

Summary
Dynamic regulation of insulin signaling and metabolic gene expression is critical to nutrient homeostasis; dysregulation of these pathways is widely implicated in insulin resistance and other disease states. While the metabolic effects of insulin are well established the components linking insulin signal transduction to a metabolic response are not as well understood. Here, we show that Cdc2-like kinase 2 (Clk2) is a novel insulin regulated suppressor of hepatic gluconeogenesis and glucose output. Clk2 protein levels and kinase activity are induced as part of the hepatic refeeding response by the Insulin/Akt pathway, where Clk2 directly phosphorylates the SR domain on PGC-1α and results in repression of gluconeogenic gene expression and hepatic glucose output. Additionally, Clk2 is down-regulated in db/db mice, re-introduction of Clk2 largely corrects glycemia. Thus, we have identified a novel function and regulation of the Clk2 kinase as a component of hepatic insulin signaling and glucose metabolism.
Introduction
Hepatic gluconeogenesis, the net production of glucose from substrate molecules, is critical for adaptation to fasting conditions and contributes to hyperglycemia in diabetes (Biddinger and Kahn, 2006; Pilkis and Granner, 1992; Quinn and Yeagley, 2005). In the fed state, insulin normalizes blood glucose levels by increasing glucose uptake in peripheral tissues and by suppressing gluconeogenesis in the liver. While the metabolic effects of insulin have been studied for decades, a mechanistic understanding of insulin signal transduction is just recently coming into focus. In the liver, most of insulin's metabolic effects are through activation of Insulin receptor, PI3K and Akt pathways. Akt can directly suppress hepatic gluconeogenesis by negatively regulating the gluconeogenic transcriptional regulators: Foxo1, PGC-1α and CRTC2 (also known as TORC2) (Dentin et al., 2007; Li et al., 2007; Matsumoto et al., 2007; Nakae et al., 2001; Puigserver et al., 2003).
The PGC-1α transcriptional coactivator is an important mediator of hepatic fasting metabolism, PGC-1α induces and maintains expression of the gluconeogenic gene program during fasted states responding to glucagon/cAMP through CRTC2 (Herzig et al., 2001; Koo et al., 2005; Rhee et al., 2003; Yoon et al., 2001). During the fed state insulin/Akt can suppress PGC-1α activity through several mechanisms. Akt can directly phosphorylate S570 in PGC-1α's SR (serine-arginine) domain (Li et al., 2007). In addition, the PGC-1α SR domain interacts with Foxo1 in an insulin/Akt dependent manner (Puigserver et al., 2003). These studies suggest that the PGC-1α SR domain acts as an insulin-responsive domain to control its gluconeogenic function.
The Cdc2-like kinases (Clk), also termed LAMMER kinases, are an evolutionary conserved family of dual-specificity CMGC kinases found in most, if not all, eukaryotes. They are putative high-level regulators of alternative splicing through phosphorylation of SR domains on splicing factors (Colwill et al., 1996b; Duncan et al., 1995; Hanes et al., 1994; Hillman et al., 2004; Lee et al., 1996; Prasad et al., 1999). However, there is an extremely limited understanding of Clk targets, function, and regulation in a biological context. Here, we show that a member of the Clk kinases, Clk2, is regulated by feeding/fasting cycles in the liver. In the refed state, hepatic Clk2 is induced by insulin/Akt signaling, where Clk2 then phosphorylates the SR domain on PGC-1α. Clk2 phosphorylation of PGC-1α causes potent repression of gluconeogenic gene expression and hepatic glucose output that leads to hypoglycemia.
Results
Feeding and insulin/Akt regulate Clk2
We and others have previously found that the SR domain is an insulin responsive region of PGC-1α (Li et al., 2007; Puigserver et al., 2003). In splicing factors hyper-phosphorylation of SR regions can modulate their function and activity (Prasad et al., 1999). In order to investigate the signalling and transcriptional metabolic effects of the feeding/insulin response, we hypothesized that the Clk kinases responsible for SR domain phosphorylation in splicing factors may be involved in PGC-1α feeding/insulin regulation. Among the Clk kinases tested, we identified Clk2 as being regulated by nutritional status. Clk2 protein had low abundance in mouse liver extracts following 24hrs of fasting however, Clk2 protein was strongly induced after 4 hours of refeeding and remained elevated at longer refeeding time points (Figure 1A). Clk2 protein levels were also regulated in gastrocnemius muscle and in the heart by fasting/feeding albeit less dramatically (Supplemental Figure 1A). Unlike the regulation of many classic feeding/fasting genes which occurs at the transcriptional level, steady state Clk2 mRNA levels were largely unchanged (Figure 1B). Notably, induction of Clk2 protein correlated with suppression of Pepck and PGC-1α gene expression.
Figure 1. Clk2 is regulated by feeding/fasting and insulin.
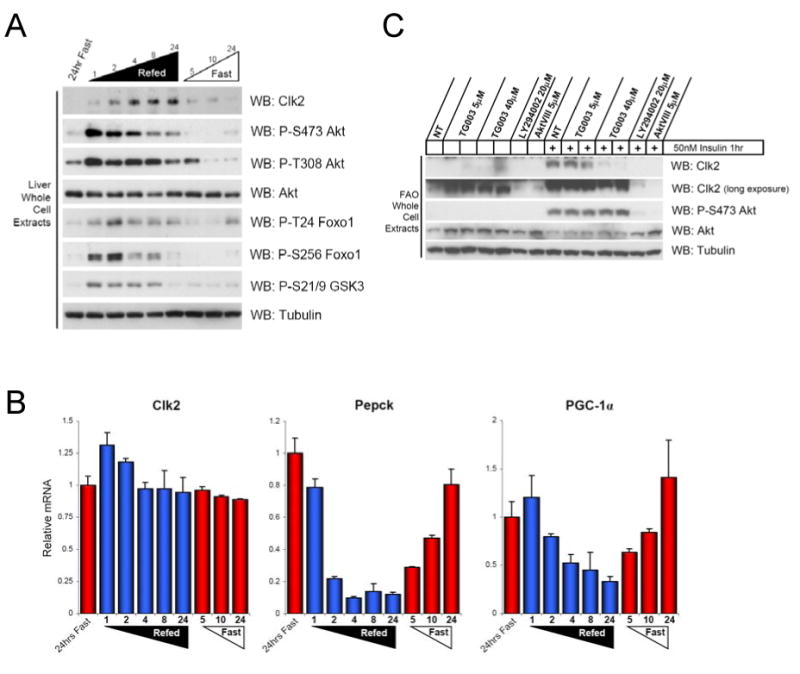
(A) Clk2 protein is induced in the mouse liver by refeeding. Western Blot analysis of liver whole cell extracts from mice sacrificed at indicated time during a feeding-fasting-refeeding time course. (B) Clk2 mRNA levels are not regulated by fasting/feeding in the mouse liver. Q-RT-PCR analysis of Clk2, Pepck, and PGC-1α gene expression from total liver RNA isolated from mice in (A). Data presented as average +/- SEM, each bar N=2-3. (D) Clk2 protein is induced by insulin downstream of PI3K and Akt. Whole cell extracts from FAO hepatoma cells grown in RPMI + 0.5%BSA for 24 hours and treated with indicated inhibitor overnight prior to insulin stimulation for 1 hour.
Since elevation of serum insulin during refeeding is a major stimulator of the hepatic refed metabolism, we tested whether insulin was sufficient to regulate Clk2 protein levels. Treatment of FAO rat hepatoma cells with insulin strongly induced Clk2 protein (Figure 1C). The insulin induction of Clk2 was blocked by treatment with the PI3K inhibitor: LY294002 and the Akt 1/2 inhibitor: Akt VIII, suggesting that the PI3K/Akt pathway mediated the effects of insulin on Clk2 protein. Additionally, intra-peritoneal injection of insulin into fasted mice similarly induced Clk2 protein (data not shown). Together, these results suggest that hepatic Clk2 protein levels are regulated by insulin during feeding/ fasting nutrient cycles.
