

Abstract
An ability to measure the biochemical parameters and structures of protein complexes at defined locations within the cellular environment would improve our understanding of cellular function. We describe widely applicable, calibrated Förster resonance energy transfer methods that quantify structural and biochemical parameters for interaction of the human estrogen receptor α-isoform (ERα) with the receptor interacting domains (RIDs) of three cofactors (SRC1, SRC2, SRC3) in living cells. The interactions of ERα with all three SRC-RIDs, measured throughout the cell nucleus, transitioned from structurally similar, high affinity complexes containing two ERαs at low free SRC-RID concentrations (<2 nm) to lower affinity complexes with an ERα monomer at higher SRC-RID concentrations (∼10 nm). The methods also showed that only a subpopulation of ERα was available to form complexes with the SRC-RIDs in the cell. These methods represent a template for extracting unprecedented details of the biochemistry and structure of any complex that is capable of being measured by Förster resonance energy transfer in the cellular environment.
Keywords: Hormones, Hormones/Steroid, Methods/Fluorescence, Protein/Protein-Protein Interactions, Receptors/Nuclear, Receptors/Steroid/Thyroid, FRET
Introduction
The dynamic processes that are fundamental to life consist of intersecting webs of interactions among biologic factors and cellular signaling pathways (1, 2). Historically, details of interaction kinetics have been measured in test tube studies that compare the level of interaction between purified factors in relationship to the amounts of those purified factors. The extent to which these quantifiers of interaction are modified when challenged by the complex environment and structure of the living cell remains an open question (3–5). For example, an ability to measure the biochemical details of estrogen action within different cellular environments may be necessary to understand tissue-specific estrogen physiology. An ability to perform cellular biochemistry also may improve our knowledge of, and treatments for, the aberrations of estrogen signaling associated with diseases such as breast or uterine cancers (6, 7). This would involve understanding how interactions of the estrogen receptors with any of a large number of cofactors (8–11) are impacted by cell environment in different tissues of the body.
Current techniques that detect interactions between factors in cells include co-immunoprecipitation, two-hybrid analysis, and Förster resonance energy transfer (FRET)2 microscopy. FRET microscopy senses when factors co-localized in a cellular domain interact in a way that brings fluorophores attached to the factors into close enough proximity to allow energy to transfer from a Donor fluorophore to an Acceptor fluorophore (12–17). All of these methods commonly are interpreted in a simplistic binary “interaction or no interaction” fashion that does not describe quantitative differences among interactions. New technologies are needed to quantify the kinetics of interactions directly in the cell, where the rate and amounts of complexes formed may be affected by the intracellular distributions of the interacting partners and by other cellular factors or cross-talking pathways.
FRET microscopy has been used to detect in cells the hormone-regulated interaction of the α isoform of the estrogen receptor (ERα) or other nuclear receptors with the receptor interacting domains (RIDs) of three cofactors (SRC1, -2, and -3) (18–20). As with all FRET measurements of interactions between factors, a large cell-to-cell scatter in the FRET measurement of ER·SRC-RID interaction has made hormone response difficult to accurately quantify (18–20). Here, we introduce calibrated FRET procedures that complete prior progress toward interpreting the scatter typical of FRET measurements as a function of the kinetic model of biochemical interaction (3, 13, 21, 22). The new analytical and calibration procedures allowed direct measurement in the cell of traditional biochemical parameters (such as the Kd and Bmax, measured in nm) for each of the three ER·SRC-RID interactions. The methods also provided comparative details about the structures of the three different ER·SRC-RID complexes and calculated the concentration of ERα available in the cell to bind each SRC-RID. The widely applicable procedures described here expand the information obtainable from FRET studies and provide previously unknown biochemical and structural details in living cells of the hormone-regulated interactions of the estrogen receptor with three different cofactors.
EXPERIMENTAL PROCEDURES
Transfection
The ERα-CFP and YFP-SRC-RID proteins were expressed from cDNAs inserted into ECFP-N1 and EYFP-C1 expression vectors (Clontech, Palo Alto, CA). For biochemical analysis, 24,000 ± 4,700 (mean ± S.D. across all studies) ERα-CFP molecules were expressed per cell. Constant level expression of this Donor-labeled factor together with variable level expression of the Acceptor-labeled factor (YFP-SRC) was achieved for transient transfections into HeLa cells by engineering the expression vectors to express ERα-CFP poorly relative to YFP-SRC-RID. Under those conditions, 70% (1.4 μg) of the total 2.0 μg of DNA in the transfection mixture consisted of expression vector for the ERα-CFP. Using a transfection reagent that delivered a consistent total amount of vector to each cell (determined in quantitative fluorescence studies in our laboratory), the amount of ERα-CFP expression vector received by each cell was relatively constant. By contrast, different amounts of expression vector for the well-expressed YFP-SRC-RID (0.2 or 0.6 μg) were combined with 0.4 or 0.0 μg of an inert plasmid (Rc/CMV, Invitrogen). 5 μl of Lipofectamine 2000 was added to each 2.0-μg mixture of DNAs. After incubation, the reagent mixtures containing 0.2 and 0.6 μg of YFP-SRC-RID were mixed. One-third of the transfection mixtures (1.3 μg of total DNA) was added to each well of a 6-well dish to which 300,000 HeLa cells grown in estrogen-stripped medium had been plated 1 day earlier onto 22 × 22-mm UV-sterilized No. 1 coverslips.
Data Collection
One day after transfection cells were treated with 10−8 m estradiol or ethanol control vehicle. Images were collected between 30 and 40 min post-treatment on a previously described microscope using a 20×/0.75 NA objective (22, 23). Initial studies conducted on cells treated with estradiol for 2 hours showed similar results. The binding at 30 min, therefore, had reached steady state, which is a prerequisite for equilibrium biochemical analysis. The Acceptor, Donor and FRET images were collected in rapid succession, with the Acceptor images collected at two integration times to capture large variations in YFP-SRC expression levels, as previously described (22, 23). Image collection was followed by previously described semi-automated procedures for background-subtraction, nuclei identification, and debris identification/elimination (22). Regions of interest containing any saturated pixels were eliminated from further analysis. ERα-CFP and all three YFP-SRC-RIDs were localized exclusively in the nucleus, which have similar thickness in different cells to minimize measurement variations originating with the use of an area-based fluorescence intensity measurement as a surrogate for a volume-based concentration measurement (3).
