

Abstract
The nonreceptor protein-tyrosine kinase c-Src is frequently overexpressed and/or activated in a variety of cancers, including those of the breast. Several heterologous binding partners of c-Src have been shown to regulate its catalytic activity by relieving intramolecular autoinhibitory interactions. One such protein, p130Cas (Cas), is expressed at high levels in both breast cancer cell lines and breast tumors, providing a potential mechanism for c-Src activation in breast cancers. The Cas-binding protein BCAR3 (breast cancer antiestrogen resistance-3) is expressed at high levels in invasive breast cancer cell lines, and this molecule has previously been shown to coordinate with Cas to increase c-Src activity in COS-1 cells. In this study, we show for the first time using gain- and loss-of-function approaches that BCAR3 regulates c-Src activity in the endogenous setting of breast cancer cells. We further show that BCAR3 regulates the interaction between Cas and c-Src, both qualitatively as well as quantitatively. Finally, we present evidence that the coordinated activity of these proteins contributes to breast cancer cell adhesion signaling and spreading. Based on these data, we propose that the c-Src/Cas/BCAR3 signaling axis is a prominent regulator of c-Src activity, which in turn controls cell behaviors that lead to aggressive and invasive breast tumor phenotypes.
Keywords: Cancer/Breast, Phosphorylation/Cytoskeletal Proteins, Phosphorylation/Kinases/Tyrosine, Signal Transduction/Adapter Proteins, Signal Transduction/Phosphotyrosine, c-Src
Introduction
In normal tissues, the nonreceptor tyrosine kinase c-Src exists as a tightly regulated molecule that is responsible for many cellular processes, including proliferation, adhesion, migration, and invasion (1). This regulation is often lost in solid tumors, including those of the breast, resulting in the transduction of signals that promote tumor progression (2). Interactions between c-Src and other proteins play an important role in the regulation of c-Src activity. Here, we investigate how altered expression of the adapter molecule BCAR3 (breast cancer antiestrogen resistance-3) coordinates with the c-Src-binding protein p130Cas (Cas)3 to regulate c-Src activity and c-Src-mediated biological processes.
BCAR3 (also known as AND-34 and NSP2) is a member of the novel Src homology 2 (SH2)-containing protein family (3) and was first identified in a screen for genes whose overexpression conferred resistance to antiestrogens (4). BCAR3 contains an SH2 domain and a guanine nucleotide exchange factor-like domain with homology to the CDC25 family of guanine nucleotide exchange factors (3, 5). Studies directed toward understanding BCAR3-mediated antiestrogen resistance have implicated the activities of Rac, phosphoinositide kinase-3, and cyclin D1 in this process (6, 7). Additionally, BCAR3 appears to promote an epithelial-mesenchymal transition in breast cancer cells (7, 8). We have recently reported that loss of BCAR3 from BT549 breast cancer cells disrupts epidermal growth factor-induced migration and invasion, coincident with a decrease in epidermal growth factor-induced tyrosine phosphorylation of its binding partner, the adapter molecule Cas (9).
Cas, also known as BCAR1 (breast cancer antiestrogen resistance-1), was initially identified as a highly tyrosine-phosphorylated protein in v-Src- and v-Crk-transformed cells (10–12). The carboxyl terminus of Cas contains not only the binding site for BCAR3 (3, 13) but also a bipartite binding site for the SH2 and SH3 domains of c-Src (14). It is through this latter interaction that Cas is able to relieve the autoinhibitory conformation of c-Src and stimulate its catalytic activity (15–17). Co-overexpression of Cas and c-Src drives Cas into complex with c-Src, leading to increased kinase activity (13, 15, 18–21). This in turn leads to phosphorylation of a variety of substrates, including Cas (18, 19). Tyrosine phosphorylation of Cas creates binding sites for downstream signaling proteins that serve to activate pathways important for cell proliferation, survival, and migration (22).
A number of breast cancer cell lines overexpress Cas and c-Src, with more invasive and aggressive cell lines also overexpressing BCAR3 (7–9, 21).4 This provides a unique cellular platform in which to study the integrated functions of these three molecules. In this study, we show for the first time that BCAR3 regulates c-Src activity and adhesion-dependent Cas phosphorylation in breast cancer cells. We further demonstrate that the coordinated activity of these proteins contributes to breast cancer cell adhesion signaling and spreading. These data, together with previous work from our group and others (6, 7, 9, 13, 23), support the idea that BCAR3 functions as an oncogenic cofactor that utilizes Cas and c-Src to promote more aggressive and invasive breast tumor phenotypes.
EXPERIMENTAL PROCEDURES
Cell Culture and Generation of MCF7 Clones Expressing Tetracycline-inducible BCAR3
COS-1 green monkey kidney cells and BT549 breast cancer cell lines were obtained from American Type Tissue Culture (Manassas, VA) and maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. Tetracycline-regulated MCF7 clones stably expressing Myc-BCAR3-pTre2-Puro were generated by transfecting plasmid DNA into “Tet-Off” MCF7 cells (Clontech) and selecting with 0.75 μg/ml puromycin. Individual clones were isolated, and regulated protein expression was verified by immunoblot and immunofluorescence in the presence or absence of 1–2 μg/ml doxycycline (Dox). Clones were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 4 mm l-glutamine, 100 μg/ml G418, and 0.75 μg/ml puromycin. Cells were grown in the presence or absence of 1–2 μg/ml of Dox for experimental analysis. The c-Src inhibitor SU6656 was purchased from Sigma and used at 10 μm where indicated.
Antibodies
Phosphotyrosine (Tyr(P)) monoclonal antibody 4G10 was purchased from Millipore (Billerica, MA). Myc monoclonal antibody 9E10 was obtained from the University of Virginia Lymphocyte Culture Center (Charlottesville, VA). CasB and BCAR3 antibodies have been described previously (9, 24, 25). c-Src monoclonal antibody 2-17 was a gift of S. J. Parsons (University of Virginia, Charlottesville, VA). c-Src pY416 polyclonal antibody was purchased from Invitrogen. FLAG M2 affinity resin, FLAG M5 monoclonal antibody, horseradish peroxidase-conjugated monoclonal β-actin antibody, and β-tubulin monoclonal antibody were purchased from Sigma. Protein G-PLUS-agarose was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Protein A-Sepharose was purchased from Amersham Biosciences and HA.11 from Covance (Princeton, NJ).
Plasmids
pRK5 constructs encoding the genes for full-length Cas and the c-Src-binding deficient Cas (P642A, Y668F/Y670F) have been described previously (21, 26), as have pFLAG3-BCAR3, pCDNA3-FLAG2AB-paxillin, and pFLAG-cortactin (15). pCDNA-c-Src was a generous gift of S. J. Parsons (University of Virginia) (27).