Insulin, Akt, and Clk2 kinase activity control Clk2 protein stability
Based on the fact that Clk2 protein levels were strongly regulated by feeding and insulin, we next investigated the mechanisms responsible by which Clk2 protein levels fluctuate during nutrient cycles. Interestingly, in addition to the PI3K/Akt dependence of Clk2 induction, treatment with TG003: a Clk2 inhibitor (Muraki et al., 2004) also blocked the insulin induction of Clk2 (Figure 1C). This suggested that Clk2 protein levels may be regulated by Clk2 kinase activity. Indeed, ectopic expression of a wild-type Clk2 construct (Clk2 Wt) resulted in a highly expressed, stable protein. In contrast, a kinase dead, ATP binding mutant of Clk2 (K192R) (Nayler et al., 1998) was very unstable (Figure 2A and 2B). To further confirm that Clk2 kinase activity regulated protein levels, we decided to test whether the stability of the K192R mutant could be rescued by co-expression with Wt Clk2. Figure 2A shows that titrating increasing amounts of HA-Clk2 Wt could completely rescue the stability of Flag-Clk2 K192R while a HA-Clk2 K192R could not, suggesting that Clk2 protein levels may be regulated by Clk2 kinase activity. Moreover, treatment with Clk inhibitor TG003 strongly decreased the stability of Wt-Clk2, and did not further decrease the stability of the K192R kinase dead mutant (Figure 2B). Stabilization of K192R by Wt Clk2 was also blocked by TG003 treatment (Supp. Figure 1B). By performing deletion mapping (data not shown) we found that the effects of TG003 on Clk2 stability were dependent on the amino acids 82-99, deletion of this region renders Clk2 protein resistant to TG003 dependent degradation (Figure 2B) without altering Clk2 kinase activity (data not shown). Further refinement of this region showed that amino acids S98/Y99 were required for TG003 dependent Clk2 degradation (Figure 2C). These results suggest that fluctuations of Clk2 protein levels during feeding/fasting cycles may be regulated by Clk2 kinase activity itself.
Figure 2. Clk2 kinase activity controls Clk2 protein stability.
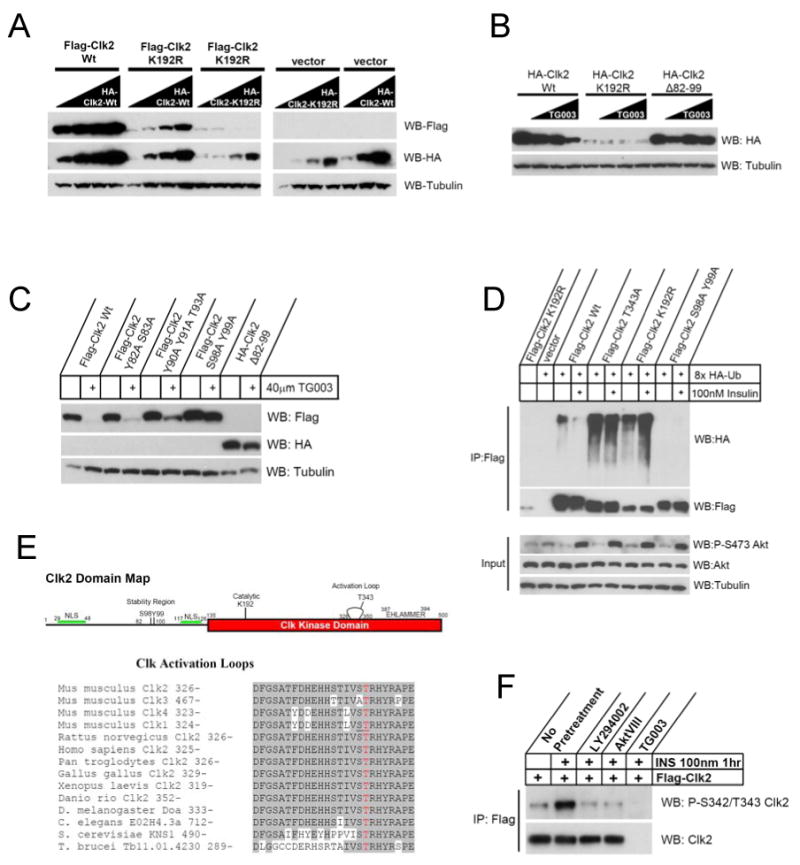
(A) Wt Clk2 rescues stability of kinase dead mutant. HEK293 cells were transfected with constant amounts of indicated Flag tagged Clk2 construct with increasing amounts of indicated HA-Clk2 construct. All transfection amounts were normalized with empty vector plasmid. (B) TG003 induces Clk2 degradation. Western blot analysis of HEK293 cells transfected with indicated Clk2 construct overnight prior to a 1 hour treatment with 0, 10, 20, or 40μM TG003. (C) TG003 degradation of Clk2 is dependent on residues S98 and Y99. Western blot analysis of HEK293 cells transfected with indicated Clk2 constructs and treated with 40μM TG003 1 hour prior to harvest. (D) Clk2 ubiquitination. Western blot analysis of whole cell extracts (input) or following Flag immunoprecipitation (IP:Flag) of H2.35 hepatocytes transfected with indicated Clk2 construct and 8xHA-ubiquitin. Cells were serum starved overnight prior to treatment with 10μM MG132 +/- 100nM insulin for 2 hours. (E) Alignment of Clk activation loops. Amino acid residues shaded in gray indicate conservation to Mus musculus Clk2. Red residue indicates T343. Residues underlined on Mus Clk1 were found to be phosphorylated in crystal structure of human Clk1 (PDB: 2VAG). (F) Phosphorylation at S342/T343 is induced upon insulin stimulation in H2.35 hepatocytes transfected with Flag-Clk2, serum starved prior to treatment with indicated inhibitor for 30 minutes prior to stimulation with insulin for 1 hour.
The regulation of Clk2 protein stability suggested that ubiquitination and proteasomal degradation might be involved. Indeed, co-expression of Flag-Clk2 with HA-tagged ubiquitin resulted in low levels of ubiquitination on Wt Clk2 (Figure 2D). In contrast, the less stable K192R mutant displayed high levels of ubiquitination and the S98A/Y99A mutant (which is more stable in the presence of TG003) showed no detectable ubiquitination (Figure 2D and Supp. Figure 1C). Interestingly, S98 and Y99 are highly conserved sites on vertebrate Clk2s (Supp. Figure 1D), however this region is specific to Clk2 and shows no conservation or homology to any region on mammalian Clk paralogues. We have also identified S98/Y99 as residues that are auto-catalytically phosphorylated (Supp. Figure 1E and F), suggesting that S98/Y99 is a conserved functional region on Clk2, potentially a degron sequence which controls Clk2 stability.
The data presented above clearly demonstrate how Clk2 protein levels could be controlled by Clk2 kinase activity. Moreover, these results hint that Clk2 kinase activity could be responding to insulin signalling to control Clk2 protein levels. Consistent with this idea, treatment of H2.35 hepatocytes with insulin or expression of myristolated Akt decreased ubiquitination of Wt Clk2 (Figure 2D and Supp. Figure 1C). However, neither insulin nor myristolated Akt had an effect on ubiquitination of Clk2 catalytic mutants K192R and T343A. In addition, ubiquitination of endogenous Clk2 was similarly regulated in primary hepatocytes. (Supp. Figure 1G). These data suggested that insulin/Akt could stabilize Clk2 protein by a similar mechanism through which Clk2 regulates its own stability. However, because insulin and myristolated Akt could not rescue the stability of a kinase dead Clk2 (Figure 2D, Supp. Figures 1C, and 1H) suggested that Clk2 kinase activity is required for the complete insulin/Akt stabilization of Clk2.