Fluorescence Analysis
YFP-SRC-RID and ERα-CFP levels were measured in fluorescence units that are affected by the instrumentation. Instrument-calibrated bleed-through corrections were implemented to determine raw background-subtracted CFP, YFP, and FRET fluorescence values within each cell nucleus. Further instrument calibrations (22, 24) were used to convert those values to the percent of CFP fluorescence lost to energy transfer (E). The CFP fluorescence intensities then were corrected for that loss of CFP fluorescence and for the calibrated ability of the equipment to detect CFP relative to YFP (3, 22, 24). Fluorescence intensities were converted to ERα and SRC-RID concentrations (in nm) after instrument calibrations that used a stable cell line expressing a consistent, measured amount of a dual-labeled CFP-AR-YFP (where AR is the androgen receptor) expressed in HeLa cells. The average number of CFP-AR-YFP molecules present in each cell was determined by comparing Western blots of whole cell lysates from 10 × 106 cells with known amounts of AR measured as the amount of [35S]methionine incorporated during in vitro translation. The average number of CFP-AR-YFP moles/cell was divided by the average volume of each cell (measured from 10 × 106 cells) and compared with the average fluorescence of CFP-AR-YFP in each stable cell to correlate fluorescence intensity (in fluorescence units) with concentration.
Biochemical Conversion
Data transformation with Equations 2–4 was done using Excel (Microsoft, Redmond, WA) using initial assumptions of ERα availability that varied, mostly in 5% increments, from 1 to 100%. Common FRET read-outs other than E will not be linear with the concentration of the ER·SRC-RID complex (3) and should not be used for the data transformations described here. The transformed data were transferred to Prism (GraphPad; San Diego, CA) from which the best-fitting curves for Equation 1 were calculated at each assumed level of availability using no fixed variables, strict convergence criteria, and no weighting. All curves shown were determined using first order kinetics. The Bmax data collected for each assumed level of ERα availability was assessed for its fit to the measured concentration of ERα-CFP, as described under “Results.” ERα availabilities were resolved reiteratively until the calculated ERα concentration agreed within 0.01 nm to the ERα concentration measured by fluorescence intensity.
RESULTS
Application of Biochemical Theory to FRET Analysis
The standard biochemical relationship for interaction between two factors, ERα and SRC, is shown in Equation 1.
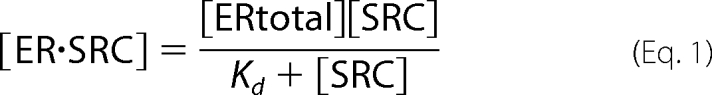 |
Equation 1 describes a curve (Fig. 1A) in which, for a fixed concentration of all ERα ([ERtotal]), the concentration of ERα bound in the complex ([ER·SRC], y axis) increases with the concentration of free SRC ([SRC], x axis) up to a maximal ER·SRC concentration (the Bmax). The free SRC concentration (x axis) at which half of the complex is formed (y = 0.5 × Bmax) represents the equilibrium dissociation constant (Kd), which is the amount of free SRC present when ERα is partitioned equally into the bound and free states. The challenge that will be addressed here is the conversion of FRET and fluorescence intensity measurements into the concentrations of bound ERα and free SRC, which then can be applied to measure biochemistry in the cell environment.
FIGURE 1.

Application of biochemical theory to FRET analysis. A, curve described by Equation 1 that depicts the concentration of ERα in an ER·SRC complex in relationship to the concentration of unbound SRC. B, representative ERα-CFP and YFP-SRC2-RID images from a typical study collected using excitation/emission wavelengths (in nm) of 431–440/455–485 and 496–505/520–550. A third image was collected (431–440/520–550) from which were subtracted the contributions of ERα-CFP and YFP-SRC2-RID. The energy transfer levels in the resulting FRET images varied with estradiol treatment and with the level of YFP-SRC expressed in each cell nucleus. FRET data points in subsequent figures also are converted, according to previously defined equipment calibrations (22, 24), from raw FRET intensity levels into the percentage of ERα-CFP lost to energy transfer.
To apply biochemical theory to FRET measurement, a relatively constant amount of ERα fused at its carboxyl terminus to CFP (fusion described in supplemental Fig. 1A) was expressed transiently in HeLa cells with variable levels of the ∼185-amino acid-long RIDs of three different SRC cofactors (25) fused at their amino termini with YFP (see supplemental Fig. 1B). The SV40 nuclear localization signal also was added to each YFP-SRC-RID fusion. This ensured that all three SRC-RIDs resided together with ERα in the nuclear compartment (Fig. 1B) from which all measurements were made. As will be described in the following sections, the amounts of energy transfer in each cell nucleus (Fig. 1B, FRET) increased in estradiol-treated cells with increasing amounts of co-expressed YFP-SRC-RID in a fashion that was consistent with biochemical theory.
The RIDs for all of SRC1, -2, and -3 were used in the current studies because energy transfer from ERα-CFP to each of the YFP-SRC-RIDs could be detected (18). By contrast, the co-expression of ERα-CFP with full-length SRC2 showed no energy transfer even when estradiol was added to the cells (not shown). The large size of the intact cofactor likely positioned the YFP too far (>80 Å) from the CFP to support energy transfer in the complex. This maximal distance constraint represents one limitation of the FRET approach, although as described below, the strict sensitivity of FRET to distance is an asset for obtaining structural information from the FRET data.
Relationship of Energy Transfer to Total Acceptor Fluorescence
For the biochemical interaction curve (Fig. 1A), a FRET measurement that is linear with the amount of complex formed can be used as a surrogate measurement of the proportion of ERα in the ER·SRC-RID complex (Fig. 1A, y axis) (3, 22). The percentage of CFP energy transferred from ERα-CFP to YFP-SRC-RID, referred to as the efficiency of energy transfer (E), is one such measurement of complex concentration. Fig. 2A shows an example of E (y axis) plotted against the fluorescence intensity of YFP-labeled SRC-RID (x axis) in each of a total of 720 nuclei in cells treated with either 10−8 m estradiol (closed boxes) or drug vehicle (open boxes). The measurements from cells treated with estradiol generally fit well to curves described by Equation 1; “goodness of fit” (R2) values were 0.76 ± 0.16, 0.82 ± 0.10, or 0.70 ± 0.12 (mean ± S.D. from three independent studies) for interactions of ERα with the SRC1, SRC2, or SRC3 RID, respectively. Runs tests, which assess whether abnormal runs of consecutive data points reside above or below the best-fitting curve, showed a statistically significant run in only one of those nine studies.
FIGURE 2.

Emax compares the structure of the complexes. A, the relationship of the percent energy transfer (E) from ERα-CFP in the ER·SRC-RID complex to the total amount of YFP-SRC2-RID (fluorescent units). This example shows measurements from a total of 720 cells treated with estradiol (closed boxes) or with vehicle (open boxes). Emax, maximal amount of ERα-CFP energy transfer at saturating YFP-SRC2-RID. B, Emax for the interaction of ERα-CFP with each of the YFP-tagged SRC1, -2, and -3 RIDs (mean ± S.D. from three independent experiments) and with ERα-YFP (nine independent experiments).