Transfection, Immunoprecipitation, and Protein Expression
Transient transfection of COS-1 cells was performed using Superfect (Qiagen, Valencia, CA) according to the manufacturer's specifications. Cells were lysed 24 h post-transfection in modified radioimmunoprecipitation assay buffer (RIPA: 150 mm/liter NaCl, 50 mm Tris (pH 7.5), 1% IGEPAL CA-630, 0.5% deoxycholate) supplemented with protease and phosphatase inhibitors (100 μm leupeptin, 1 mm phenylmethylsulfonyl fluoride, 0.15 unit/ml aprotinin, and 1 mm sodium orthovanadate). Protein concentrations were determined using the bicinchoninic acid assay kit (Pierce). A BCAR3-specific small interfering RNA (siRNA) (5′-AAAUCAACCGGACAGUUCU-3′) was synthesized to target human but not murine BCAR3 (Dharmacon, Lafayette, CO). ON-TARGETplus SMARTpool siRNAs targeted to human BCAR3 were also purchased from Dharmacon. BT549 cells were treated with 20 μm control nontargeting siRNAs or BCAR3-specific siRNAs using Oligofectamine (Invitrogen) transfection reagent, as described previously (28), or the Lipofectamine RNAiMax (Invitrogen) reverse transfection method per the manufacturer's specifications. Cells were grown for 48–72 h before lysis in RIPA buffer. Immunoprecipitations were performed as described previously (13, 15). Proteins were resolved by SDS-PAGE, transferred to nitrocellulose, immunoblotted with the indicated antibodies, and detected by horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin (Amersham Biosciences) followed by enhanced chemiluminescence (PerkinElmer Life Sciences) or horseradish peroxidase substrate luminol reagent (Millipore).
Cell Adhesion to Fibronectin and Microscopy
10-cm tissue culture dishes and glass coverslips were coated with 20 μg/ml fibronectin (Sigma) for 1 h or overnight at 4 °C and then washed with phosphate-buffered saline. BT549 cells were transfected with control or BCAR3-specific siRNAs as described above. Cells were grown for 72 h and then trypsinized, counted, and left in suspension for 90 min in complete growth media containing serum. Suspension cells were then either lysed or plated onto tissue culture plates and fibronectin-coated coverslips in complete growth media for the indicated lengths of time. Cells plated onto coverslips were washed twice in phosphate-buffered saline, fixed with 3% paraformaldehyde for 20 min, washed, and mounted onto slides. In parallel, cells were plated immediately following transfection and analyzed 72 h later as cycling adherent cells. For the c-Src inhibitor studies, dimethyl sulfoxide (DMSO) or 10 μm SU6656 was added to the cells during the 90-min suspension and subsequent plating steps. ImageJ software (National Institutes of Health) was used to calculate cell area using the freehand tracing tool. 100+ cells were counted per time point for at least three independent experiments.
Densitometry and Statistical Analysis
Densitometry was performed using the Bio-Rad GS-800 densitometer (Bio-Rad) and quantified using ImageQuant TL 2005 (Amersham Biosciences). A two-tailed Student's t test was used for comparisons between the various sample sets. Statistical significance was defined at ≥95% confidence interval or p value ≤0.05. Bar graphs represent the mean ± S.D.
RESULTS
Cas/c-Src Interactions Are Required for BCAR3-dependent Enhancement of Cas-mediated c-Src Kinase Activity
To assess the molecular requirements for c-Src activation by Cas and BCAR3, COS-1 cells were transfected with plasmids encoding c-Src, Myc-tagged Cas, and/or FLAG-tagged BCAR3. In each case, plasmids encoding FLAG-tagged cortactin were also transfected to provide an exogenous substrate for c-Src activity in the transfected population. Twenty four hours post-transfection, cells were lysed, and the phosphorylation of cortactin was examined using phosphotyrosine-specific antibodies. Cells overexpressing Cas exhibited increased activation of c-Src, as measured by the increased phosphorylation of cortactin, compared with cells expressing only vector (Fig. 1A, top panel, lanes 1 and 2). In contrast, no increase in cortactin phosphorylation was observed when BCAR3 was expressed (Fig. 1A, top panel, lane 3). However, when BCAR3 was co-expressed with Cas, phosphorylation of cortactin was even greater than in the presence of Cas alone (Fig. 1A, top panel, lane 4). These data confirm our previous finding that BCAR3 expression enhances Cas-dependent c-Src activity (13). Similar results were obtained using paxillin as an exogenous substrate (Fig. 1B, top panel, lanes 2 and 5), demonstrating that the enhancement in c-Src substrate phosphorylation observed in the presence of BCAR3 and Cas was not restricted to cortactin.
FIGURE 1.

BCAR3 augments Cas-mediated enhancement of c-Src activity. A, co-overexpression of Cas and BCAR3 enhances Cas-dependent c-Src activity. COS-1 green monkey kidney cells were transfected with plasmids encoding wild type c-Src, FLAG-cortactin, pRK5 vector (V), Myc-Cas (C), and/or FLAG-BCAR3. FLAG immune complexes were generated from cell lysate 24 h post-transfection and immunoblotted with Tyr(P) and FLAG-specific antibodies (top two panels). Whole cell lysates were immunoblotted with Myc and FLAG antibodies (bottom two panels). IP, immunoprecipitation. B, BCAR3 requires Cas/c-Src binding to augment c-Src activity. COS-1 green monkey kidney cells were transfected with plasmids encoding wild type c-Src, FLAG-paxillin, pRK5 vector (V), Myc-Cas (C), Myc-Cas-TM (TM), and/or FLAG-BCAR3. Cells were lysed 24 h post-transfection, and proteins were analyzed as described above. Data shown are representative of 2–4 independent experiments.
To determine whether the augmentation of Cas-dependent c-Src activity by both Cas and BCAR3 required interactions between Cas and c-Src, a construct of Cas containing three amino acid substitutions (P642A, Y668F/Y670F) in the c-Src-binding site (14) was utilized in experiments similar to those described above. This Cas mutant, termed Cas-TM for “triple mutant,” is significantly impaired in binding to c-Src (21). As expected, phosphorylation of the c-Src substrate paxillin was observed under conditions of Cas overexpression, and this was increased significantly when both Cas and BCAR3 were co-overexpressed (Fig. 1B, top panel, lanes 2 and 5). In contrast, expression of Cas-TM either alone or in combination with BCAR3 failed to promote paxillin phosphorylation (Fig. 1B, top panel, lanes 3 and 6). These data indicate that the interaction between Cas and c-Src is required for both Cas- and BCAR3/Cas-dependent activation of c-Src.