Activation loop phosphorylation is a common mechanism regulating enzymatic activity of kinases (Nolen et al., 2004). In fact, mapping of Clk2 phosphorylation sites identified a phospho-peptide that corresponded to the activation loop of Clk2 (Supp. Figure 1E), but the exact phosphorylated residue was ambiguous. Interestingly, in the deposited Protein Data Bank crystal structure of the Human Clk1 catalytic domain (PDB:2VAG) the activation loop was dually phosphorylated on residues which correspond to S342 and T343 on mouse Clk2 (Pike, 2007) (Figure 2E). Clk activation loops are very highly conserved throughout evolution (Figure 2E) suggesting that phosphorylation may be a conserved point of regulation (Farkas et al., 2009). In order to test if any residues on Clk2's activation loop had a functional role in activity, we constructed point mutations of all phosphorylate-able activation loop residues and only T343 effected Clk2 activity (Supp. Figures 2A and B). Stability of the T343A mutant protein resembled that of the K192R (Figure 2D, Supp. Figures 1C, and 2C) and in an in vitro assay the kinase activity of the T343A mutant was 20% that of Wt Clk2 (Supp. Figure 2D) while the S342A mutation did not have an effect on kinase activity or stability (Supp. Figures 2C and D). Moreover, in vitro both Clk2 and Akt phosphorylated the Clk2 catalytic domain (Supp. Figure 2E) and Akt2 could specifically phosphorylate the T343 residue (Supp. Figure 2F). Using an antibody toward phospho-S342/T343 Clk2, this site was regulated by insulin in hepatocytes (Figure 2F). Additionally, Akt and Clk2 physically interacted as demonstrated by co-immunoprecipitation experiments with ectopically expressed and endogenous proteins (Supp. Figures 2G and H) and in vitro Akt specifically interacted with the Clk2 kinase domain (Supp. Figure 2I). Together, these results suggest that T343 phosphorylation (by Akt and/or Clk2 itself) on the Clk2 activation loop is an important regulatory mechanism controlling Clk2 kinase activity and stability in response to insulin and feeding.
Clk2 phosphorylates and represses PGC-1α
The fact that Clk kinases phosphorylate SR regions (Bullock et al., 2009; Colwill et al., 1996b; Duncan et al., 1998; Prasad et al., 1999; Prasad and Manley, 2003), PGC-1α contains an insulin-responsive SR domain, and is counter-regulated to Clk2 by fasting/feeding in the liver prompted us to test if Clk2 could directly phosphorylate PGC-1α SR domain. Clk2 robustly phosphorylated full length GST-PGC-1α in vitro (Figure 3A), mapping experiments showed that Clk2 exclusively phosphorylated the SR domain on PGC-1α (Supp. Figure 3A). In addition, overexpression of Clk2 was sufficient to phosphorylate PGC-1α on the SR domain as recognized by phospho-Akt-substrate antibodies and by a shift in PGC-1α mobility (Figure 3B). Importantly, insulin treatment resulted in increased PGC-1α SR domain phosphorylation which was largely blocked by Clk2 shRNA (Figure 3C and Supp. Figure 3B). In primary hepatocytes, Clk2 expression led to high levels of PGC-1α phosphorylation that were not further augmented by insulin treatment (Supp. Figure 3B). In vitro analysis showed that Clk2 phosphorylates the PGC-1α SR domain and catalyzes auto-phosphorylation with similar efficacy (Supp. Figure 3C). Together, these results indicate that Clk2 kinase mediates the insulin responsive phosphorylation of the PGC-1α SR domain.
Figure 3. Clk2 phosphorylates and represses PGC-1α transcriptional coactivator.
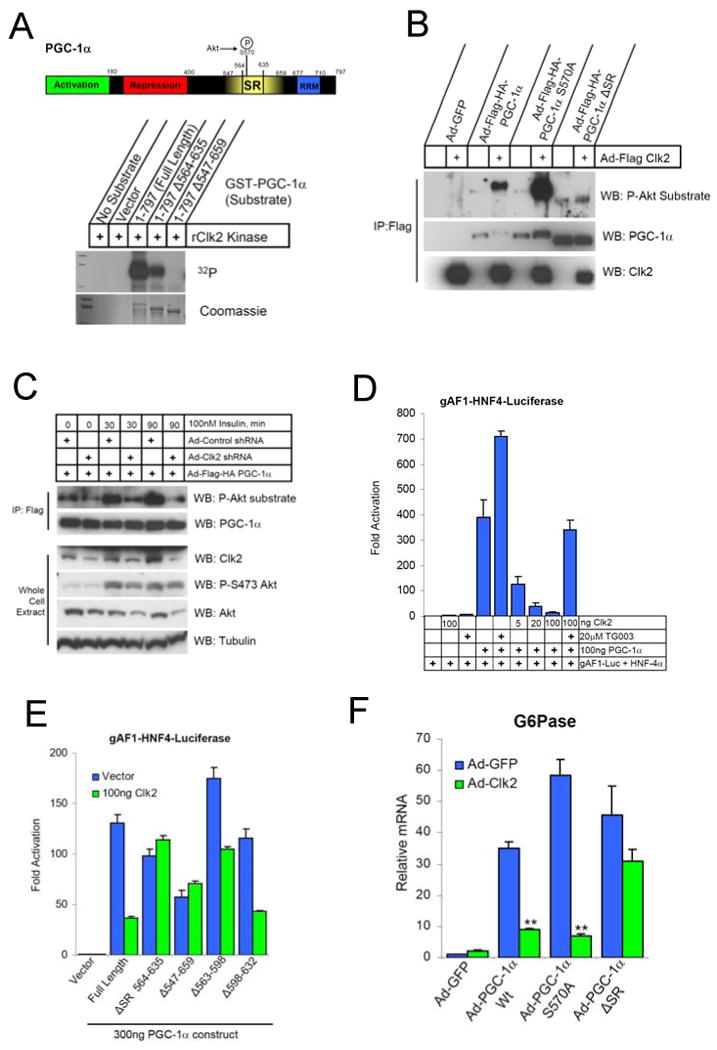
(A) Domain map and in vitro phosphorylation of PGC-1α with indicated deletion by purified recombinant Clk2 kinase (rClk2) in the presence of γ32P-ATP reaction conditions described in Experimental Procedures. (B) Clk2 induces PGC-1α SR domain phosphorylation recognized by P-Akt substrate antibodies. Western blot analysis of Flag immunoprecipitates from Primary Hepatocytes infected with indicated adenoviruses, all viral loads were normalized with Ad-GFP. (C) Clk2 shRNA block insulin induction of PGC-1α phosphorylation. Western blot analysis of flag immunoprecipitates (IP:Flag) and whole cell extracts of H2.35 hepatocytes infected with indicated adenoviruses. Cells were serum starved overnight prior to insulin stimulation as indicated. PGC-1α transcription co-activation assay in (D) HE3K293 cells and (E) H2.35 hepatocytes using HNF-4α and gAF1-luiferase reporter performed as described in Experimental Procedures. (F) Q-RT-PCR analysis of G6Pase expression in total RNA from primary hepatocytes infected with indicated adenoviruses. Data is presented as average +/- SEM. Significance determined by unpaired two-tailed Students T-Test, ** P<0.01.
To investigate the functional consequence of Clk2 phosphorylation on PGC-1α, we performed luciferase based transcriptional reporter assays. PGC-1α strongly activates the HNF4α gAF-1 response element (Puigserver et al., 2003; Rhee et al., 2003) however, co-expression of Wt Clk2, but not kinase dead mutants, repressed PGC-1α activity in a dose-dependent manner (Figure 3D, Supp. Figures 2A, and 2B). Clk2 inhibition was reversed by treatment with the Clk2 inhibitor TG003 (Figure 3D). Clk2 could also repress PGC-1α activation of Foxo1 on the IRS response element (Supp. Figure 3D), as well as repress the Pepck promoter (Supp. Figure 3E). Interestingly, Clk2 did not repress the PGC-1α transcriptional activation of the NRF-1 transcription factor (Supp. Figure 3F) suggesting Clk2 repression of PGC-1α may be transcription factor specific.