By contrast, in the absence of ligand (two independent studies each), R2 values were 0.14 ± 0.07, 0.24 ± 0.00, or 0.28 ± 0.11 (mean ± range) for the SRC1, -2, and -3 RIDs with runs tests showing that 3 of the 6 curves deviated significantly from those poorly fitting curves. Thus, estradiol addition resulted in a sufficient increase in the cell-to-cell uniformity in FRET levels such that the data from all cells could fit well to a single binding curve. The consistent and higher levels of energy transfer upon estradiol addition may have resulted from an enhanced ability of ERα to interact with each SRC-RID and/or a more favorable repositioning of the fluorophores in a pre-existing ER·SRC-RID complex. As described below, these well-fitting curves provided measurements necessary to define the structure of the estradiol-bound ER·SRC-RID complex and provided starting values to extract biochemical kinetic parameters in classical terms.
Conformation of ERα Complexes with the SRC1, -2, and -3 RIDs
For cells treated with estradiol, the maximal energy transfer at saturating SRC-RID levels (Fig. 2A, Emax) was extrapolated from the well-fitting curves. The Emax measurements were consistent across three independent experiments for the RIDs of each of SRC1, -2, and -3 (Fig. 2B). The Emax is distinct from the Bmax, discussed later, in that the maximal level of energy transfer is affected by the distance between, and orientations of, the fluorescent proteins in the ER·SRC-RID complex (26–28). The Emax, therefore, is a quantifier of the structure of the complex rather than a biochemical quantifier (3). The Emax values determined for all three SRC-RIDs were not statistically different from each other (p > 0.05). For comparison, the Emax determined for the estradiol-induced dimer interaction of ERα-CFP and ERα-YFP was determined in parallel (Fig. 2B) and was considerably higher (p < 0.001).
From the Emax measurements, the distances between the dipoles of CFP and YFP in the ER·SRC-RID or ER·ER complexes were calculated as 62.1 ± 1.1 Å (ER·SRC1-RID complex), 60.5 ± 1.6 Å (ER·SRC2-RID), 61.1 ± 2.1 Å (ER·SRC3-RID), and 53.2 ± 1.2 Å (ER·ER). Thus, 7 Å differences in the distances between the fluorescent proteins in the ER·SRC-RID and ER·ER complexes are detected with high statistical confidence. Those distance measurements are based upon a distance of 49.2 Å at 50% energy transfer from CFP to YFP (29) and the inverse association of energy transfer with the sixth power of the distance between the fluorophores (26–28). The distance estimates are provided to demonstrate the exquisite sensitivity of the FRET measurement to conformation. However, dipole orientation also affects energy transfer (26–28), and the apparent similar positions of the fluorescent proteins in all three ER·SRC-RID complexes may be erroneous if structural constraints orient CFP and YFP differently within each complex.
Dependence of ER·SRC-RID Complex Formation on AF-2/LXXLL Interaction
One purpose of these studies was to compare and contrast biochemical measurements made within the cell to those previously measured by traditional in vitro methods. We first established if the ER·SRC-RID complexes detected in the cell by FRET were analogous to the ER·SRC-RID interactions that have been characterized in vitro. Mutations known to reduce ER·SRC-RID interaction in vitro (25, 30, 31) were introduced into ERα-CFP and the YFP-SRC-RIDs. Because the FRET data points obtained with those mutants did not fit well to the binding curve, quantitative comparisons of the extent of each mutant's effect on ER·SRC-RID complex formation depended on defining methods that did not rely on curve-fitting.
For interactions of wild-type ERα and each SRC-RID, a strong and consistent FRET signal could be detected from cells expressing close to saturating levels of YFP-SRC-RID (Fig. 2). Therefore, FRET measurements were compared only from cells expressing more YFP-SRC-RID than needed to achieve half of Emax for the estradiol-dependent interaction between the wild-type factors. Fig. 3A shows one example of a scatterplot depicting individual FRET measurements collected from ERα-containing cells expressing only this high level of SRC2-RID. In the absence of estradiol, energy transfer from wild-type ERα-CFP to wild-type YFP-SRC2-RID (Fig. 3A, open boxes) was greater on average than zero FRET (neg, defined as the measurement noise from the negative control expressing ERα-CFP in the absence of YFP-SRC). FRET increased strongly when the cells were treated with estradiol (Fig. 3A, closed boxes). The interactions of wild-type ERα with wild-type SRC2-RID (or the SRC1 and SRC3 RIDs, not shown) fell into three general levels of energy transfer defined by visual inspection of all studies (dotted lines, Fig. 3A): no interaction typical of the negative control (E < 1.75%), low level interaction typical of that observed in the absence of estradiol (1.75 > E < 6.25%), and high level interaction typical of estradiol treatment (E > 11.25%).
FIGURE 3.

ERα interaction with the SRC2-RID is disrupted by mutants within the AF-2 and LXXLL motifs. A, scatterplot of E measured in individual cells expressing wild-type (wt) ERα-CFP and high levels (>1 Kd) of YFP-SRC2-RID. B, histograms of percentage of cells expressing ERα-CFP and high levels of wild-type YFP- SRC1, -2, or -3 RID that fall into the indicated levels of energy transfer. Also shown are the effects on those histograms of mutants disrupting: C and D, ERaf2, ERα K362A mutant inhibiting activation function-2 within the ERα SRC-LXXLL-interacting surface; E and F, ERmono, ERα L504A/L508A/L511A mutant reducing ERα dimerization; G and H, SRClxxll, SRC LXXLL mutated to AXXAA. E2, estradiol-treated cells; neg, zero FRET collected from cells expressing ERα-CFP only. The examples shown in A, C, E, and F were collected in a single study and thus show the same negative control. Data were collected from three or two independent experiments for estradiol-treated (mean ± S.D.) or vehicle-treated (mean ± range) cells, respectively.
The percentage of all cells with E in each level and in a fourth intermediate level (6.25 > E < 11.25%) was quantified from multiple studies of wild-type ERα-CFP with YFP-SRC1, SRC2, or SRC3 RID and plotted as a histogram (Fig. 3B). As an example, for ERα interaction with SRC2-RID in the absence of hormone (open triangles), 41% of cells showed no interaction (E < 1.75%), 50% of cells showed low level interaction (1.75 > E < 6.25%), 9% of cells showed moderate level interaction (6.25 > E < 11.25%), and 0% of cells showed high level interaction (E > 11.25%). The distribution of data observed for cells in the absence of estradiol was well above the measurement noise of the negative control cells (Fig. 3B, neg). Upon the addition of estradiol (Fig. 3B, closed triangles), the distribution of energy transfer levels changed dramatically to <1, <1, 5, and 94% in the lowest to highest energy transfer categories.