Loss of Endogenous BCAR3 Expression in Breast Cancer Cells Attenuates c-Src Activity, Cas Tyrosine Phosphorylation, and the Association between Cas and c-Src
Although we show above that Cas and BCAR3 regulate c-Src activity in ectopic expression systems, these regulatory pathways have not yet been investigated in the context of endogenously expressed proteins. Many breast tumors and cancer cell lines that exhibit aggressive and invasive phenotypes express high levels of Cas, BCAR3, and c-Src, and these proteins can be found in complex with one another (see Fig. 2 for endogenous protein complexes in BT549 cells) (7–9, 21, 29–34). In contrast, cells representative of earlier stage breast cancers generally express BCAR3 at very low or undetectable levels, despite the fact that expression of Cas and c-Src is often high (7–9). This dichotomy provides an excellent system with which to examine the effect of BCAR3 expression on Cas/c-Src interactions and c-Src activity under physiological conditions.
FIGURE 2.
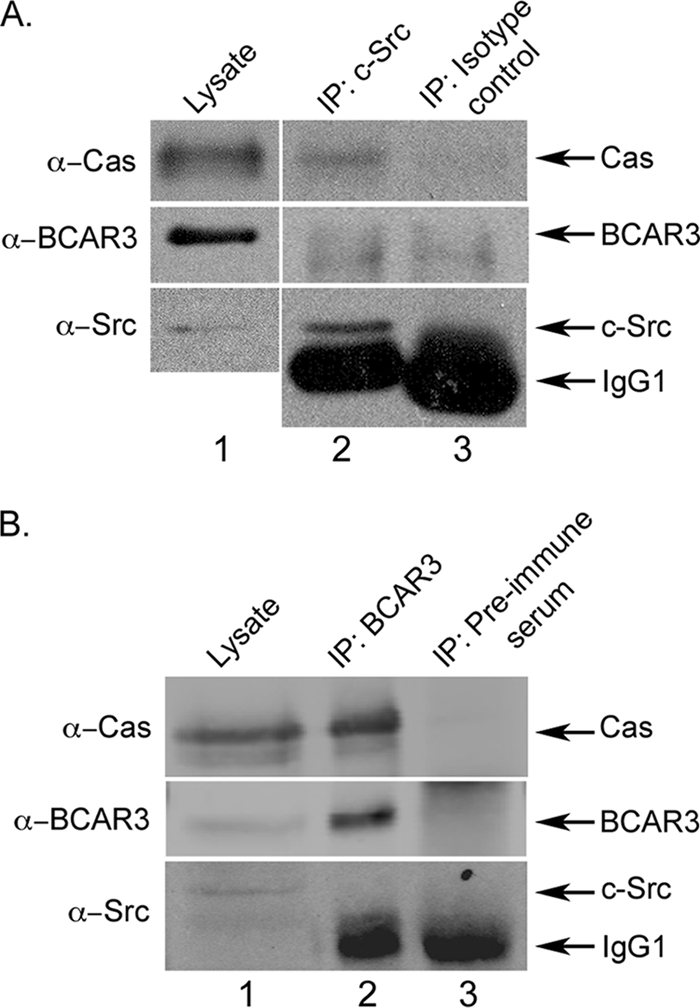
Two distinct pools of Cas exist in BT549 cells. A, Cas and c-Src are present in a molecular complex. 400 μg of BT549 cell lysate were incubated with c-Src monoclonal antibody 2-17 (lane 2) or isotype-matched monoclonal antibody HA.11 as a control (lane 3). Immune complexes were collected and immunoblotted with the indicated antibodies. For comparison, 20 μg of cell lysate were immunoblotted (lane 1). The vertical separation indicates different exposure times between lysate and immunoprecipitation (IP) lanes. B, Cas and BCAR3 are present in a molecular complex. BCAR3 or control (preimmune serum) immune complexes were generated as described above from 350 μg of BT549 cell lysate, collected on protein A-agarose, and immunoblotted with the indicated antibodies (lanes 2 and 3). For comparison, 30 μg of cell lysate were immunoblotted (lane 1).
To determine whether BCAR3 is important for the regulation of c-Src activity in cells expressing high endogenous levels of this protein, BCAR3 expression was reduced by RNA interference in BT549 cells. Following transfection with BCAR3-targeted siRNA oligonucleotides, BCAR3 expression was consistently reduced by 82–99%, as measured by densitometry (Fig. 3A, top panel). Depletion of BCAR3 correlated with a concomitant reduction in c-Src autophosphorylation at tyrosine 419 (Tyr-419 is the autophosphorylation site on human c-Src, equivalent to Tyr-416 in chicken), a marker for c-Src activity (Fig. 3A, 2nd panel and associated graph).
FIGURE 3.

Depletion of BCAR3 in BT549 breast cancer cells disrupts c-Src activation, Cas phosphorylation, and Cas/c-Src interactions. A, BT549 cells depleted for BCAR3 have decreased c-Src activity. BT549 breast cancer cells were transfected with nontargeting or BCAR3-specific siRNA duplexes and grown for 48 h. Whole cell lysate was immunoblotted with the indicated antibodies. The mean relative Tyr(P)-419 (α-pY419) phosphorylation was determined from four independent experiments. B, BT549 cells depleted for BCAR3 have decreased Cas phosphorylation. Cas immune complexes were generated from 72-h siRNA-treated BT549 cells and immunoblotted with antibodies specific to Tyr(P) and Cas (top two panels). Cell lysate was immunoblotted for BCAR3 to verify loss of BCAR3 expression (bottom panel). The mean relative Cas phosphorylation was determined from four independent experiments. C, BT549 cells depleted for BCAR3 have decreased Cas/c-Src interactions. c-Src immune complexes were generated from 72-h siRNA-treated BT549 cells and immunoblotted with Cas and c-Src antibodies (top panel). Whole cell lysate was also immunoblotted with BCAR3 antibodies (bottom panel). The mean Cas levels in Cas-c-Src immune complexes were determined from five independent experiments. Two-tailed Student's t tests were conducted for comparison between siControl and siBCAR3 samples for A–C. Bars indicate standard deviation; asterisk indicates significant difference from the mean at ≥95% confidence interval. Within all panels, exposure time is the same; the white vertical lines denote noncontiguous sample lanes.
As Cas is a well established c-Src substrate, we next investigated whether the decrease in c-Src activity exhibited by cells depleted for BCAR3 was accompanied by a loss in tyrosine phosphorylation of Cas. Cas was immunoprecipitated from lysates derived from cells treated with control or BCAR3-targeted siRNAs, and the resultant complexes were immunoblotted with a Tyr(P) antibody. Cas tyrosine phosphorylation was reduced by an average of 70% under conditions of BCAR3 knockdown (Fig. 2B). Similar results were obtained when BCAR3 was depleted using a second pool of four independent BCAR3-specific siRNAs, confirming that the decrease in Cas phosphorylation was due to depletion of BCAR3 (supplemental Fig. 1, A–C). These results in cycling cells extend our previous study, which showed a reduction in epidermal growth factor-dependent Cas tyrosine phosphorylation under conditions of BCAR3 depletion (9).