As Clk2 strongly and exclusively phosphorylated the SR domain on PGC-1α, we determined whether this region was required for Clk2 repression of PGC-1α. A PGC-1α allele lacking the SR domain (ΔSR) had transcriptional activity comparable to full length, however ΔSR is resistant to Clk2 repression (Figure 3E). The entire SR domain on PCG-1α was involved in Clk2 mediated repression as PGC-1α constructs lacking smaller regions of the SR domain were still potently repressed by Clk2 (Figure 3E and Supp. Figure 3G). Large scale mutation analysis of the phosphorylate-able residues (S, T and Y) in the SR domain to alanine rendered PGC-1α similarly resistant to Clk2 repression (Supp. Figures 3H and I).
We next tested whether Clk2 repression of PGC-1α had an effect on endogenous gluconeogenic target genes. As expected, adenovirus-mediated expression of PGC-1α strongly induced the gluconeogenic gene G6Pase in primary hepatocytes (Figure 3F). Co-infection of Clk2 adenovirus with PGC-1α potently repressed this induction, but not the PGC-1α ΔSR allele (Figure 3F). Moreover, Clk2 shRNAs (Supp. Figure 4A) further induced PGC-1α dependent G6Pase and Pepck gene expression in primary hepatocytes (Figure 4A). These results indicate that Clk2 kinase is a repressor of PGC-1α transcriptional activity on gluconeogenic genes through phosphorylation of the PGC-1α SR domain.
Figure 4. Clk2 represses gluconeogenic gene expression in primary hepatocytes.
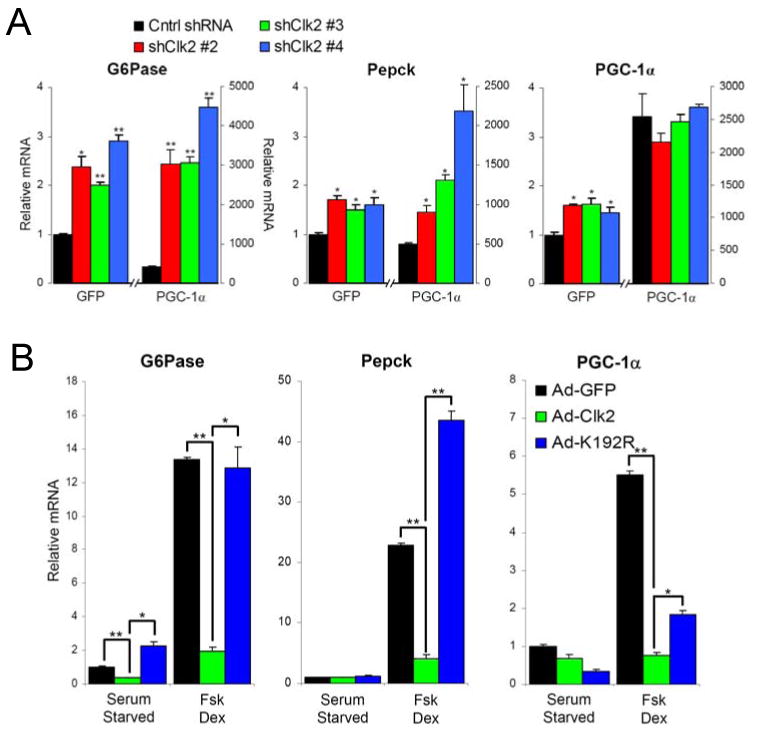
(A) Clk2 shRNA induces gluconeogenic gene expression. Q-RT-PCR analysis of total RNA from primary hepatocytes infected with GFP or PGC-1α and Control shRNA or Clk2 shRNA adenoviruses. Cells were grown for 2 days post infection in DMEM + 10%FBS. Each bar N=4. (B) Clk2 expression suppresses gluconeogenic gene expression. Q-RT-PCR analysis of total RNA from primary hepatocytes infected with GFP, Clk2, or Clk2 K192R adenoviruses. 24 hours after infection Cells were switched to starvation media 6 hours prior to treatment with 10μM forskolin and 1 μM dexamethasome for 1.5 hours. Each bar N=4. All data is presented as average +/- SEM. Experiments performed at least 3 times with similar results. Significance determined by two-tailed unpaired Students T-Test * denotes P<0.05, ** P<0.01.
Clk2 suppresses hepatic gluconeogenesis
To gain a clearer understanding of how Clk2 functions in the context of feeding/fasting hormonal regulation of hepatic glucose metabolism, we performed Clk2 gain-of-function experiments. Treatment of primary hepatocytes with forskolin and dexamethasone (Fsk + Dex) mimics the fasting action of glucagon and glucocorticoids respectively strongly inducing the gluconeogenic gene program: Pepck, G6Pase and PGC-1α (Quinn and Yeagley, 2005). While expression of Clk2 only had modest effects in unstimulated/serum starved primary hepatocytes, it almost completely blunted the Fsk+Dex induction of gluconeogenic genes (Figure 4B). Importantly, Clk2 kinase activity was required to repress gluconeogenic genes as expression of Clk2 K192R did not have a large effect (Figure 4B), however we did observe that K192R stabilizes endogenous PGC-1α protein in primary hepatocytes (Supp. Figure 4B). Similar results with Clk2 and K192R were observed on overexpressed PGC-1α (which mimics many of the effects of Fsk + Dex on gene expression) in primary hepatocytes (Supp. Figure 4C and D). Overall Clk2 acts as a general suppressor of PGC-1α activity in primary hepatocytes, suppressing PGC-1α induction of genes involved in many pathways included β–oxidation and mitochondrial respiration, however by far Clk2's most potent effects are on regulation of gluconeogenic gene expression (Supp. Figure 4E).
These studies were broadened in vivo where modest adenoviral mediated Clk2 expression (3-4 fold over endogenous fed Clk2 protein levels) in the liver (Supp. Figure 5A) resulted in significant fasting hypoglycemia (Figure 5A). Clk2 also strongly repressed hepatic glucose output as measured by an intra-peritoneal pyruvate tolerance test (Figure 5B) which correlated with reduced gluconeogenic gene expression of Pepck, G6Pase, and PGC-1α (Figure 5C). Clk2 expression also resulted in lower levels of endogenous PGC-1α protein, which migrated with an upward shift in an SDS PAGE (Supp. Figure 5B), similar to the shift in PGC-1α caused by Clk2 expression in primary hepatocytes (Supp. Figure 3B and 4D). These in vivo effects of Clk2 are largely dependent on PGC-1α: co-infection of PGC-1α shRNA and Clk2 were not additive nor were they synergistic, suggesting that the effects of Clk2 on hepatic gluconeogenic gene expression require PGC-1α (Figure 5D). Next we used Clk2 shRNA adenovirus to test the in vivo requirement of Clk2 in the refeeding repression of gluconeogenesis. Mice infected with Clk2 shRNA have impaired refeeding suppression of gluconeogenic gene expression which is most significant after 4 and 9 hours of refeeding (Figure 5E). Taken together, these results indicate that Clk2 is a potent repressor of hepatic gluconeogenesis. Moreover, as Clk2 is induced by insulin/Akt in the refed state suggests that Clk2 may be an important component of the physiologic refed response by suppressing gluconeogenesis.
Figure 5. Clk2 represses hepatic glucose output and causes hypoglycemia in vivo.