This histogram analysis showed that the estradiol-regulated interactions of wild-type ERα with the wild-type SRC1-RID (Fig. 3B, boxes), wild-type SRC2-RID (triangles), and wild-type SRC3-RID (diamonds) all were affected strongly by the K362A mutation in ERα (Figs. 3, C and D). The K362A mutation disrupts the ERα “activation function-2” (AF2) surface that mediates interaction with the SRC RIDs (31). In the absence of estradiol, the weak interaction of all three SRCs with ERα was virtually eliminated by the K362A mutation. The stronger interaction in the presence of estradiol was substantially decreased, but not eliminated, by the K362A mutation.
An ERα mutant (L504A/L508A/L511A) that strongly reduces ERα dimerization (32) blocked estradiol-independent interaction only with SRC1 and had more modest effects on the other estradiol-regulated interactions with the three SRCs (Figs. 3, E and F). Detailed cellular biochemical analyses of this mutant (see later in text) reveal how ERα dimerization affects the extent and types ER·SRC-RID complexes formed in a cell.
Three conserved LXXLL motifs present in the RIDs (see supplemental Fig. 1B) mediate in vitro binding of each SRC-RID to the ERα AF-2 (25). Mutation of all three LXXLL motifs in the SRC1-RID eliminated both the estradiol-independent and -dependent SRC1 interaction with ERα (Figs. 3, G and H). Mutation of two LXXLL motifs in the SRC2-RID previously reported to participate in the in vitro interaction with ERα (30) reduced, but did not eliminate, interaction. Overall, the mutational analysis showed that ER·SRC-RID interactions detected by FRET in living cells generally depended on the same AF-2/LXXLL interaction sites characterized in vitro.
Measurement of Kd and Bmax
Measurement of the kinetics of ERα interaction with the SRC1, -2, or -3 RIDs in the cell required the development of methods and calibrations to convert the y and x axes of the initial data plots (Fig. 2A) from E and total YFP-SRC-RID fluorescence intensity into molar concentrations of the bound ERα and the free SRC-RID (as in Fig. 1A). Measured YFP and CFP fluorescence levels (corrected for the percentage of CFP lost to energy transfer) first were converted into the molar concentrations of SRC-RID and ERα in the cell using fluorescence:molar calibrations defined from a stable cell line expressing a known amount of YFP and CFP (see “Experimental Procedures”). These measurements reflect the concentrations of all SRC-RID (i.e. bound and free, [SRCtotal]) and all ERα ([ERtotal]) in the cell.
Within each cell the proportion of ERα bound with SRC-RID is equivalent to the amount of E measured in that cell divided by the Emax extrapolated from all cell measurements. Knowing the total concentration of ERα ([ERtotal]) from the CFP fluorescence, the concentration of ERα in the ER·SRC-RID complex in each cell (desired y axis of the curve) can be determined by multiplying the proportion of ERα bound by the total concentration of ERα (Equation 2).
ERavail refers to the proportion of ERα present in the cell that is available to bind to SRC-RID. As shown below, ERavail can be limited by events in the complex cellular environment. This contrasts with in vitro binding studies in which all ERα generally is considered to be available for binding.
The concentration of free (not bound) SRC-RID (desired x axis) also is necessary to extrapolate biochemical kinetic values from the FRET data. The YFP fluorescence data provides information about the total concentration (both bound and free) of SRC such that the concentration of free SRC could be determined as shown in Equation 3.
The concentration of SRC-RID bound in the complex will be equivalent to the concentration of bound ERα multiplied by the stoichiometry of SRC-RID relative to ERα in the complex. Thus, the concentration of free SRC-RID will be determined by Equation 4,
where N is the number of SRC-RID molecules in a complex relative to ERα molecules.
The concentrations of bound ERα and free SRC-RID were determined for each data point using the above equations. Because the binding curve (Equation 1) is for the interaction of variable SRC-RID amounts to a constant amount of ERα, the total ERα concentration used in the data transformation for each cell was that averaged from all cells within an experiment. An example (Fig. 4A) shows the data transformation for the same estradiol-treated cells shown in Fig. 2A, assuming that all ERα was available to bind the SRC RIDs at 1:1 ER·SRC-RID stoichiometry. The transformed data do not fit to the best-fitting curve described by Equation 1 and were skewed above the curve at low free SRC-RID concentrations (Fig. 4B, Residuals). When the data transformation is correct, the Bmax derived from the transformed curve also should agree with the average ERα concentration measured directly in the cells by ERα-CFP fluorescence (12.0 nm), which was not observed for this data transformation (27.1 nm ER·SRC-RID, from Fig. 4A).
FIGURE 4.

Transformation of FRET data into biochemical curves. A, Equations 2–4 were used to transform the data from the estradiol-treated cells shown in Fig. 2A assuming that all ERα was available in the cell to interact with SRC-RID at saturation. B, residuals show the extent to which each data point lies above or below the poor-fitting curve of panel A. C, the same data were transformed under variable assumptions of ERα availability or ER·SRC-RID stoichiometry until the corrected data were consistent with the total concentration of ERα-CFP measured in the cells. D, the residuals were more consistently distributed around the curve with the corrected data.
The data transformations, therefore, were repeated with a range of ERα availability or stoichiometry assumptions to identify data transformations that agreed with the measured level of ERα. For the example provided, the Bmax of 4.41 nm ER·SRC-RID, obtained when 36.7% of the ERα was assumed to be available to bind SRC-RID at 1:1 stoichiometry (Fig. 4C), was consistent with the 12.0 nm total ERα measured (4.41/0.367). Note that this curve also was consistent with an interaction of 73.4% of available ERα at a stoichiometry of 1 SRC-RID per 2 ERα. With this data transformation, a slight skewing of the data points above the curve was observed at very low SRC-RID concentrations (Fig. 4D), which is discussed later.
Biochemistry and Structure of ERα Complexes with the SRC1, -2, and -3 RIDs
Reiterative solving of the Bmax using variable ERα availability was repeated for three independent studies for each of the SRC1, SRC2, and SRC3 RIDs collected in estradiol-treated cells. The Bmax, Kd, and ERα availability were averaged from the three studies (Fig. 5A). The concentration of ER·SRC-RID at saturation (Bmax) averaged ∼5 nm for all three SRCs, which was approximately half of the measured intracellular ERα concentrations (11.3, 12.4, and 11.7 nm). This could indicate a 2:1 stoichiometry of ERα to SRC-RID in the ER·SRC-RID complex in living cells. However, Bmax values as high as 6 nm ER·SRC-RID were obtained at ERα concentrations as low as 8 nm (Fig. 5B). This suggested that the average stoichiometry of ERα and SRC-RID was less than 2:1 in the cellular complex, at least under the conditions of SRC-RID excess at which Bmax is measured.