Cas is an effective activator of c-Src catalytic activity through its ability to bind to c-Src and relieve its autoinhibitory conformation (15). We therefore hypothesized that the decreases in c-Src activity and Cas tyrosine phosphorylation observed under BCAR3-depleted conditions might be the result of a reduction in c-Src/Cas association. To determine whether this was the case, c-Src was immunoprecipitated from lysates generated from BT549 cells treated with control or BCAR3-targeted siRNAs, and the resultant complexes were immunoblotted for Cas. As expected, Cas was seen to associate with c-Src in cells treated with nontargeting siRNA oligonucleotides (Fig. 3C, lane 1, and supplemental Fig. 2A). In cells depleted of BCAR3, c-Src/Cas association was reduced by ∼60% (Fig. 3C, lane 2). This result supports our hypothesis that the observed loss in c-Src activity is a consequence of decreased Cas association.
Ectopic Expression of BCAR3 in Breast Cancer Cells Induces c-Src Activity and Cas Tyrosine Phosphorylation
MCF7 cells express low to undetectable levels of BCAR3 and serve as a model for early stage breast cancer (8, 9). To examine the effect of BCAR3 overexpression in this background, MCF7 cell lines were generated that express BCAR3 under a tetracycline-regulated promoter (Tet-Off). BCAR3 expression can be turned off in these cells by the addition of 2 μg/ml Dox (Fig. 4A, top panel). To determine whether BCAR3 expression alters c-Src activity in this cell system, c-Src was immunoprecipitated from lysates and immunoblotted for Tyr-419 phosphorylation. Phosphorylation of Tyr-419 was elevated almost 2-fold under conditions of BCAR3 overexpression (Fig. 4B), demonstrating that BCAR3 is capable of inducing c-Src activity when overexpressed in breast cancer cells. To determine whether this increase in c-Src activity correlated with an increase in BCAR3-dependent Cas phosphorylation, Cas was immunoprecipitated from lysates isolated from cells grown in the presence (endogenous levels of BCAR3) or absence (overexpressed BCAR3) of Dox, and the resultant complexes were immunoblotted with an antibody recognizing Tyr(P). Indeed, Cas tyrosine phosphorylation was increased by 3-fold under conditions of BCAR3 overexpression (Fig. 4C, compare lanes 1 and 3). Because BCAR3 expression in MCF7 cells has been reported to induce phosphorylation of Cas on serine residues (8), we confirmed the specificity of the Tyr(P) antibody for phosphotyrosine by preincubating the antibody with O-phospho-l-tyrosine- or O-phospho-l-serine-conjugated bovine serum albumin prior to using it for immunoblotting. The Cas Tyr(P) signal was lost under conditions of preincubation with the Tyr(P), but not the Ser(P), inhibitor (supplemental Fig. 3). Finally, to test whether the increase in Cas phosphorylation was directly due to c-Src activity, the cells were cultured for 24 h in the presence or absence of 10 μm of the c-Src family kinase inhibitor SU6656 (30). SU6656 treatment of BCAR3-overexpressing cells reduced overall Cas phosphorylation and abrogated the BCAR3-dependent increase in Cas phosphorylation (Fig. 4C, lanes 2 and 4). These data confirm that the increase in Cas phosphorylation in cells overexpressing BCAR3 is due to increased c-Src activity.
FIGURE 4.
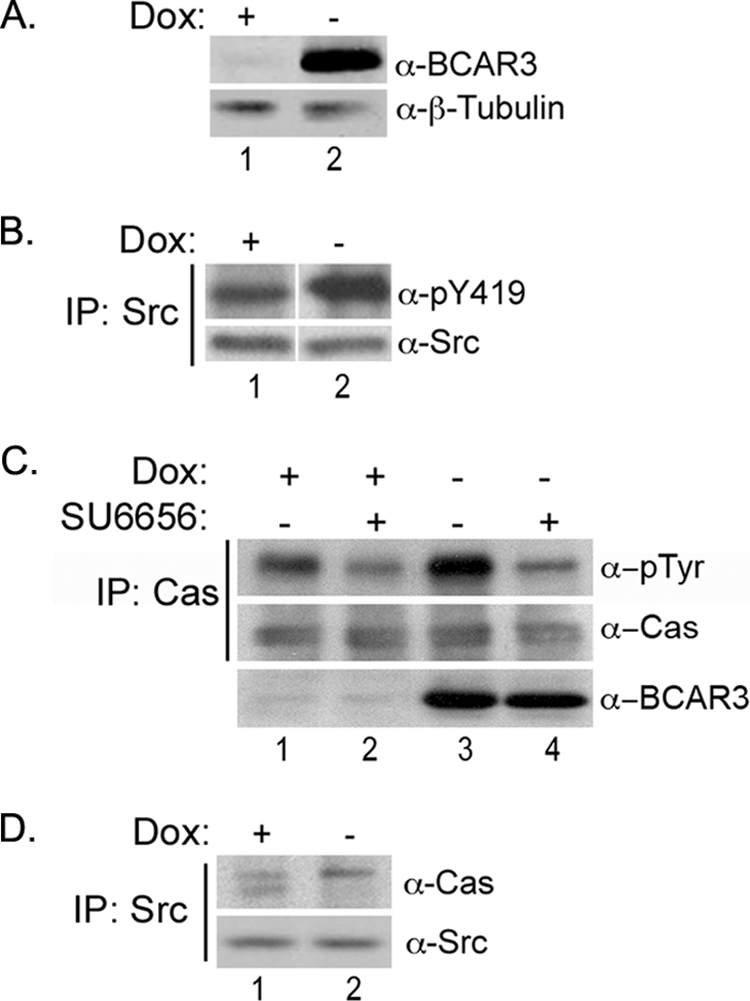
BCAR3 overexpression in MCF7 breast cancer cells increases c-Src activity and Cas phosphorylation. A, Dox-regulated expression of BCAR3 in Tet-Off MCF7 cells. MCF7 cells overexpressing BCAR3 under control of a tetracycline-responsive promoter were grown in the presence or absence of 2 μg/ml Dox for 72 h. Whole cell lysate was immunoblotted with antibodies specific to BCAR3 and β-tubulin. B, BCAR3 overexpressing cells exhibit increased c-Src activation. c-Src immune complexes were generated from MCF7 cells grown in the presence or absence of 2 μg/ml Dox for 72 h and immunoblotted with antibodies specific for Tyr(P)-419 and total c-Src. Tyr(P)-419 was increased by an average of 1.9 ± 0.43-fold in three-independent experiments (p = 0.02). The white vertical line denotes noncontiguous sample lanes; the exposure times for the +/− Dox samples in each immunoblot is the same. C, BCAR3-dependent Cas phosphorylation is mediated by Src family kinases. MCF7-B6 cells grown in the presence or absence of Dox for 48 h were treated with vehicle (DMSO) or 10 μm SU6656 and grown for an additional 24 h. Cas immune complexes were generated from lysate and immunoblotted with antibodies specific to Tyr(P) and Cas (top two panels). Whole cell lysate was also immunoblotted with BCAR3 antibodies (bottom panel). Cas phosphorylation increased by an average of 2.9 ± 0.64-fold (p = 0.009) in the presence of overexpressed BCAR3. Data shown are representative of four independent experiments. D, BCAR3 expression regulates Cas/c-Src interactions. c-Src immune complexes were generated from MCF7 cells grown in the presence or absence of 2 μg/ml Dox for 72 h and immunoblotted with Cas- and c-Src-specific antibodies.