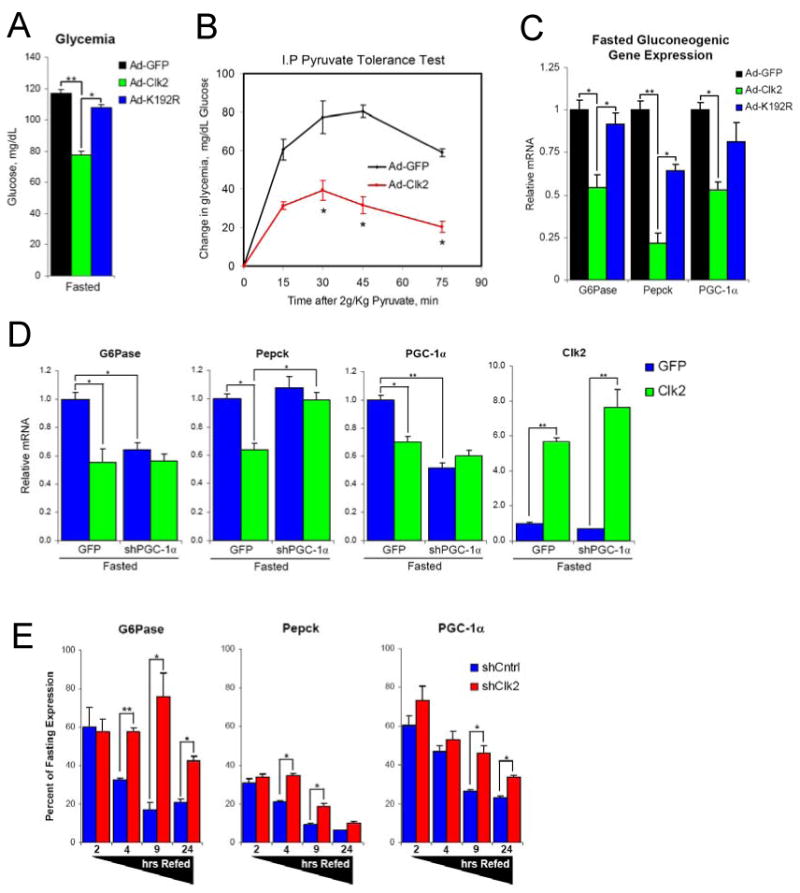
(A) Hepatic Clk2 expression causes fasting hypoglycemia. Glycemia from mice infected with GFP, Clk2 or Clk2 K192R adenoviruses following a 9 hour fast. Each bar N=6-9. (B) Clk2 suppresses hepatic glucose production in a pyruvate tolerance test. Intra-peritoneal (I.P.) pyruvate tolerance test from mice infected with GFP (N=4) or Clk2 (N=5) adenoviruses. Mice fasted overnight prior to injection of 2g/kg sodium pyruvate. Data is presented as change in glycemia following pyruvate injection. (C) Clk2 suppresses hepatic gluconeogenic gene expression in vivo. Q-RT-PCR analysis of gluconeogenic gene expression from total liver RNA from mice infected with GFP (N=5), Clk2 (N=6), or Clk2 K192R (N=3) adenoviruses following a 9 hour fast. (D) Clk2 suppression of gluconeogenesis requires PGC-1α. Q-RT-PCR analysis of gene expression from mice infected with indicated adenovirus, fasted overnight prior to sacrifice, each bar N=4. (E) Hepatic Clk2 knock-down impairs refeeding suppression of gluconeogenic gene expression. Q-RT-PCR analysis of gene expression presented as percent of fasting expression. Data presented is average of multiple experiments, for each virus and feeding condition: fasted (N=12), 2hr refed (N=4), 4hr refed (N=12), 9hr refed (N=4), 24hr refed (N=7). All data is presented as average +/- SEM. Significance determined by two-tailed unpaired Students T-Test * denotes P<0.05, ** P<0.01.
Clk2 expression normalizes db/db diabetic phenotype
Elevation of hepatic glucose output in type II diabetic patients as well as in animal models of obesity and diabetes due to insulin resistance is well documented (Biddinger and Kahn, 2006; Quinn and Yeagley, 2005). Since Clk2 is a suppressor of PGC-1α on gluconeogenic gene expression and hepatic glucose production, we hypothesized that dysregulation of Clk2 could potentially be an underlying mechanism of increased hepatic glucose output in a diabetic state. We decided to test this hypothesis using the leptin resistant db/db mouse model. As expected, in the fed state db/+ heterozygous control mice (Leprdb/+) mice showed high levels of Clk2 protein in the liver, however the diabetic and obese db/db mice (Leprdb/Leprdb) showed a striking down regulation of hepatic Clk2 protein (Figure 6A). Upon a 12hr fast Clk2 protein was downregulated in both control and db/db mice, however still at lower levels in the db/db mice. This led us to ask if we could rescue hyperglycemia in db/db mice by re-introduction of hepatic Clk2. Consistent with the effects observed in Balb/c mice (Figure 5) hepatic Clk2 expression in db/db mice dramatically reduced glucose production from pyruvate, to a level similar to control db/+ mice (Figure 6B) which correlated with reduced fasting gluconeogenic gene expression (Figure 6C) and lower levels of PGC-1α protein which also displayed a shift in mobility (Supp. Figure 5C). Interestingly and similar to endogenous Clk2 (Figure 6A), we observed that expressed Clk2 was also at lower levels in db/db mice compared controls (Supp. Figure 5D), despite elevated Clk2 mRNA (Supp. Figure 5E). This suggested to us that Clk2 stability may be dysregulated in db/db mice. Indeed, immunoprecipitated Clk2 from db/db mice displays a much higher degree of steady state ubiquitination than Clk2 from db/+ mice (Supp. Figure 5F). Taken together, these data show that Clk2 is down regulated in the leptin resistant db/db diabetic and this correlates with elevated gluconeogenic activity. Moreover, re-introduction of Clk2 in the livers of db/db mice dramatically restores the glucose phenotype to near control levels.
Figure 6. Hepatic Clk2 expression normalizes db/db diabetic phenotype.
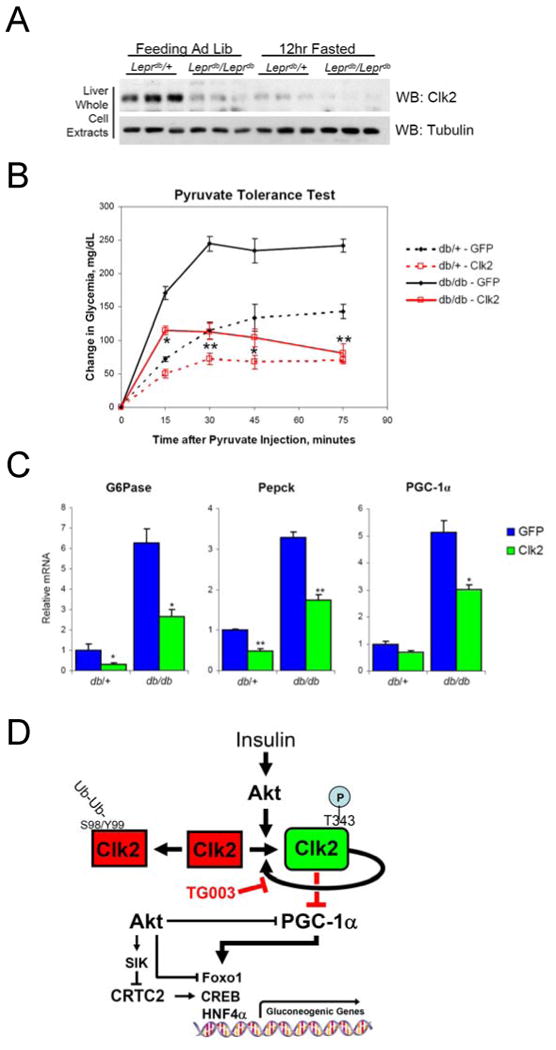
(A) Clk2 protein is down regulated in db/db mice. Western blot analysis of liver whole cell extracts from heterozygous control mice (Leprdb/+) or homozygous db/db mice (Leprdb/Leprdb) sacrificed during the feeding or 12hour fasted. (B) Pyruvate tolerance test from db/db or db/+ mice infected with GFP or Clk2 adenovirus. Mice were fasted overnight prior to intraperitioneal administration of 2g/Kg pyruvate. Data presented as average +/- SEM, db/db mice each line N=4, db/+ mice each line N=2. Significance indicated is determined between db/db GFP vs. db/db Clk2. (C) Q-RT-PCR analysis of gluconeogenic gene expression from total liver RNA from db/+ or db/db mice following an overnight fast. Data presented as average +/- SEM, db/db mice each bar N=4, db/+ mice each bar N=2. (D) Proposed model depicted regulation of Clk2 kinase activity, stability, and suppression of PGC-1α and gluconeogenesis. Significance determined by two-tailed unpaired Students T-Test * P<0.05, ** P<0.01.