FIGURE 5.

Biochemistry of ER·SRC-RID complex formation. A, Bmax, Kd, and ERα availability measured for ERα interaction with SRC1, -2, or -3 RID in estradiol-treated cells (mean ± S.D., three independent experiments). B, the Bmax levels, which measure the concentration of ERα in an ER·SRC-RID complex, do not vary with ERα-CFP concentration in individual experiments (±S.E. for the curve fit within an experiment).
Studies of SRC1, -2, and -3 RID interactions with the dimerization-deficient ERα mutant further indicated that the ER·SRC-RID complex could form in the absence of dimerization of ERα. This mutant disrupts ERα dimerization (32) through the interface identified by crystallography (33). As with the wild-type ERα, consistent Emax measurements, albeit lower than that for wild-type ERα, could be defined for interaction of this “monomeric” ERα with all three SRC-RIDs (Figs. 6, A and B). The ability to define Emax enabled cellular biochemical measurements to be determined for SRC-RID interactions with the monomeric ERα (Fig. 6C). The maximum amount of ER·SRC-RID complexes formed (the Bmax) with SRC1 or SRC2 was the same for the wild-type and monomeric ERα, whereas the Bmax was lower for SRC3 binding to the monomeric ERα. Overall, interaction with the monomeric ERα further demonstrated that ERα could form complexes with all SRC-RIDs at less than 2:1 ER·SRC-RID stoichiometry in the cell environment.
FIGURE 6.

Mutation of ERα dimer interface minimally affects overall ER·SRC-RID complex formation. A, Emax determination for the monomeric ERα mutant (closed boxes) in comparison to wild-type ERα collected in parallel (open boxes; same example shown in Fig. 2A). B, Emax and C, Bmax, Kd and ERα availability measured for interaction of the monomeric ERα mutant with SRC1, -2, and -3 RID (mean ± range, two independent experiments).
Stoichiometry and Affinity of the ER·SRC-RID Complexes Vary with SRC-RID Concentration
Comparison of the biochemical and structural parameters measured for the wild-type and monomeric ERα helped to define further details about the ER·SRC-RID complexes formed within the cell. The binding affinities measured for interaction of the SRC1, SRC2, and SRC3 RIDs with wild-type ERα (Kd values of 4.9, 2.6, and 2.8 nm, respectively) were stronger than those measured with the monomeric ERα (Kd values of 9.7, 4.2, and 4.4 nm). The lower affinity measured with the monomeric ERα presumably was a direct result of the inability to form a dimer. However, if the hypothesis is correct that the wild-type and mutant ERα both interact as a monomer with the SRC RIDs (at least at saturating SRC-RID concentrations), then the Kd values of interaction with the wild-type ERα and monomeric ERα should have been the same. We, therefore, examined whether the binding curves of wild-type ERα with the SRC-RIDs showed any evidence for an ERα dimer-containing complex at subsaturating SRC-RID levels.
The best-fitting curves for all data sets were calculated under the assumption of uniform, first order interaction kinetics across all SRC-RID concentrations. The curve-fits and data transformations also assumed that the ER·SRC-RID complexes formed at low SRC-RID concentrations provided the same level of energy transfer as the complexes at high concentrations (3). If those assumptions were true, then the binding curves should fit equally well to the data points at low and high SRC-RID concentrations.
Visual inspection of the binding curves for SRC-RID interactions with the wild-type ERα (see Fig. 7A for an example of one SRC3-RID study) showed at the lowest SRC-RID concentrations, a tendency for the best-fitting curve to lie to the right of the actual data points. This deviation was most evident when plotting the distance of each data point above or below the curve in relationship to free SRC-RID concentration (Fig. 7B, residuals). To examine if this deviation was consistently observed, the residuals for interactions with the wild-type ERα were averaged across multiple studies in relationship to SRC-RID concentrations (Fig. 7C) grouped as (a) less than one Kd, (b) between the Kd and 2× Kd, (c) from 2–3× the Kd, and (d) greater than 3× the Kd. This analysis was repeated for SRC-RID interactions with the monomeric ERα (Figs. 7, D–F). Evidence for the second complex was observed consistently at sub-Kd concentrations for the interaction of wild-type ERα with all SRC-RIDs (Fig. 7C), after which the data points were distributed normally around the curve. The high affinity component was diminished for SRC-RID interactions with the monomeric ERα (Fig. 7F), suggesting that ERα dimerization contributes to the second component of the binding data.
FIGURE 7.

A high affinity component of the ER·SRC-RID complex is dependent on the ERα dimer interface. A, the binding curve and B, residuals from that binding curve for the interaction of wild-type (wt) ERα with wild-type SRC3-RID show a strong tendency for the data points to lie above the curve at very low SRC-RID concentrations. mut, mutant. C, the residuals for data points at low SRC-RID concentrations (<1 Kd) are elevated consistently for six total studies (mean ± S.D. of the average residuals in each of two studies each for the SRC-1, -2, and -3 RIDs). D–F, analysis of SRC-RID interaction with the monomeric ERα mutant collected in parallel suggests that the high affinity component of ER·SRC-RID interaction is dependent upon an ability to form a dimer.
High Affinity ER·SRC3-RID Interaction
The data sets captured for each of the SRC1 (Fig. 8A), SRC2 (Fig. 8B) and SRC3 (Fig. 8C) RIDs showed that each SRC-RID behaved uniquely with respect to the complex found at low SRC-RID concentrations (filled bars). SRC3-RID showed the strongest elevation above the curve, suggesting that the actual Kd of the wild-type ER·SRC3-RID complex was considerably lower (higher affinity) than the 2.8 nm estimated from the binding curve. Similarly the Kd of the ER·SRC2-RID complex is likely to be moderately lower than the 2.6 nm measured, whereas the Kd of the ER·SRC1-RID complex is likely to be closer to the 4.9 nm measured. When examined with the monomeric ERα (Fig. 8, open bars), the binding curves fit well for interaction of ERα with SRC1-RID and SRC3-RID; the monomeric ERα mutant did not reduce the deviation in the binding kinetics of the ER·SRC2-RID complex as strongly.
FIGURE 8.