Because enhancement of c-Src activity by BCAR3 in COS-1 cells required an interaction between Cas and c-Src, and given the decrease in c-Src/Cas association exhibited by BT549 cells depleted for BCAR3 (Fig. 3C), we next investigated whether BCAR3 regulates c-Src/Cas interactions in MCF7 cells. Interestingly, the absolute amount of Cas present in c-Src immune complexes was not dramatically altered under conditions of BCAR3 overexpression (Fig. 4D). However, in contrast to control conditions where multiple species of Cas were seen to associate with c-Src, virtually all of the Cas in complex with c-Src in BCAR3-overexpressing MCF7 cells exhibited a retarded electrophoretic mobility (Fig. 4D, lane 2). This slower migrating form of Cas has been reported previously to represent a highly phosphorylated species of Cas (12, 29), consistent with the increase in Cas tyrosine phosphorylation seen in cells overexpressing BCAR3 (Fig. 4C). Together with the loss-of-function studies in BT549 cells described above, these data support a role for BCAR3 in regulating c-Src activity and Cas tyrosine phosphorylation in breast cancer cells via its ability to influence c-Src/Cas interactions.
BCAR3 Regulates Cell Spreading on Fibronectin and Adhesion-mediated Cas Tyrosine Phosphorylation
Because BCAR3 was found to regulate c-Src activity and Cas tyrosine phosphorylation in cycling, continuously adherent cells, we next investigated whether it played a similar role in response to adhesion and spreading on extracellular matrix, a biological process that is controlled by Cas and c-Src signaling (31, 35). BT549 cells were transiently transfected with nontargeting or BCAR3-specific siRNAs and cultured for 72 h. Cells were trypsinized, left in suspension for 90 min, and then plated on fibronectin-coated tissue culture dishes or coverslips for 30–240 min. The area of cells on the fibronectin-coated coverslips was determined for each time point as described under “Experimental Procedures.” At 30 min, control cells expressing BCAR3 had begun to spread, and this continued through 240 min (Fig. 5, A, top panel, and B, black bars). BCAR3-depleted cells exhibited delayed adherence and spreading at all time points compared with control cells (Fig. 5, A, bottom panel, and B, gray bars). This defect in spreading was also seen using alternate BCAR3 siRNAs, confirming that the defect in cell spreading was induced by the loss of BCAR3 expression in these cells (supplemental Fig. 1D).
FIGURE 5.

Loss of BCAR3 expression disrupts cell spreading and adhesion-mediated Cas phosphorylation. A and B, BT549 cells depleted for BCAR3 exhibit decreased cell spreading on fibronectin. BT549 cells were transfected with nontargeting or BCAR3-specific siRNA duplexes and grown for 72 h. Cells were left in suspension for 90 min and then plated on fibronectin-coated coverslips in complete growth media with 10% fetal bovine serum for the indicated lengths of time in normal growth media. A coverslip from cycling, adherent cells was also examined. Mean cell area from three independent experiments was determined by using ImageJ software. Bars indicate standard deviation; asterisk indicates significant difference from the mean at ≥95% confidence interval. C, BT549 cells depleted for BCAR3 exhibit decreased adhesion-mediated Cas phosphorylation. BT549 cells were transfected and treated as described above. Cas immune complexes were generated from lysate and immunoblotted with Tyr(P) and Cas antibodies (top two panels). Whole cell lysate was also immunoblotted with antibodies specific to BCAR3 (bottom panel). Data shown are representative of four independent experiments. Within all panels, exposure time is the same; the white vertical line denotes noncontiguous sample lanes. IP, immunoprecipitation.
Because Cas becomes tyrosine-phosphorylated in response to cell adhesion (18, 29, 35), we next investigated whether changes in adhesion-dependent Cas phosphorylation coincided with the delay in spreading in BCAR3-depleted cells. Cas was immunoprecipitated from lysates generated under the treatment conditions described above and immunoblotted with a Tyr(P) antibody to measure Cas phosphorylation. Tyrosine phosphorylation of Cas in control cells was seen to increase over time in an adhesion-dependent manner, peaking at 120 min (Fig. 5C, top panel, lanes 1–5). A similar, albeit significantly reduced, adhesion-dependent increase in Cas phosphorylation was seen in cells depleted for BCAR3 (Fig. 5C, top panel, lanes 7–11). These differences in phosphorylation were also observed in continuously adherent cells grown on fibronectin (Fig. 5C, top panel, lanes 6 and 12), confirming our earlier observations in cells plated on plastic (Fig. 3B). These data show that BCAR3 expression is required for efficient adhesion-mediated Cas phosphorylation as well as cell spreading in BT549 cells.
BCAR3 Regulates Cell Spreading through c-Src-dependent and -independent Pathways
As BCAR3 was shown to be involved in c-Src signaling (Fig. 4, B and C), we next investigated whether loss of c-Src activity could be responsible for the defect in spreading exhibited by cells depleted of BCAR3. BT549 cells were transiently transfected with nontargeted or BCAR3-specific siRNAs and cultured for 72 h. Cells were then trypsinized, left in suspension in the presence or absence of 10 μm SU6656 for 90 min, and then plated on fibronectin-coated coverslips in the continued presence or absence of SU6656 for 30–240 min. Consistent with our earlier observations, knockdown of BCAR3 resulted in a significant impairment in spreading compared with control cells at all time points (Fig. 6, A, 1st and 2nd rows, and B, black and gray bars). If BCAR3 regulates cell spreading through its ability to enhance c-Src activity, we reasoned the following: 1) inhibition of c-Src in control cells should phenocopy BCAR3 knockdown, and 2) the combined knockdown of BCAR3 and c-Src inhibition should have no greater effect than either treatment alone. Cells treated with SU6656 did in fact show a delay in spreading, albeit to a lesser degree than BCAR3-depleted cells (Fig. 6, A, 3rd rows, and 6B, black striped bars). However, combined BCAR3 knockdown and SU6656 treatment resulted in a delay in spreading that was significantly greater than what was observed for either condition alone (Fig. 6B, gray striped bars). With the caveat that c-Src inhibition and BCAR3 knockdown may not have been complete, these results suggest that c-Src activity may be only partially required for BCAR3-dependent cell spreading and vice versa. Moreover, the fact that combined c-Src inhibition and BCAR3 depletion did not completely abrogate cell spreading suggests the involvement of additional pathways(s) that may be independent of both of these molecules.
