase A. J.; Heath C.; Rahemtulla A.; Cross N. C. (2004) Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia 18, 962–966. [DOI] [PubMed] [Google Scholar]
- Pardo O. E.; Latigo J.; Jeffery R. E.; Nye E.; Poulsom R.; Spencer-Dene B.; Lemoine N. R.; Stamp G. W.; Aboagye E. O.; Seckl M. J. (2009) The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 69, 8645–8651. [DOI] [PubMed] [Google Scholar]
- Lyons J. F.; Wilhelm S.; Hibner B.; Bollag G. (2001) Discovery of a novel Raf kinase inhibitor. Endocr. Relat. Cancer 8, 219–225. [DOI] [PubMed] [Google Scholar]
- Rinehart J.; Adjei A. A.; Lorusso P. M.; Waterhouse D.; Hecht J. R.; Natale R. B.; Hamid O.; Varterasian M.; Asbury P.; Kaldjian E. P.; Gulyas S.; Mitchell D. Y.; Herrera R.; Sebolt-Leopold J. S.; Meyer M. B. (2004) Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 22, 4456–4462. [DOI] [PubMed] [Google Scholar]
- Molina G.; Vogt A.; Bakan A.; Dai W.; Queiroz de Oliveira P.; Znosko W.; Smithgall T. E.; Bahar I.; Lazo J. S.; Day B. W.; Tsang M. (2009) Zebrafish chemical screening reveals an inhibitor of Dusp6 that expands cardiac cell lineages. Nat. Chem. Biol. 5, 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum-Finney B.; Xu L.; Brashem-Stein C.; Nourigat C.; Flowers D.; Bakkour S.; Pear W. S.; Bernstein I. D. (2000) Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat. Med. 6, 1278–1281. [DOI] [PubMed] [Google Scholar]
- Hitoshi S.; Alexson T.; Tropepe V.; Donoviel D.; Elia A. J.; Nye J. S.; Conlon R. A.; Mak T. W.; Bernstein A.; van der Kooy D. (2002) Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 16, 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford P. J.; Lan Y.; Jiang R.; Lindsell C.; Weinmaster G.; Gridley T.; Kelley M. W. (1999) Notch signalling pathway mediates hair cell development in mammalian cochlea. Nat. Genet. 21, 289–292. [DOI] [PubMed] [Google Scholar]
- Conlon R. A.; Reaume A. G.; Rossant J. (1995) Notch1 is required for the coordinate segmentation of somites. Development 121, 1533–1545. [DOI] [PubMed] [Google Scholar]
- Lawson N. D.; Scheer N.; Pham V. N.; Kim C. H.; Chitnis A. B.; Campos-Ortega J. A.; Weinstein B. M. (2001) Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128, 3675–3683. [DOI] [PubMed] [Google Scholar]
- McDaniell R.; Warthen D. M.; Sanchez-Lara P. A.; Pai A.; Krantz I. D.; Piccoli D. A.; Spinner N. B. (2006) NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 79, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Krantz I. D.; Deng Y.; Genin A.; Banta A. B.; Collins C. C.; Qi M.; Trask B. J.; Kuo W. L.; Cochran J.; Costa T.; Pierpont M. E.; Rand E. B.; Piccoli D. A.; Hood L.; Spinner N. B. (1997) Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat. Genet. 16, 243–251. [DOI] [PubMed] [Google Scholar]
- Oda T.; Elkahloun A. G.; Pike B. L.; Okajima K.; Krantz I. D.; Genin A.; Piccoli D. A.; Meltzer P. S.; Spinner N. B.; Collins F. S.; Chandrasekharappa S. C. (1997) Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet. 16, 235–242. [DOI] [PubMed] [Google Scholar]
- Joutel A.; Corpechot C.; Ducros A.; Vahedi K.; Chabriat H.; Mouton P.; Alamowitch S.; Domenga V.; Cecillion M.; Marechal E.; Maciazek J.; Vayssiere C.; Cruaud C.; Cabanis E. A.; Ruchoux M. M.; Weissenbach J.; Bach J. F.; Bousser M. G.; Tournier-Lasserve E. (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383, 707–710. [DOI] [PubMed] [Google Scholar]
- Bulman M. P.; Kusumi K.; Frayling T. M.; McKeown C.; Garrett C.; Lander E. S.; Krumlauf R.; Hattersley A. T.; Ellard S.; Turnpenny P. D. (2000) Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 24, 438–441. [DOI] [PubMed] [Google Scholar]