High affinity ERα complexes with the SRC1, -2, or -3 RIDs. Analysis of the extent to which the residuals deviate from the best-fitting curve for ERα interaction with A, SRC1-RID, B, SRC2-RID, and C, SRC3-RID. Interactions were measured with the wild-type (wt, filled bars) or monomeric ERα mutant (mut, open bars). Data represent the mean ± range of the average residuals in two independent studies each for the SRC-1, -2 and -3 RIDs.
Given the possible two-component nature of ER·SRC-RID complex formation, the data also were solved using best-fitting curves with two components, A and B, as shown in Equation 5.
Two-site binding assumptions provided Bmax or Kd determinations that were in most cases illogical (e.g. first order Kd < 0 and second component Bmaxs that are extremely large and not supported by the data). This likely reflects the problem that each data point at the lower SRC-RID concentrations consists of different proportions of high affinity 2:1 ER·SRC-RID complexes and multiple 1:1 complexes with different characteristic energy transfer levels (see “Discussion”). A future challenge will be to devise higher dimensional computational processes that conduct data transformations to define the best-fitting solution under all possible progressions of mixed stoichiometries and structures (3).
DISCUSSION
Cellular Biochemistry
A goal of cellular biochemistry has been to define and quantify cellular events in terms of the biochemical parameters typically measured in vitro (3, 13, 21, 22). Ideally, detailed interaction kinetics could be measured in the cellular environment with the same level of precision possible in vitro. We developed calibrated FRET techniques that allow traditional biochemical parameters to be discerned in living cells. The techniques can be adapted to measure the biochemistry and structure of any interacting complex directly in living cells, provided that the interacting partners can be tagged with fluorescent proteins at positions permissive for energy transfer within the complex.
Fluorophore distance requirements place some limitations on the interactions that can be studied by FRET. In the current example, energy transfer could be examined only with the receptor-interacting domains of the large (160 kDa) SRC co-factors, and it remains uncertain whether measurements on this non-functional complex accurately reflect the dynamic processes mediated by a full-length SRC (34). The cellular biochemistry measurements also represent the average of all interactions within the cellular domain measured (the nucleus) and do not resolve biochemical parameters unique to submicroscopic cellular or gene-specific microenvironments. Still, the ability to define precise global biochemical details with a cellular domain will be useful for the investigation of biologic action.
Interactions of ERα and SRC-RIDs in the Cell
The novel cellular biochemical and structural capabilities provided previously unknown details of ER·SRC complex formation and confirmed, in the cell environment, some prior understandings of ER binding to the SRCs. In the absence of hormone, energy transfer of ERα-CFP to all three YFP-SRC-RIDs was clearly above background and was substantially increased upon estradiol addition (Figs. 2 and 3). Estradiol induction of energy transfer may be consistent with a direct induction of ERα-CFP interaction with the three YFP-SRC-RIDs. Alternatively, estradiol activation of energy transfer could originate from an estradiol-regulated re-positioning of “helix 12” of ERα (33, 35) to bring CFP into a position within the ER-CFP·YFP-SRC-RID complex more optimal for consistent, measurable levels of energy transfer to YFP. FRET data alone cannot distinguish between those possibilities.
Mutational analyses (Fig. 3) showed that the interactions measured in the cell by FRET were dependent upon the interaction surfaces in ERα and in the SRC-RIDs previously shown by in vitro studies to contribute to the estradiol induction of ER·SRC-RID interaction (25, 30, 31). This suggests that the estradiol-regulated increase in FRET in the cell may originate with increased cellular interaction of ERα with the SRC-RIDs. Although that conclusion remains subject to the limitations of the FRET approach, the studies showed more conclusively that the ERα complexes formed with the SRC1, SRC2, and SRC3 RIDs in the cell had similar molecular dependences (Fig. 3) and conformations (Emax, Fig. 2). These global similarities were consistent with evidence for some functional redundancies among the SRCs in knock-out mouse studies, although those same studies also clearly show SRC-specific functions (6, 36–38).
The Kd values of the interactions showed that ERα interacts in the cell with all SRC RIDs at mildly different relative affinities (Figs. 5 and 8). The binding preference (SRC3 ≥ SRC2 > SRC1) was slightly different than the relative affinities reported for in vitro binding of ERα to the SRC RIDs (SRC3 > SRC1 ≥ SRC2) (34). For SRC1-RID binding to ERα in the cells, the Kd determined here (5 nm) is similar to the 5–30 nm reported from prior in vitro competition or direct binding studies (34, 39, 40). Thus, the affinity for at least SRC1-RID interaction with ERα was not grossly affected by cellular factors.
The comparison of the Bmax measurements with the total concentration of ERα within the cell demonstrated that the cellular environment restricted the amount of ERα available to bind to any of the SRC1, -2, or -3 RIDs (Fig. 5B). It is conceivable that the 5–6 nm limit defined here may be a direct result of a dependence of the ER·SRC-RID complex on some modification of ERα and/or SRC or a reliance on an additional limiting factor. The limit also could reflect functional limitations imposed by the cell. For example, the ER·SRC complexes undergo rapid association/dissociation cycles at the promoters of specific genes (41–43), which imply that the transcriptional process itself will cause a subgroup of ERα to be in the unbound state at any given time. This ability to measure the maximal binding capacity within the cell is a unique feature of the cellular biochemistry approach. Thus, cellular biochemistry can examine the effects of cellular events or conditions on multiple facets of biologic response.
Dynamic Composition of the ER·SRC-RID Complex
Prior in vitro studies of interaction between increasing amounts of ERα with full-length SRCs and SRC RIDs indicated that the formation of stable ER·SRC complexes proceeds first through a transient, intermediate ER·SRC complex (34). Here, the elevation in data points above the curve specifically for the wild-type ERα at low SRC-RID concentrations (Figs. 7 and 8) also suggested that a high affinity SRC-RID complex forms first with a dimeric wild-type ERα, which at higher SRC concentrations progressively shifts toward 1:1 ER·SRC-RID complexes. Increases of only ∼10,000 bound and free SRC-RID molecules were sufficient to alter the balance from predominantly 2:1 to 1:1 ER·SRC-RID complexes. Thus, the composition of ER·SRC complexes could be different, for example, in the subpopulation of breast tumors with high levels of expression of SRC3 (also called amplified in breast cancer 1, AIB1) (44–46). The ability to define and distinguish the different types of complexes capable of forming within different tumor environments may assist in identifying the types of complexes best targeted for novel breast cancer therapies.