FIGURE 6.

BCAR3 regulates both c-Src-dependent and -independent signaling pathways in response to adhesion and cell spreading. A, BT549 cells were transfected with nontargeting or BCAR3-specific siRNA duplexes and grown for 72 h. Cells were left in suspension for 90 min in the presence of DMSO or 10 μm SU6656 and then plated onto fibronectin-coated coverslips in complete growth media with 10% fetal bovine serum for the indicated lengths of time in normal growth media with or without SU6656. B, mean cell area was calculated from four independent experiments. Two-tailed Student's t tests were conducted for comparison between the various sample sets. Bars indicate standard deviation. *, #, and ⋀ indicate significant difference from the mean versus control DMSO, control SU6656, and siBCAR3 DMSO, respectively, at ≥95% confidence interval.
DISCUSSION
In this study, we present evidence for the regulation and function of an important signaling network in breast cancer cells involving three proteins, Cas, c-Src, and BCAR3. Previously, we showed that the co-overexpression of BCAR3, Cas, and c-Src induces c-Src activity in untransformed monkey kidney cells (COS-1) (13). This study expands upon this finding by demonstrating that, in BT549 and MCF7 breast cancer cell lines, BCAR3 can regulate c-Src/Cas interactions, c-Src activity, c-Src-dependent Cas tyrosine phosphorylation, and cell spreading. Collectively, these studies establish BCAR3 as a key regulator of signaling events involving Cas and c-Src in the setting of breast cancer cells and for the first time provide insight into why breast cancer cells with elevated BCAR3 expression may exhibit more aggressive behaviors.
Molecular Dynamics of the c-Src/Cas/BCAR3 Signaling Axis
The ability of BCAR3 and Cas to regulate c-Src activity provides a mechanism through which c-Src signaling can be both temporally and spatially controlled within the cell. We postulate that BCAR3, which helps to localize Cas to membrane-proximal sites (8, 9, 13), may enhance Cas-dependent c-Src activity by bringing Cas into proximity of membrane-bound c-Src (Fig. 7). Subsequent association of Cas with c-Src would then relieve the autoinhibitory conformation of c-Src, leading to increased catalytic activity in these regions of the cell. Support for this model comes from the loss-of-function and gain-of-function studies presented above, which show that BCAR3 expression directly impacts c-Src/Cas interactions and Cas tyrosine phosphorylation. Moreover, we show that this is largely dependent on c-Src activity because treatment with the c-Src inhibitor SU6656 blocked the BCAR3-dependent increase in Cas phosphorylation (Fig. 4C). Interestingly, it does not appear that c-Src, Cas, and BCAR3 need to be present in a single molecular complex to regulate c-Src activity and functions because BCAR3 was not detected in c-Src-Cas complexes (Fig. 2A) and c-Src was similarly not present in BCAR3-Cas complexes (Fig. 2B). These data suggest that there are two distinct pools of Cas, one that is associated with c-Src and one that is associated with BCAR3. Based on these findings, we propose that BCAR3 brings Cas into close proximity with c-Src, where it subsequently promotes association with, and activation of, this kinase. It is this dynamic relationship between pools of Cas that initiates the c-Src/Cas/BCAR3 signaling axis.
FIGURE 7.

Proposed mechanism of BCAR3-mediated enhancement of c-Src activation, cell spreading, and adhesion. Step 1, cytosolically localized Cas is recruited to membrane-proximal regions by BCAR3 through an indirect or direct interaction with BCAR3. Once at the membrane (step 2), Cas interacts with and activates c-Src. c-Src activation results in the phosphorylation of Cas, which leads to the recruitment of substrate domain binding partners such as Crk (step 3). Step 4, Cas/Crk interactions activate downstream signaling pathways important for cell spreading, migration, and invasion. BCAR3-dependent and BCAR3-independent c-Src signaling can also activate other signaling pathways important for cell proliferation and survival in the presence of antiestrogens. Step 5, BCAR3 itself activates an unidentified c-Src-independent pathway important for cell spreading.
Although we propose that the recruitment of Cas by BCAR3 to membrane-proximal sites is an important factor in the activation of c-Src by BCAR3, it may not be the sole determinant. Indeed, we previously showed in rat embryo fibroblasts that a carboxyl-terminal deletion mutant of BCAR3 could induce Cas accumulation at lamellipodia without promoting Cas-dependent c-Src activity (13). The carboxyl terminus of BCAR3 has two known activities that may contribute to its ability to regulate c-Src activity. First, it contains the sequences necessary for Cas binding. However, although Cas-BCAR3 complexes have been detected in a number of breast cancer cell lines (Fig. 2B) (7–9), we showed previously that BCAR3 was still able to promote activation of c-Src through a mutant of Cas that could not bind to BCAR3 (13). Second, the carboxyl terminus of BCAR3 contains sequences shown to be important for activating a number of small GTPases, including Rap1 (3, 23). Interestingly, constitutively active Rap1 can substitute for BCAR3 in the activation of c-Src in the COS-1 system (13).
The regulation of c-Src activity by Cas and BCAR3 appears to be under tight control. Thus, overexpression of BCAR3 to very high levels in MCF7 cells (∼35-fold above the level of BCAR3 endogenously expressed in BT549 cells; data not shown) resulted in only a modest 2-fold increase in c-Src autophosphorylation (Fig. 4B). We propose that the pool of Cas capable of activating c-Src under these conditions may be a limiting factor, and any BCAR3 expressed above a certain threshold would therefore have little added effect. This model is supported by the finding that the total level of Cas associated with c-Src was not significantly changed in MCF7 cells overexpressing BCAR3 (Fig. 4D). However, there was a marked qualitative difference in the Cas found in this complex, indicative of a heavily phosphorylated Cas species (8, 12, 29). It is not clear whether this difference is a cause or an effect of the increased c-Src activity present under these conditions. Regardless, we predict that this modified complex has an increased potential for transducing signals necessary for adhesion signaling, spreading, and migration.