Discussion
Insulin is a crucial component of glucose homeostasis by its action in many tissues, the liver perhaps being the most central. One of the key metabolic actions of insulin is repression of hepatic gluconeogenesis. In this regard, we have identified the Clk2 protein kinase as a novel insulin regulated suppressor of hepatic gluconeogenesis. In response to insulin signaling through PI3K/Akt, Clk2 kinase activity is induced by phosphorylation of the activation loop residue T343. Once Clk2 kinase activity is induced it undergoes auto-phosphorylation which enhances Clk2 protein stability. Clk2 can then phosphorylate the SR domain on PGC-1α causing repression of PGC-1α transcriptional activity on gluconeogenic genes and reducing hepatic glucose output (model outlined in Figure 6C).
Regulation of Clk2 by Nutritional Status and Insulin
The Clk family of kinases show a very high degree of evolutionary conservation, most if not all eukaryotes have at least one Clk kinase. Despite the prevalence of Clks very little is known about their activity, regulation, and function. Here, we present the first detailed mechanistic insights of regulation and biological function of a mammalian Clk. Thus, Clk2 is subject to at least two levels regulation: an initial stimulating phosphorylation on the activation loop and by Clk2 kinase activity itself. In response to insulin, Akt phosphorylates the activation loop of the inactive and unstable Clk2 protein. Phosphorylation of the activation loop induces Clk2 kinase activity which is required for Clk2 stability. Once induced and stable, Clk2 can sustain activity and possibly amplify the initial signal by auto-phosphorylation of its activation loop without the continual need for Akt or another kinase/signal. Indeed, earlier work on the Clks has shown that they are strongly auto-catalytic and suggested that this is a crucial aspect of Clk activity (Duncan et al., 1997; Lee et al., 1996; Nayler et al., 1998; Prasad and Manley, 2003), but have lacked a detailed mechanism of regulation.
PGC-1α SR domain phosphorylation
Among the first descriptions of Clks relates to their substrate specificity for regions rich in serine-arginine residues (SR domains) (Colwill et al., 1996a; Colwill et al., 1996b; Lee et al., 1996; Nikolakaki et al., 2002). The SR domain on PGC-1α is a well documented regulatory region and has been shown to mediate PGC-1α's insulin responsiveness via interaction with Foxo1 transcription factor as well as containing an Akt phosphorylation site (Li et al., 2007; Puigserver et al., 2003). Taking this into account, the identification of Clk2 as an insulin dependent PGC-1α SR domain kinase further confirmed the regulatory function of the SR region. However, mechanistic basis of how the SR domain regulates PGC-1α activity remains elusive. We have observed that the SR domain's most potent regulatory effects are on PGC-1α's co-activation of nuclear receptors and Foxo1 and does not appear to be a general repression of PGC-1α on all transcription factors. Many nuclear proteins that contain SR domains are known to interact with other proteins via these domains. In fact, phosphorylation of SR domains has been shown to alter their protein binding patterns (Huang et al., 2004; Ngo et al., 2005), it is possible that phosphorylation of PGC-1α's SR domain affects interaction with specific transcriptional components depending on transcription factors and target promoters.
Another interesting aspect of this work is the implication of alternative splicing. The Clks are putative regulators of alternative splicing through phosphorylation of SR domains (Stojdl and Bell, 1999). Notably, the Clk2s are known to regulate the alternative splicing of their own transcripts (Duncan et al., 1997). Indeed, we have observed that alternative splicing of Clk2 mRNA is highly regulated in a consistent manner with the regulation presented here (Rodgers and Puigserver, unpublished results). Additionally, PGC-1α has been implicated in splicing via its SR and RNA recognition (RMM) domains (Knutti and Kralli, 2001; Monsalve et al., 2000). However, precise role of PGC-1α in alternative splicing has not yet been documented, one interesting hypothesis is that SR phosphorylation by Clk2 could regulate binding of specific RNAs on PGC-1α's SR and RRM domains. We are currently exploring possibilities of PGC-1α and Clk2 cooperation in regulated alternative splicing events.
Akt and Clk2 in refed hepatic glucose metabolism
Insulin suppression of hepatic gluconeogenesis is largely dependent on the PI3K and Akt kinase pathways. Recent work has shown that Akt can directly impinge on gluconeogenesis by negative regulation of the CRTC2, Foxo1, and PGC-1α transcriptional regulators (Dentin et al., 2007; Li et al., 2007; Nakae et al., 2001; Puigserver et al., 2003). Our identification of Clk2 as an insulin regulated kinase which suppresses gluconeogenesis, may at first suggest a redundant function with that of Akt. However, we believe our data imply a separate and potentially quantitative role of Clk2 and Akt in repression of gluconeogenesis in the fed state. It is well established that Akt activity is induced very rapidly following insulin stimulation, where it acutely suppresses the activity of CRTC2, Foxo1, and PGC-1α. However, it is known that hepatic gluconeogenesis continues well into the later stages of refeeding (Radziuk and Pye, 2001), suggesting that the suppressive effects of insulin on gluconeogenesis are not immediate and that gluconeogenesis requires several hours to be quantitatively suppressed. We propose that Clk2 acts as a second and perhaps more powerful phase of refeeding repression on gluconeogenesis. Thus, following initial insulin stimulated phosphorylation by Akt, it takes several hours for Clk2 protein levels to build up and achieve full induction, which strongly correlates with the increasing suppression of Pepck gene expression during refeeding. Consistent with this, in vivo knock-down of Clk2 impairs refeeding suppression of gluconeogenesis particularly at the later stages (>4 hours) when Clk2 levels are elevated, but has no affect in the initial suppression of gluconeogenesis (Figure 1A and 5E) which is mostly likely due to immediate activation of Akt pathways. It is interesting to note that we observe decreased levels of Akt phosphorylation and phosphorylation of Akt targets at these later stages of refeeding. This suggests to us that Akt activity is downregulated after prolonged refeeding, however gluconeogenic targets display continued and increased suppression (Figure 1A and B). Clearly, Akt cannot be the only suppressive signal at these stages of refeeding, we hypothesize that Clk2 is a major gluconeogenic suppressor at these later stages. It is also interesting to note that glycolysis and lipogenesis, other hallmarks of hepatic feeding metabolism, are not immediately induced upon refeeding but take several hours to achieve maximal expression (Horton et al., 2002; Kalaany and Mangelsdorf, 2006; Postic et al., 2007), a pattern of expression mirroring that of Clk2 induction. While insulin is widely considered the major stimulus of glycolytic and lipogenic pathways, the refeeding expression pattern suggests that Akt is not a direct activator (Semple et al., 2009), the role of Clk2, if any, in these pathways is an active area of research.
Clk2 regulation and implications for metabolic diseases
Our findings that Clk2 is down-regulated in the livers of db/db mice supplies a disease context which further supports our model of Clk2 being a suppressor of hepatic gluconeogenesis. The db/db mice display elevated hepatic glucose output and hyperglycemia and it has been shown that PGC-1α and CRTC2 are both elevated in db/db livers (Dentin et al., 2007; Yoon et al., 2001). Notably, knock-down of PGC-1α in livers of db/db mice is sufficient to significantly reduce blood glucose levels (Koo et al., 2004) suggesting that hyper-activity of hepatic PGC-1α is a major defect in this insulin resistant model. Our results suggest that activation of Clk2 in a hyper-glycemic and insulin resistant state would reduce hepatic glucose output and improve systemic glucose levels.
