

Summary
Micro-RNA (miR) are increasingly recognized as critical regulators of tissue-specific patterns of gene expression. CD4+ T cells lacking miR-155, for example, exhibit bias towards Th2 differentiation, indicating that the absence of individual miR could alter CD4+ T cell differentiation. We now show that MiR-155 is induced upon T cell activation and that it promotes Th1 differentiation when over-expressed in activated CD4+ T cells. Antagonism of miR-155 leads to induction of IFN-γRα, and a functional miR-155 target site is identified within the 3′ UTR of IFN-γRα. These results identify IFN-γRα as a second miR-155 target in T cells and suggest that miR-155 contributes to Th1 differentiation in CD4+ T cells by inhibiting IFN-γ signaling.
Keywords: CD4+ T Cells, Cell Differentiation, Cytokines
Introduction
CD4+ T cells orchestrate diverse modes of immune responses by differentiating into distinct helper subsets, characterized by unique patterns of cytokine secretion [1]. Th1 cells, which secrete the signature cytokine IFN-γ, and Th2 cells, which secrete the signature cytokine IL-4, were the first two subsets to be characterized. More recently, Th17 cells, which secrete IL-17, were identified as a third helper T cell subset. Dysregulated differentiation of CD4+ T cells can lead to defects in the development of pathogen-specific immune responses, and can also result in lymphocyte-mediated disease [1]. The differentiation of CD4+ T cells into different subsets is strongly influenced by cytokine signaling and by the expression of subset specific transcription factors [1]. More recently, micro-RNA (miR) have also been implicated in the process of CD4+ T cell differentiation [2, 3].
In CD4+ T cells, deficiency of Dicer, a nuclease essential to miR biogenesis, leads to diminished proliferation and increased cell death upon activation [3]. Dicer-deficient CD4+ T cells are also pre-disposed towards differentiation into IFN-γ-producing Th1 cells [3]. Subsequent studies have begun to characterize the roles of individual miR in CD4+ T cells. Expression data derived from Northern blot screening and miR micro-array technology have led to functional studies that have identified a role for miR-181 in modulating T cell antigen receptor sensitivity [4], miR-150 in the regulation of the transcription factor c-Myb [5, 6], and miR-155 in the differentiation of CD4+ T cells into Th1 cells and the development of regulatory T cells [7-13].
The specific gene regulatory networks by which miR influence CD4+ T cell differentiation remain largely undefined. MiR-155 is the only miR known to influence CD4+ T cell subset differentiation and its only known targets in CD4+ T cells are c-Maf and SOCS1. We selected three miR, miR-155, miR-150, and miR-146a, thought to play an important role in lymphocyte activation and differentiation and examined their expression patterns over time in naïve CD4+ T cells that were activated and cultured in Th1-inducing, Th2-inducing, and unbiased conditions. We then examined the ability of these miR to influence Th1/Th2 differentiation when over-expressed during CD4+ T cell activation. MiR-155 over-expression was found to bias CD4+ T cells towards Th1 differentiation, while antagonism of miR-155 was found to bias the cells towards Th2 differentiation. MiR-155 antagonism in activated CD4+ T cells resulted in increased expression of the IFN-γ receptor alpha chain (IFN-γRα). We subsequently identified the message of IFN-γRα as an additional target of miR-155 in Th1 cells that could account for enhanced Th1 differentiation. Our results suggest that miR-155 inhibits IFN-γ responsiveness in differentiating CD4+ T cells and promotes Th1 differentiation.
Results and Discussion
Changes in the expression of individual miR during CD4+ T cell differentiation
To evaluate changes in the expression of individual miR during the differentiation of naïve CD4+ T cells into Th1 and Th2 cells, the expression of miR-155, miR-146a, and miR-150 was evaluated by Northern blot analysis of RNA extracted from CD4+ T cells cultured under unbiased, Th1-inducing, and Th2-inducing cytokine conditions (Fig. 1). These three miR were chosen for further study because they exhibit three different patterns of expression in T cells. MiR-155, encoded by the bic gene, is rapidly induced in both B and T cells upon activation [14-16]. CD4+ T cells from bic deficient mice exhibit preferential Th2 differentiation upon activation, thought to be in part secondary to the increased expression of the Th2-associated transcription factor c-Maf [7, 8]. MiR-146a has the most discordant expression between Th1 cells and Th2 cells observed to date with the observation of much higher expression in fully differentiated Th1 clones than in fully differentiated Th2 clones [10]. MiR-150 has been shown to be down-regulated on lymphocyte activation, and to target the transcription factor c-Myb [5, 6].
Figure 1.
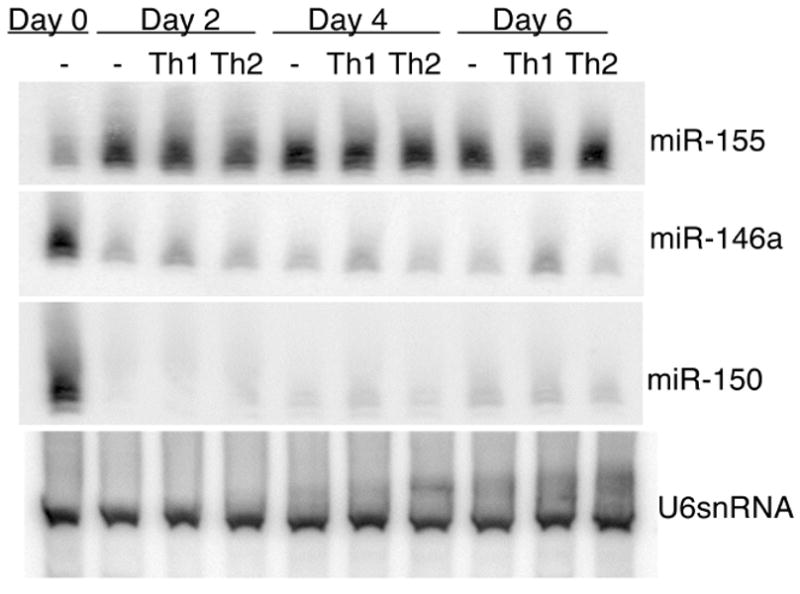
Analysis of miR-155, miR-146a and miR-150 expression by Northern blot. CD4+ T cells from BALB/c mice were stimulated in vitro, and cultured in unbiased, Th1-, or Th2-polarizing conditions. On the second, fourth and sixth days of culture cells were collected for RNA isolation and subsequent Northern blotting. The blot was probed for miR as indicated and for U6snRNA as a loading control. Data is representative of three independent experiments.