- Reedijk M.; Odorcic S.; Chang L.; Zhang H.; Miller N.; McCready D. R.; Lockwood G.; Egan S. E. (2005) High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 65, 8530–8537. [DOI] [PubMed] [Google Scholar]
- Weng A. P.; Ferrando A. A.; Lee W.; Morris J. P. t.; Silverman L. B.; Sanchez-Irizarry C.; Blacklow S. C.; Look A. T.; Aster J. C. (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271. [DOI] [PubMed] [Google Scholar]
- Dovey H. F.; John V.; Anderson J. P.; Chen L. Z.; de Saint Andrieu P.; Fang L. Y.; Freedman S. B.; Folmer B.; Goldbach E.; Holsztynska E. J.; Hu K. L.; Johnson-Wood K. L.; Kennedy S. L.; Kholodenko D.; Knops J. E.; Latimer L. H.; Lee M.; Liao Z.; Lieberburg I. M.; Motter R. N.; Mutter L. C.; Nietz J.; Quinn K. P.; Sacchi K. L.; Seubert P. A.; Shopp G. M.; Thorsett E. D.; Tung J. S.; Wu J.; Yang S.; Yin C. T.; Schenk D. B.; May P. C.; Altstiel L. D.; Bender M. H.; Boggs L. N.; Britton T. C.; Clemens J. C.; Czilli D. L.; Dieckman-McGinty D. K.; Droste J. J.; Fuson K. S.; Gitter B. D.; Hyslop P. A.; Johnstone E. M.; Li W. Y.; Little S. P.; Mabry T. E.; Miller F. D.; Audia J. E. (2001) Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 76, 173–181. [DOI] [PubMed] [Google Scholar]
- Hayes S.; Nelson B. R.; Buckingham B.; Reh T. A. (2007) Notch signaling regulates regeneration in the avian retina. Dev. Biol. 312, 300–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N.; Tanigaki K.; Tsuji M.; Yabe D.; Ito J.; Honjo T. (2006) Inhibition of Notch/RBP-J signaling induces hair cell formation in neonate mouse cochleas. J. Mol. Med. 84, 37–45. [DOI] [PubMed] [Google Scholar]
- Rao S. S.; O’Neil J.; Liberator C. D.; Hardwick J. S.; Dai X.; Zhang T.; Tyminski E.; Yuan J.; Kohl N. E.; Richon V. M.; Van der Ploeg L. H. T.; Carroll P. M.; Draetta G. F.; Look A. T.; Strack P. R.; Winter C. G. (2009) Inhibition of NOTCH signaling by γ secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. Cancer Res. 69, 3060–3068. [DOI] [PubMed] [Google Scholar]
- Tarassishin L.; Yin Y. I.; Bassit B.; Li Y.-M. (2004) Processing of Notch and amyloid precursor protein by γ-secretase is spatially distinct. Proc. Natl. Acad. Sci. U.S.A. 101, 17050–17055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering R. E.; Cornejo M.; Davis T. N.; Del Bianco C.; Aster J. C.; Blacklow S. C.; Kung A. L.; Gilliland D. G.; Verdine G. L.; Bradner J. E. (2009) Direct inhibition of the NOTCH transcription factor complex. Nature 462, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q. L.; Nichols J.; Chambers I.; Smith A. (2003) BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell 115, 281–292. [DOI] [PubMed] [Google Scholar]
- Ludwig T. E.; Levenstein M. E.; Jones J. M.; Berggren W. T.; Mitchen E. R.; Frane J. L.; Crandall L. J.; Daigh C. A.; Conard K. R.; Piekarczyk M. S.; Llanas R. A.; Thomson J. A. (2006) Derivation of human embryonic stem cells in defined conditions. Nat. Biotechnol. 24, 185–187. [DOI] [PubMed] [Google Scholar]
- Ying Q.-L.; Wray J.; Nichols J.; Batlle-Morera L.; Doble B.; Woodgett J.; Cohen P.; Smith A. (2008) The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Do J. T.; Zhang Q.; Yao S.; Yan F.; Peters E. C.; Schöler H. R.; Schultz P. G.; Ding S. (2006) Self-renewal of embryonic stem cells by a small molecule. Proc. Natl. Acad. Sci. U.S.A. 103, 17266–17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T.; Yamamoto M.; Sato A.; Sakano S.; Takahashi Y. (2008) Indole derivatives sustain embryonic stem cell self-renewal in long-term culture. Biosci. Biotechnol. Biochem. 72, 1242–1248. [DOI] [PubMed] [Google Scholar]