While we present data showing that insulin strongly stabilizes Clk2, it is presently unclear how Clk2 is negatively regulated. Considering the nature of Clk2 auto-regulation it implies that there is a negative regulatory pathway which opposes Clk2 during fasting and in diabetic models. Consistent with this we observer lower expression of endogenous as well as ectopic Clk2 in the db/db diabetic mice which also displays elevated levels of ubiquitination. This may suggest that hyperactivity of the ubiquitin pathway/ligase responsible of Clk2 degradation is causal. We have also shown that Clk2 ubiquitination can be regulated by insulin, an alternative explanation is that the insulin resistance itself is enough to dysregulate Clk2 protein stability. Currently, the identification of factors and pathways which control Clk2 degradation is under investigation and might provide important therapeutical targets to treat metabolic diseases.
Experimental Procedures
Animals Procedures
All mouse experiments (except where noted) were performed with 6-10 week old male Balb/c mice purchased from Taconic. Seven to nine week old male B6.Cg Leprdb/Leprdb and heterozygous control mice were purchased from Jackson Laboratory. Adenovirus infections were performed by tail vein injection of 0.5×109 infectious particles per mouse. Double infections were performed using a total of 1.0×109 virus particles; all infection amounts were corrected using GFP adenovirus. Glycemia was measured by tail bleeds using an Ascencia Elite (Bayer). Pyruvate tolerance tests were performed by fasting animals prior to intraperitoneal injection of 2g/kg sodium pyruvate dissolved in PBS. Animals were fed a standard diet (by mass: 22.5% protein, 11.8% fat, 52% carbohydrate) were sacrificed 5-7 days after infection by CO2 asphyxiation; the livers were removed and snap frozen on liquid nitrogen until processing. Data from mouse experiments is the combination of at least 3 independent experiments performed with at least 3 mice per condition. All animals work was performed in accordance to Dana-Farber Cancer Institute animal care and use policies.
Cell Culture
Mouse primary hepatocytes were isolated using liver digest media (Invitrogen) detailed protocol available upon request. Adenovirus infections were performed using 1.0×107 infectious particles per well of a 6-well plate for 2.5 hours in Maintenance Media (DMEM 0.2% BSA, 1nM insulin, 2% Na-pyruvate, Pen/Strep). Cells were grown in starvation media (DMEM 0.2% BSA, 2% Na-pyruvate, Pen/Strep) for 6 hours prior to insulin/fsk/dex treatments. Cells were harvested 1-2 days after infection as indicated.
FAO hepatoma cells were cultured in F-12 media 5% FBS, Pen/Step. Adenovirus infections were performed using 1.0×108 infectious particles per well of a 6-well plate for 3 hours.
H2.35 SV-40 immortalized hepatocytes were cultured in DMEM 5% FBS, 5mM Glucose, 0.2μM Dex, Pen/Strep. Adenovirus infections were performed using 1.0×108 infectious particles per well of a 6-well plate for 4 hours. Transfections were performed with Lipofectamine LTX (Invitrogen) according to manufacturer's recommendations.
HEK 293 cells were cultured in DMEM 10% CCS, Pen/Strep. Transfections were performed with Polyfect (Qiagen) according to manufacturer's recommendations.
We obtained Bovine Insulin (I0516), Forskolin (F3917), and Dexamethasome (D4902) from Sigma. LY294002 was obtained from Caymen Chemical, MG132 from Andwin Scientific, U0126 from BIOMOL, Akt VIII, TG003, and Lactocystin from CalBiochem.
Transcriptional Reporter Assays
HEK 293 or H2.35 cells were grown and transfected as described above. All transfection amounts were corrected with empty vector plasmid DNA. Cells were grown for 24 hours post transfection, harvested using 1× Reporter Lysis Buffer (Promega). Luciferase counts were measured using Luciferase Substrate (BD Pharmingen) on a Fluorostar Optima (BMG). All experiments were performed in at least two independent trials in duplicate-triplicate.
Recombinant Kinase Assays
Recombinant Akt2 kinase (Cell Signaling) experiments were carried out according to manufacturer's recommendations in Akt Kinase Buffer with 0.2mM ATP. Recombinant Clk2 was made by cloning the Clk2 cDNA into pGex 5x-2 and expressed in BL-21 bacteria (Promega) followed by IPTG induction and purification using glutathione agarose (roche). Where indicated γ32P-ATP was added at a 1:100 (hot:cold) molar ratio.
Constructs
Clk2 cDNA was cloned from mouse liver cDNA into pcDNA 3.1 (Invitrogen) Flag or HA tagged expression vectors. All adenoviruses were constructed using the pAd-Track/pAd-Easy system. Flag-Clk2 and Flag-Clk2 K192R cDNA's are expressed by a CMV promoter. shRNA adenoviruses are expressed by a U6 promoter: shControl (ccttcgattccctcaaagaca) shClk2#2 (ccttcgatttcctcaaagaca), shClk2#3 (gagaacagcagttaccgaa), shClk2#4 (cggatgatcagaaagacaa). PGC-1α shRNA adenovirus was utilized as previously described (Koo et al., 2004). All adenoviruses were purified by CsCl ultracentrifugation and titered by serial dilution/infection and quantifying GFP expressing cells.
RT-PCR
Total RNA was isolated from cells or pulverized liver using Trizol (Invitrogen). For Q-RT-PCR analysis cDNA was synthesized using random primers and High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Gene expression analysis was performed using a BioRad IQ5 and HotStart-IT SYBER mix (USB). All gene expression data corrected to 36B4 expression and normalized using ΔΔCT method. All gene expression experiments performed in at least 2 independent experiments each performed duplicate or triplicate. All primer sequences available upon request.
Cell Extracts
Whole cell extracts were prepared using RIPA buffer with 1 mM PMSF, 1× Protease Inhibitors (Roche), 30mM glycerol 2-phosphate, 50mM NaF, and 2mM Na orthovanadate. Liver whole cell extracts were prepared using a TissueLyzer (Qiagen) in RIPA buffer. Liver nuclear extracts were prepared as described (Rodgers and Puigserver, 2007).
Immunoprecipitations
Flag immunoprecipitations were performed using anti-Flag-M2-Agarose (Sigma) in RIPA buffer. Akt/Clk2/Ubiquitin co-immunoprecipitations were performed by harvesting the cells in 1% Triton X-100, 20mM Tris-Cl pH 7.5, 150mM NaCl, 1mM EDTA, 1mM EGTA, 2.5mM Na-pyrophosphate, 1mM glycerol 2-phosphate, 1mM Na-orthovandate, 1mM PMSF, and 1× protease inhibitors (Roche). Immunoprecipitations were performed with indicated antibody with anti-IgG agarose (True Blot, eBiosciences), protein A/G agarose (roche), anti-flag-agarose (Sigma), anti-HA-agarose (Sigma) for 3 hours at 4°C, immunoprecipates were washed at least 3 times with lysis buffer and resolved by SDS-PAGE.
Antibodies
Rabbit anti-Clk2 was a gift from Axel Ullrich. Rabbit anti-TORC2 (CRTC2) was a gift from Marc Montminy. Rabbit anti-Phospho-S570 PGC-1α was a gift from Morris Birnbaum. Anti-Clk2 and anti-phospho S342/T343A from Cell Signaling. We obtained M2-Flag-HRP (Sigma), HA-HRP (Roche), P-S256 Foxo1 and Tubulin (Upstate), Actin (Abchem), and PGC-1α (H300 Santa Cruz and 4C1.3 Calbiochem). Akt, P-T308 Akt, P-S473 Akt, P-T24 Foxo1, Foxo1, P-S21/9 GSK3, P-S133 CREB, P-Akt Substrate, and ubiquitin antibodies from Cell Signaling.