As discussed above, the Cas that is found in association with c-Src in BCAR3-expressing MCF7 cells is exclusively the slower migrating species, indicative of hyperphosphorylation. Makkinje et al. (8) have recently shown that Cas becomes phosphorylated on serine residues in the presence of BCAR3. Although these authors failed to detect tyrosine phosphorylation on Cas by mass spectroscopy in the presence of overexpressed BCAR3, our studies clearly show that tyrosine phosphorylation of Cas is elevated as a function of BCAR3 expression in these cells (Fig. 3B and Fig. 4C). We have not evaluated whether serine phosphorylation of Cas is also elevated under these conditions in our system. However, the increased tyrosine phosphorylation on Cas that was observed as a function of BCAR3 expression in MCF7 cells coincided with an increase in Cas-Crk complexes (data not shown), which become established through interactions between the SH2 domain of Crk and phosphorylated tyrosine residues in the substrate binding domain of Cas (37, 38). Cas tyrosine phosphorylation has also been reported to be elevated when the BCAR3 family member SHEP1 was constitutively targeted to the membrane, a process that required c-Src activity (32).
Biological Activities of the c-Src/Cas/BCAR3 Signaling Axis
The data presented in this study establish that c-Src, Cas, and BCAR3 function together in a coordinated network when all three proteins are endogenously expressed in breast cancer cells. Each of these proteins has been implicated in a wide variety of cellular processes in breast cancer cells, including cell proliferation, survival, adhesion signaling, and migration (7–9, 22, 36). In this study, we have focused on cell spreading as a component of adhesion signaling and migration dynamics, processes that, when deregulated, can contribute to aggressive and invasive tumor phenotypes. We show for the first time that BCAR3 regulates the kinetics of cell spreading on fibronectin. Coincident with slower spreading in BCAR3-depleted cells, Cas tyrosine phosphorylation, but not overall Cas expression, was reduced. These data suggest that the regulation of Cas phosphorylation by BCAR3 may contribute to cell spreading dynamics in cells expressing this molecule. Cas-null mouse embryo fibroblasts show a similar spreading defect (35), and expression of a construct of Cas that lacks the heavily phosphorylated substrate binding domain fails to rescue the spreading and migratory defects (39). As mentioned above, tyrosine phosphorylation of Cas within the substrate binding domain provides binding sites for the small adapter molecule Crk; this interaction in turn regulates cell adhesion, spreading, and migration through the activation of the small GTPases Rap1 and Rac1 (37, 38). Interestingly, enforced expression of BCAR3 in MCF7 cells mediates an increase in the formation of Cas-Crk complexes (data not shown), while knockdown of BCAR3 in BT549 cells results in a reduction in Cas-Crk complexes (9). Cas/Crk coupling is thus likely to be an important downstream consequence of BCAR3-dependent c-Src activity and Cas phosphorylation, which in turn contributes to cell spreading, migration, and invasion (Fig. 7).
We hypothesize that the regulation of c-Src activity by BCAR3 contributes in part to the defect in spreading observed when BCAR3 is depleted from breast cancer cells. As was the case for BCAR3 depletion, inhibition of c-Src activity by SU6656 was shown to delay spreading (Fig. 6). However, modulation of c-Src activity may not be the only mechanism through which BCAR3 functions to regulate cell spreading because combined c-Src inhibition and BCAR3 depletion resulted in a significantly greater, albeit modest, delay in cell spreading relative to c-Src inhibition alone. Similarly, it appears from our data that c-Src may function to control cell spreading through a mechanism that is in part independent of BCAR3. Finally, pathways independent of both BCAR3 and c-Src are likely to play some role in controlling this cellular process because combined depletion/inhibition of these molecules failed to completely block cell spreading.
Although this study has focused on the role of BCAR3 in Cas and c-Src signaling during cell spreading, it is important to note that this signaling axis may also be exploited by breast cancer cells for other biological activities. For example, both Cas and BCAR3 induce antiestrogen resistance in estrogen receptor-positive breast cancer cells (4, 6, 7, 21, 40–42), and Cas and c-Src play a role in chemoresistance (43, 44). Thus, the expression of BCAR3 and Cas, and the subsequent activation of c-Src-dependent signaling pathways, may be one mechanism that drives the oncogenic potential, therapeutic response, and metastatic spread of breast tumor cells. Further investigation into the consequences of BCAR3 and Cas expression and their effect(s) on c-Src activity may aid in the development of novel therapeutic strategies for patients whose breast tumors overexpress these proteins.
Supplementary Material
Acknowledgments
We thank the members of the laboratory and Drs. Jim Casanova, Deborah Lannigan, Ulrike Lorenz, and Margaret Shupnik for their helpful comments and suggestions.
This work was supported, in whole or in part, by National Institutes of Health Training Grant T32 CA009109 (to N. R. S., M. S. G., and R. S. S.) and Research Grant R01 CA096846 (to A. H. B.). This work was also supported by Department of Defense Breast Cancer Research Program Grants BC043365 (to N. R. S.) and BC050339 (to R. S. S.) and Ruth L. Kirschstein National Research Service Award CA130168 (to M. S. G.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
N. R. Schuh, M. S. Guerrero, R. S. Schrecengost, and A. H. Bouton, unpublished data.
- Cas
- Crk-associated substrate
- SH
- Src homology
- Dox
- doxycycline
- siRNA
- small interfering RNA.