- Borowiak M.; Maehr R.; Chen S.; Chen A. E.; Tang W.; Fox J. L.; Schreiber S. L.; Melton D. A. (2009) Small molecules efficiently direct endodermal differentiation of mouse and human embryonic stem cells. Cell Stem Cell 4, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Borowiak M.; Fox J. L.; Maehr R.; Osafune K.; Davidow L.; Lam K.; Peng L. F.; Schreiber S. L.; Rubin L. L.; Melton D. (2009) A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 5, 258–265. [DOI] [PubMed] [Google Scholar]
- Warashina M.; Min K. H.; Kuwabara T.; Huynh A.; Gage F. H.; Schultz P. G.; Ding S. (2006) A synthetic small molecule that induces neuronal differentiation of adult hippocampal neural progenitor cells. Angew. Chem., Int. Ed.. 45, 591–593. [DOI] [PubMed] [Google Scholar]
- Sadek H.; Hannack B.; Choe E.; Wang J.; Latif S.; Garry M. G.; Garry D. J.; Longgood J.; Frantz D. E.; Olson E. N.; Hsieh J.; Schneider J. W. (2008) Cardiogenic small molecules that enhance myocardial repair by stem cells. Proc. Natl. Acad. Sci. U.S.A. 105, 6063–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K.; Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Takahashi K.; Tanabe K.; Ohnuki M.; Narita M.; Ichisaka T.; Tomoda K.; Yamanaka S. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Okita K.; Ichisaka T.; Yamanaka S. (2007) Generation of germline-competent induced pluripotent stem cells. Nature 448, 313–317. [DOI] [PubMed] [Google Scholar]
- Kaji K.; Norrby K.; Paca A.; Mileikovsky M.; Mohseni P.; Woltjen K. (2009) Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M.; Nagaya M.; Utikal J.; Weir G.; Hochedlinger K. (2008) Induced pluripotent stem cells generated without viral integration. Science 322, 945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K.; Nakagawa M.; Hyenjong H.; Ichisaka T.; Yamanaka S. (2008) Generation of mouse induced pluripotent stem cells without viral vectors. Science 322, 949–953. [DOI] [PubMed] [Google Scholar]
- Huangfu D.; Maehr R.; Guo W.; Eijkelenboom A.; Snitow M.; Chen A. E.; Melton D. A. (2008) Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 26, 795–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T. S.; Hanna J.; Zhang X.; Ku M.; Wernig M.; Schorderet P.; Bernstein B. E.; Jaenisch R.; Lander E. S.; Meissner A. (2008) Dissecting direct reprogramming through integrative genomic analysis. Nature 454, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Desponts C.; Do J. T.; Hahm H. S.; Scholer H. R.; Ding S. (2008) Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell 3, 568–574. [DOI] [PubMed] [Google Scholar]
- Lyssiotis C. A.; Foreman R. K.; Staerk J.; Garcia M.; Mathur D.; Markoulaki S.; Hanna J.; Lairson L. L.; Charette B. D.; Bouchez L. C.; Bollong M.; Kunick C.; Brinker A.; Cho C. Y.; Schultz P. G.; Jaenisch R. (2009) Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc. Natl. Acad. Sci. U.S.A. 106, 8912–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N.; Hochedlinger K. (2009) TGF-β signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Curr. Biol. 19, 1718–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.; Ambasudhan R.; Yuan X.; Li W.; Hilcove S.; Abujarour R.; Lin X.; Hahm H. S.; Hao E.; Hayek A.; Ding S. (2009) A chemical platform for improved induction of human iPSCs. Nat. Methods 6, 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]