Mass Spectrometry
Protein from Coomassie stained gel bands was in-gel reduced with DTT, cysteine residues were alkylated with iodoacetamide, and the protein was digested using either trypisn or chymotrypsin(Shevchenko et al., 1996). The generated peptide mixtures were subjected to microcapillary liquid chromatrography tandem mass spectrometry on an LTQ Orbitrap (Thermo Fisher Scientific) essentially as described elsewhere (Haas et al., 2006). MS/MS spectra were assigned by searching them against the GST-Clk2 protein sequence using the SEQUEST algorithm and allowing Ser, Thr, and Tyr residues to be phosphorylated (Eng, 1994). The precursor ion tolerance for the search was set to ±2 Da and no enzyme specificity constratints were applied. Peptide assignments were filtered to be consistent with the enzyme specificity of the protease use for the digest and to have a mass deviation of less than 7 ppm. Additional filtering through SEQUEST scores XCorr and dCn was followed by manual validation of phosphopeptide MS/MS spectra.
Supplementary Material
Acknowledgments
We would like to thank: Axel Ullrich, Marc Montminy, Morris Birnbaum, and Alexander Banks for their donation of reagents which were of critical importance to this work. Susan Keezer and Qingyuan Ge at Cell Signaling for development of Clk2 and P-Clk2 antibodies. Roderick Bronson, Bruce M. Spiegelman, Lewis C. Cantley, and Nathanael S. Gray and members of their labs for discussion. A special thanks is given to all members of the Puigserver lab: Christine Chin for technical assistance, Timothy Kelly, John Dominy, John Tom Cunningham, and Francisca Vazquez for their critical reading of this manuscript. A portion of this work was funded by an American Heart Association Pre-Doctoral Fellowship (JTR). Charles A. King Trust and Bank of America Co-Trustee (WH). These studies were supported in part by an Ellison Medical Foundation New Scholar Award, American Diabetes Association, US Department of Defense and NIH RO1, DK069966 (to PP).
Footnotes
Author Contributions: J.T.R. designed and performed experiments, analyzed data, and wrote the manuscript. W.H. and S.P.G performed M.S. experiments and analyzed M.S. data. P.P. designed experiments, analyzed data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annual review of physiology. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- Bullock AN, Das S, Debreczeni JE, Rellos P, Fedorov O, Niesen FH, Guo K, Papagrigoriou E, Amos AL, Cho S, et al. Kinase Domain Insertions Define Distinct Roles of CLK Kinases in SR Protein Phosphorylation. Structure. 2009;17:352–362. doi: 10.1016/j.str.2008.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, Fu XD. SRPK1 and Clk/Sty protein kinases show distinct substrate specificities for serine/arginine-rich splicing factors. The Journal of biological chemistry. 1996a;271:24569–24575. doi: 10.1074/jbc.271.40.24569. [DOI] [PubMed] [Google Scholar]
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. The EMBO journal. 1996b;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, Yates J, 3rd, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- Duncan PI, Howell BW, Marius RM, Drmanic S, Douville EM, Bell JC. Alternative splicing of STY, a nuclear dual specificity kinase. The Journal of biological chemistry. 1995;270:21524–21531. doi: 10.1074/jbc.270.37.21524. [DOI] [PubMed] [Google Scholar]
- Duncan PI, Stojdl DF, Marius RM, Bell JC. In vivo regulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Molecular and cellular biology. 1997;17:5996–6001. doi: 10.1128/mcb.17.10.5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan PI, Stojdl DF, Marius RM, Scheit KH, Bell JC. The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Experimental cell research. 1998;241:300–308. doi: 10.1006/excr.1998.4083. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR., 3rd An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Farkas R, Kovacikova M, Liszekova D, Beno M, Danis P, Rabinow L, Chase BA, Raska I. Exploring some of the physico-chemical properties of the LAMMER protein kinase DOA of Drosophila. Fly. 2009;3 doi: 10.4161/fly.7831. [DOI] [PubMed] [Google Scholar]
- Haas W, Faherty BK, Gerber SA, Elias JE, Beausoleil SA, Bakalarski CE, Li X, Villen J, Gygi SP. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- Hanes J, von der Kammer H, Klaudiny J, Scheit KH. Characterization by cDNA cloning of two new human protein kinases. Evidence by sequence comparison of a new family of mammalian protein kinases. Journal of molecular biology. 1994;244:665–672. doi: 10.1006/jmbi.1994.1763. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Hillman RT, Green RE, Brenner SE. An unappreciated role for RNA surveillance. Genome Biol. 2004;5:R8. doi: 10.1186/gb-2004-5-2-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of clinical investigation. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9666–9670. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annual review of physiology. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- Knutti D, Kralli A. PGC-1, a versatile coactivator. Trends in endocrinology and metabolism: TEM. 2001;12:360–365. doi: 10.1016/s1043-2760(01)00457-x. [DOI] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. doi: 10.1038/nature03967. [DOI] [PubMed] [Google Scholar]
- Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- Lee K, Du C, Horn M, Rabinow L. Activity and autophosphorylation of LAMMER protein kinases. The Journal of biological chemistry. 1996;271:27299–27303. doi: 10.1074/jbc.271.44.27299. [DOI] [PubMed] [Google Scholar]
- Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell metabolism. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. Molecular cell. 2000;6:307–316. doi: 10.1016/s1097-2765(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, Sumi K, Yomoda J, Murray MV, Kimura H, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. The Journal of biological chemistry. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. The Journal of clinical investigation. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayler O, Schnorrer F, Stamm S, Ullrich A. The cellular localization of the murine serine/arginine-rich protein kinase CLK2 is regulated by serine 141 autophosphorylation. The Journal of biological chemistry. 1998;273:34341–34348. doi: 10.1074/jbc.273.51.34341. [DOI] [PubMed] [Google Scholar]
- Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD, Ghosh G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Molecular cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Nikolakaki E, Du C, Lai J, Giannakouros T, Cantley L, Rabinow L. Phosphorylation by LAMMER protein kinases: determination of a consensus site, identification of in vitro substrates, and implications for substrate preferences. Biochemistry. 2002;41:2055–2066. doi: 10.1021/bi011521h. [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Molecular cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Pike ACW, Bullock A, Fedorov O, Pilka ES, Ugochukwu E, Von Delft F, Edwards A, Arrowsmith CH, Weigelt J, Sundstrom M, Knapp S. CRYSTAL STRUCTURE OF DI-PHOSPHORYLATED HUMAN CLK1 IN COMPLEX WITH A NOVEL SUBSTITUTED INDOLE INHIBITOR (PDB ID: 2vag) 2007 [Google Scholar]
- Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annual review of physiology. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annual review of nutrition. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Molecular and cellular biology. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad J, Manley JL. Regulation and substrate specificity of the SR protein kinase Clk/Sty. Molecular and cellular biology. 2003;23:4139–4149. doi: 10.1128/MCB.23.12.4139-4149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Quinn PG, Yeagley D. Insulin regulation of PEPCK gene expression: a model for rapid and reversible modulation. Current drug targets. 2005;5:423–437. doi: 10.2174/156800805774912962. [DOI] [PubMed] [Google Scholar]
- Radziuk J, Pye S. Hepatic glucose uptake, gluconeogenesis and the regulation of glycogen synthesis. Diabetes Metab Res Rev. 2001;17:250–272. doi: 10.1002/dmrr.217. [DOI] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, Spiegelman BM. Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple RK, Sleigh A, Murgatroyd PR, Adams CA, Bluck L, Jackson S, Vottero A, Kanabar D, Charlton-Menys V, Durrington P, et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. The Journal of clinical investigation. 2009;119:315–322. doi: 10.1172/JCI37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Analytical chemistry. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Bell JC. SR protein kinases: the splice of life. Biochemistry and cell biology = Biochimie et biologie cellulaire. 1999;77:293–298. [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.