Different Emax values were observed for 1:1 ER·SRC-RID complexes formed at high SRC-RID levels with the monomeric ERα (Emax, ∼10%) and wild-type ERα (Emax, ∼20%) (Figs. 2B and 6B). An explanation for these structural differences in the wild-type and mutant ERα complexes is suggested by the crystallographic structure (47) of the estradiol-bound ERα dimer (Fig. 9A, each monomer is in light or dark blue) in a complex with estradiol (gray) and two LXXLL peptides (light and dark brown). In vitro binding studies with isolated peptides representing each of the three LXXLL motifs found in each of the three SRC-RIDs indicated that ERα binds with highest affinity to the second LXXLL motif of all SRCs (39) (Fig. 9A, LXXLL-2). For all three SRCs, ERα binding to LXXLL-1 is uniformly poorer. Interaction with LXXLL-3 is below the sensitivity of the in vitro binding assays (39) but sufficient to co-crystallize with ERα (47). The high affinity interaction of the ERα-CFP dimer with a single YFP-SRC-RID, thus, would position the two CFPs (Fig. 9, cyan ovals) to transfer energy at different levels (gray arrows) to the single YFP positioned closest to the lower affinity, first LXXLL motif.
FIGURE 9.

ERα interaction with the SRC RIDs. A, crystallographic structure from Pike and co-workers (47) of the ERα dimer (light or dark blue) bound to estradiol (gray) and to two LXXLL peptides (light and dark brown). The approximate locations of CFP and YFP are indicated as cyan and green ovals. Energy transfer from CFP to YFP is indicated by gray arrows. Amino acids of unknown structure connecting different structural domains are indicated as dotted lines. The locations of amino acids mutated within the AF-2/LXXLL interface are shown as yellow (K362A) or lighter yellow (LXXLL, only the underlined Ls are displayed, as the intermediate leucine lies over top of, and would block the view of, Lys-362). Also highlighted are those amino acids mutated in the dimer interface (yellow) and their interacting amino acids (lighter yellow). The structures were obtained and displayed using the NCBI-Entrez publicly accessible three-dimensional structure data base (MMDB code 2033B; PDB code 1GWR) (49, 50). B and C, hypothetical levels of energy transfer when a single ERα monomer interacts with a single LXXLL peptide.
If the 1:1 complexes formed with wild-type ERα arise through a high affinity 2:1 ER·SRC-RID intermediate complex, then the resulting complexes will be equally distributed between two different 1:1 ER·SRC-RID conformations (Figs. 9, B and C). The maximal energy transfer levels determined, therefore, will be an average of the FRET levels of those two structures. By contrast, interaction of each SRC-RID with the monomeric ERα mutant will favor the binding of high affinity LXXLL-2, which will result in lower energy transfer levels per ERα molecule. Indeed, the SRC2 mutant that retains only an intact, poor affinity LXXLL-1 motif (Figs. 3, G and H) did not bind ERα well enough to extrapolate binding curves.
The cellular biochemistry curves, therefore, can be much more complex than those formed in vitro with isolated, defined factors. Limitations associated with any biochemical measurement, such as the assumption that all data points will behave uniformly with respect to a single curve, also may be problematic if individual cells in different states behave differently. Still the techniques shown here provide a template for the measurement of biochemistry directly in living cells and for the identification of instances in which biochemistry in the cell is different from that in vitro. The capability to measure biochemical parameters for interactions within cells together with automated data collection and analysis capabilities now available with high throughput microscopy (5, 48) opens the door to a wide-scale, rapid, and comprehensive screening for cellular events impacting biochemical action. The advent of the cellular biochemistry approach is expected to dramatically improve our understandings of the complex webs of biochemical pathways that regulate cell function.
Supplementary Material
This work was supported by the Congressionally Directed Medical Research Programs (Grant PC040777) and by the National Institutes of Health (Grant R01 DK54345) with infrastructure support from the National Institutes of Health (Grant P30 DK63720).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- FRET
- Förster resonance energy
- ER
- estrogen receptor
- RID
- receptor interacting domains
- AR
- androgen receptor
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein.
REFERENCES
- 1.Kitano H. (2002) Science 295, 1662–1664 [DOI] [PubMed] [Google Scholar]
- 2.Liu A. P., Fletcher D. A. (2009) Nat. Rev. Mol. Cell Biol. 10, 644–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H., Puhl H. L., Ikeda S. R. (2007) J. Biomed. Opt. 12, 054011 [DOI] [PubMed] [Google Scholar]
- 4.You X., Nguyen A. W., Jabaiah A., Sheff M. A., Thorn K. S., Daugherty P. S. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 18458–18463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giuliano K. A., Johnston P. A., Gough A., Taylor D. L. (2006) Methods Enzymol. 414, 601–619 [DOI] [PubMed] [Google Scholar]
- 6.Xu J., Li Q. (2003) Mol. Endocrinol. 17, 1681–1692 [DOI] [PubMed] [Google Scholar]
- 7.Pearce S. T., Jordan V. C. (2004) Crit. Rev. Oncol. Hematol. 50, 3–22 [DOI] [PubMed] [Google Scholar]
- 8.Acevedo M. L., Kraus W. L. (2004) Essays Biochem. 40, 73–88 [DOI] [PubMed] [Google Scholar]
- 9.Glass C. K., Rose D. W., Rosenfeld M. G. (1997) Curr. Opin. Cell Biol. 9, 222–232 [DOI] [PubMed] [Google Scholar]
- 10.McKenna N. J., Cooney A. J., DeMayo F. J., Downes M., Glass C. K., Lanz R. B., Lazar M. A., Mangelsdorf D. J., Moore D. D., Qin J., Steffen D. L., Tsai M. J., Tsai S. Y., Yu R., Margolis R. N., Evans R. M., O'Malley B. W. (2009) Mol. Endocrinol. 23, 740–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lonard D. M., O'Malley B. W. (2007) Mol. Cell 27, 691–700 [DOI] [PubMed] [Google Scholar]