REFERENCES
- 1.Finn R. S. (2008) Ann. Oncol. 19, 1379–1386 [DOI] [PubMed] [Google Scholar]
- 2.Summy J. M., Gallick G. E. (2003) Cancer Metastasis Rev. 22, 337–358 [DOI] [PubMed] [Google Scholar]
- 3.Gotoh T., Cai D., Tian X., Feig L. A., Lerner A. (2000) J. Biol. Chem. 275, 30118–30123 [DOI] [PubMed] [Google Scholar]
- 4.van Agthoven T., van Agthoven T. L., Dekker A., van der Spek P. J., Vreede L., Dorssers L. C. (1998) EMBO J. 17, 2799–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu Y., Brush J., Stewart T. A. (1999) J. Biol. Chem. 274, 10047–10052 [DOI] [PubMed] [Google Scholar]
- 6.Felekkis K. N., Narsimhan R. P., Near R., Castro A. F., Zheng Y., Quilliam L. A., Lerner A. (2005) Mol. Cancer Res. 3, 32–41 [PubMed] [Google Scholar]
- 7.Near R. I., Zhang Y., Makkinje A., Vanden Borre P., Lerner A. (2007) J. Cell. Physiol. 212, 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makkinje A., Near R. I., Infusini G., Vanden Borre P., Bloom A., Cai D., Costello C. E., Lerner A. (2009) Cell. Signal. 21, 1423–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrecengost R. S., Riggins R. B., Thomas K. S., Guerrero M. S., Bouton A. H. (2007) Cancer Res. 67, 6174–6182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reynolds A. B., Kanner S. B., Wang H. C., Parsons J. T. (1989) Mol. Cell. Biol. 9, 3951–3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuda M., Mayer B. J., Fukui Y., Hanafusa H. (1990) Science 248, 1537–1539 [DOI] [PubMed] [Google Scholar]
- 12.Sakai R., Iwamatsu A., Hirano N., Ogawa S., Tanaka T., Mano H., Yazaki Y., Hirai H. (1994) EMBO J. 13, 3748–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riggins R. B., Quilliam L. A., Bouton A. H. (2003) J. Biol. Chem. 278, 28264–28273 [DOI] [PubMed] [Google Scholar]
- 14.Nakamoto T., Sakai R., Ozawa K., Yazaki Y., Hirai H. (1996) J. Biol. Chem. 271, 8959–8965 [DOI] [PubMed] [Google Scholar]
- 15.Burnham M. R., Bruce-Staskal P. J., Harte M. T., Weidow C. L., Ma A., Weed S. A., Bouton A. H. (2000) Mol. Cell. Biol. 20, 5865–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xing L., Ge C., Zeltser R., Maskevitch G., Mayer B. J., Alexandropoulos K. (2000) Mol. Cell. Biol. 20, 7363–7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pellicena P., Miller W. T. (2001) J. Biol. Chem. 276, 28190–28196 [DOI] [PubMed] [Google Scholar]
- 18.Hamasaki K., Mimura T., Morino N., Furuya H., Nakamoto T., Aizawa S., Morimoto C., Yazaki Y., Hirai H., Nojima Y. (1996) Biochem. Biophys. Res. Commun. 222, 338–343 [DOI] [PubMed] [Google Scholar]
- 19.Vuori K., Hirai H., Aizawa S., Ruoslahti E. (1996) Mol. Cell. Biol. 16, 2606–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabodi S., Moro L., Baj G., Smeriglio M., Di Stefano P., Gippone S., Surico N., Silengo L., Turco E., Tarone G., Defilippi P. (2004) J. Cell Sci. 117, 1603–1611 [DOI] [PubMed] [Google Scholar]
- 21.Riggins R. B., Thomas K. S., Ta H. Q., Wen J., Davis R. J., Schuh N. R., Donelan S. S., Owen K. A., Gibson M. A., Shupnik M. A., Silva C. M., Parsons S. J., Clarke R., Bouton A. H. (2006) Cancer Res. 66, 7007–7015 [DOI] [PubMed] [Google Scholar]
- 22.Defilippi P., Di Stefano P., Cabodi S. (2006) Trends Cell Biol. 16, 257–263 [DOI] [PubMed] [Google Scholar]
- 23.Rufanova V. A., Alexanian A., Wakatsuki T., Lerner A., Sorokin A. (2009) J. Cell. Physiol. 219, 45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harte M. T., Hildebrand J. D., Burnham M. R., Bouton A. H., Parsons J. T. (1996) J. Biol. Chem. 271, 13649–13655 [DOI] [PubMed] [Google Scholar]
- 25.Bouton A. H., Burnham M. R. (1997) Hybridoma 16, 403–411 [DOI] [PubMed] [Google Scholar]
- 26.Burnham M. R., Harte M. T., Bouton A. H. (1999) Mol. Carcinog. 26, 20–31 [DOI] [PubMed] [Google Scholar]
- 27.Tice D. A., Biscardi J. S., Nickles A. L., Parsons S. J. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 1415–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riggins R. B., Schrecengost R. S., Guerrero M. S., Bouton A. H. (2007) Cancer Lett. 256, 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nojima Y., Morino N., Mimura T., Hamasaki K., Furuya H., Sakai R., Sato T., Tachibana K., Morimoto C., Yazaki Y., et al. (1995) J. Biol. Chem. 270, 15398–15402 [DOI] [PubMed] [Google Scholar]
- 30.Blake R. A., Broome M. A., Liu X., Wu J., Gishizky M., Sun L., Courtneidge S. A. (2000) Mol. Cell. Biol. 20, 9018–9027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cary L. A., Klinghoffer R. A., Sachsenmaier C., Cooper J. A. (2002) Mol. Cell. Biol. 22, 2427–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dail M., Kalo M. S., Seddon J. A., Côté J. F., Vuori K., Pasquale E. B. (2004) J. Biol. Chem. 279, 41892–41902 [DOI] [PubMed] [Google Scholar]
- 33.Schlaepfer D. D., Broome M. A., Hunter T. (1997) Mol. Cell. Biol. 17, 1702–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai D., Clayton L. K., Smolyar A., Lerner A. (1999) J. Immunol. 163, 2104–2112 [PubMed] [Google Scholar]
- 35.Honda H., Nakamoto T., Sakai R., Hirai H. (1999) Biochem. Biophys. Res. Commun. 262, 25–30 [DOI] [PubMed] [Google Scholar]
- 36.Ishizawar R., Parsons S. J. (2004) Cancer Cell. 6, 209–214 [DOI] [PubMed] [Google Scholar]
- 37.Feller S. M. (2001) Oncogene 20, 6348–6371 [DOI] [PubMed] [Google Scholar]
- 38.Chodniewicz D., Klemke R. L. (2004) Biochim. Biophys. Acta 1692, 63–76 [DOI] [PubMed] [Google Scholar]
- 39.Huang J., Hamasaki H., Nakamoto T., Honda H., Hirai H., Saito M., Takato T., Sakai R. (2002) J. Biol. Chem. 277, 27265–27272 [DOI] [PubMed] [Google Scholar]
- 40.Brinkman A., van der Flier S., Kok E. M., Dorssers L. C. (2000) J. Natl. Cancer Inst. 92, 112–120 [DOI] [PubMed] [Google Scholar]
- 41.van der Flier S., Chan C. M., Brinkman A., Smid M., Johnston S. R., Dorssers L. C., Dowsett M. (2000) Int. J. Cancer 89, 465–468 [DOI] [PubMed] [Google Scholar]
- 42.Dorssers L. C., van Agthoven T., Brinkman A., Veldscholte J., Smid M., Dechering K. J. (2005) Breast Cancer Res. 7, R82–R92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shah A. N., Gallick G. E. (2007) Anticancer Drugs 18, 371–375 [DOI] [PubMed] [Google Scholar]
- 44.Ta H. Q., Thomas K. S., Schrecengost R. S., Bouton A. H. (2008) Cancer Res. 68, 8796–8804 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.