- 12.Day R. N., Schaufele F. (2005) Mol. Endocrinol. 19, 1675–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon G. W., Berry G., Liang X. H., Levine B., Herman B. (1998) Biophys. J. 74, 2702–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koushik S. V., Chen H., Thaler C., Puhl H. L., 3rd, Vogel S. S. (2006) Biophys. J. 91, L99–L101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyawaki A., Tsien R. Y. (2000) Methods Enzymol. 327, 472–500 [DOI] [PubMed] [Google Scholar]
- 16.Tsien R. Y. (2003) Nat. Rev. Mol. Cell Biol. Suppl:SS16–21 [PubMed] [Google Scholar]
- 17.Day R. N., Schaufele F. (2008) J. Biomed. Opt. 13, 031202 [DOI] [PubMed] [Google Scholar]
- 18.Bai Y., Giguére V. (2003) Mol. Endocrinol. 17, 589–599 [DOI] [PubMed] [Google Scholar]
- 19.Llopis J., Westin S., Ricote M., Wang Z., Cho C. Y., Kurokawa R., Mullen T. M., Rose D. W., Rosenfeld M. G., Tsien R. Y., Glass C. K., Wang J. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 4363–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weatherman R. V., Chang C. Y., Clegg N. J., Carroll D. C., Day R. N., Baxter J. D., McDonnell D. P., Scanlan T. S., Schaufele F. (2002) Mol. Endocrinol. 16, 487–496 [DOI] [PubMed] [Google Scholar]
- 21.Hoppe A., Christensen K., Swanson J. A. (2002) Biophys. J. 83, 3652–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kofoed E. M., Guerbadot M., Schaufele F. (2008) J. Biomed. Opt. 13, 031207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Padron A., Li L., Kofoed E. M., Schaufele F. (2007) Mol. Endocrinol. 21, 49–61 [DOI] [PubMed] [Google Scholar]
- 24.Chen H., Puhl H. L., 3rd, Koushik S. V., Vogel S. S., Ikeda S. R. (2006) Biophys. J. 91, L39–L41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heery D. M., Kalkhoven E., Hoare S., Parker M. G. (1997) Nature 387, 733–736 [DOI] [PubMed] [Google Scholar]
- 26.Förster T. (1948) Ann. Phys. 6, 54–75 [Google Scholar]
- 27.Förster T. (1959) Discuss Faraday Soc. 27, 1–17 [Google Scholar]
- 28.Stryer L., Haugland R. P. (1967) Proc. Natl. Acad. Sci. U.S.A. 58, 719–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patterson G. H., Piston D. W., Barisas B. G. (2000) Anal. Biochem. 284, 438–440 [DOI] [PubMed] [Google Scholar]
- 30.Ding X. F., Anderson C. M., Ma H., Hong H., Uht R. M., Kushner P. J., Stallcup M. R. (1998) Mol. Endocrinol. 12, 302–313 [DOI] [PubMed] [Google Scholar]
- 31.Feng W., Ribeiro R. C., Wagner R. L., Nguyen H., Apriletti J. W., Fletterick R. J., Baxter J. D., Kushner P. J., West B. L. (1998) Science 280, 1747–1749 [DOI] [PubMed] [Google Scholar]
- 32.Valentine J. E., Kalkhoven E., White R., Hoare S., Parker M. G. (2000) J. Biol. Chem. 275, 25322–25329 [DOI] [PubMed] [Google Scholar]
- 33.Shiau A. K., Barstad D., Loria P. M., Cheng L., Kushner P. J., Agard D. A., Greene G. L. (1998) Cell 95, 927–937 [DOI] [PubMed] [Google Scholar]
- 34.Cheskis B. J., McKenna N. J., Wong C. W., Wong J., Komm B., Lyttle C. R., O'Malley B. W. (2003) J. Biol. Chem. 278, 13271–13277 [DOI] [PubMed] [Google Scholar]
- 35.Brzozowski A. M., Pike A. C., Dauter Z., Hubbard R. E., Bonn T., Engström O., Ohman L., Greene G. L., Gustafsson J. A., Carlquist M. (1997) Nature 389, 753–758 [DOI] [PubMed] [Google Scholar]
- 36.Xu J., Liao L., Ning G., Yoshida-Komiya H., Deng C., O'Malley B. W. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 6379–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu J., Qiu Y., DeMayo F. J., Tsai S. Y., Tsai M. J., O'Malley B. W. (1998) Science 279, 1922–1925 [DOI] [PubMed] [Google Scholar]
- 38.Gehin M., Mark M., Dennefeld C., Dierich A., Gronemeyer H., Chambon P. (2002) Mol. Cell. Biol. 22, 5923–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bramlett K. S., Wu Y., Burris T. P. (2001) Mol. Endocrinol. 15, 909–922 [DOI] [PubMed] [Google Scholar]
- 40.Margeat E., Poujol N., Boulahtouf A., Chen Y., Müller J. D., Gratton E., Cavailles V., Royer C. A. (2001) J. Mol. Biol. 306, 433–442 [DOI] [PubMed] [Google Scholar]
- 41.Shang Y., Hu X., DiRenzo J., Lazar M. A., Brown M. (2000) Cell 103, 843–852 [DOI] [PubMed] [Google Scholar]
- 42.Métivier R., Penot G., Hübner M. R., Reid G., Brand H., Kos M., Gannon F. (2003) Cell 115, 751–763 [DOI] [PubMed] [Google Scholar]
- 43.Becker M., Baumann C., John S., Walker D. A., Vigneron M., McNally J. G., Hager G. L. (2002) EMBO Rep. 3, 1188–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.List H. J., Reiter R., Singh B., Wellstein A., Riegel A. T. (2001) Breast Cancer Res. Treat. 68, 21–28 [DOI] [PubMed] [Google Scholar]
- 45.Anzick S. L., Kononen J., Walker R. L., Azorsa D. O., Tanner M. M., Guan X. Y., Sauter G., Kallioniemi O. P., Trent J. M., Meltzer P. S. (1997) Science 277, 965–968 [DOI] [PubMed] [Google Scholar]
- 46.Osborne C. K., Bardou V., Hopp T. A., Chamness G. C., Hilsenbeck S. G., Fuqua S. A., Wong J., Allred D. C., Clark G. M., Schiff R. (2003) J. Natl. Cancer Inst. 95, 353–361 [DOI] [PubMed] [Google Scholar]
- 47.Wärnmark A., Treuter E., Gustafsson J. A., Hubbard R. E., Brzozowski A. M., Pike A. C. (2002) J. Biol. Chem. 277, 21862–21868 [DOI] [PubMed] [Google Scholar]
- 48.Carpenter A. E. (2007) Nat. Chem. Biol. 3, 461–465 [DOI] [PubMed] [Google Scholar]
- 49.Chen J., Anderson J. B., DeWeese-Scott C., Fedorova N. D., Geer L. Y., He S., Hurwitz D. I., Jackson J. D., Jacobs A. R., Lanczycki C. J., Liebert C. A., Liu C., Madej T., Marchler-Bauer A., Marchler G. H., Mazumder R., Nikolskaya A. N., Rao B. S., Panchenko A. R., Shoemaker B. A., Simonyan V., Song J. S., Thiessen P. A., Vasudevan S., Wang Y., Yamashita R. A., Yin J. J., Bryant S. H. (2003) Nucleic Acids Res. 31, 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y., Addess K. J., Chen J., Geer L. Y., He J., He S., Lu S., Madej T., Marchler-Bauer A., Thiessen P. A., Zhang N., Bryant S. H. (2007) Nucleic Acids Res. 35, D298–D300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson J. D., Higgins D. G., Gibson T. J. (1994) Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.