MiR-155 and miR-150 were found to be induced and repressed, respectively, at 2 days following CD4+ T cell activation (Fig. 1). MiR-146a was found to be down-regulated 2 days following activation, with expression in Th1 inducing conditions slightly higher than that seen in unbiased or Th2 inducing conditions. For all three miR examined, changes in expression occurred within 2 days after CD4+ T cell activation, and subsequently, expression levels remained constant through the course of the primary stimulation (Fig. 1).
MiR-155 over-expression or antagonism alters Th1/Th2 differentiation
To evaluate the functional consequences of their over-expression in a CD4+ T cell differentiation assay, bicistronic retroviruses containing the primary miR sequence, including 250 bases each of 3′ and 5′ endogenous flanking sequence, of miR-155, miR-146a, or miR-150, followed by the codons of GFP were constructed. The retroviruses were then used to transduce activated CD4+ T cells (Fig. 2A, B, C). Two of the miR evaluated, miR-150 and miR-146a, did not significantly influence CD4+ T cell differentiation in this assay (Fig. 2B, C). Over-expression of miR-155 led to increased Th1 differentiation (Fig. 2A), a result that is complementary to the observed bias towards Th2 differentiation in CD4+ T cells lacking miR-155 [7, 8]. We used an antagomir, a modified antisense RNA oligomer shown to specifically reduce miR activity in vivo [17], to antagonize the activity of miR-155 and evaluate the the effects of an acute loss of miR-155 function (as opposed to constitutive deficiency) on CD4+ T cell differentiation. In agreement with the described Th2 bias seen in CD4+ T cells from bic deficient mice, we observed a bias towards Th2 differentiation in CD4+ T cells cultured in the presence of antagomir (Fig 2D).
Figure 2.
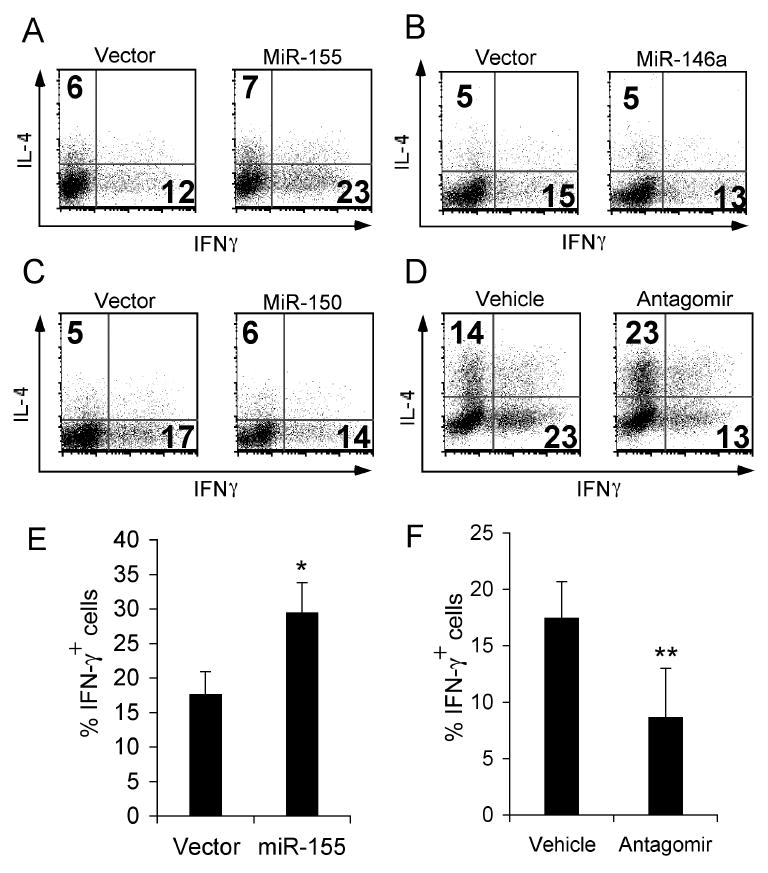
Over-expression of miR-155, miR-146a and miR-150 and antagonism of miR-155 in a CD4+ T cell Th1/Th2 differentiation assay. CD4+ T cells from C57BL/6 mice were stimulated and cultured in unbiased conditions. Cells were transduced with retrovirus encoding GFP and (A) miR-155, (B) miR-146a, or (C) miR-150 36 hours after plating. (D) Cells were cultured in the presence of an antagomir directed against miR-155 or vehicle control. On the fourth day of culture cells were re-stimulated with PMA and ionomycin and production of IL-4 and IFN-γ was measured by intracellular cytokine staining and flow cytometry. Plots shown in A, B, and C are gated on CD4+ GFP+ (transduced) cells. Plots in A, B, C, and D are representative of four independent experiments. Summary of data from A (E) and D (F), data show mean ± SEM from four independent experiments. *p<0.01 versus vector or **p<0.05 versus vehicle; Student's two-tailed t-test.
While the data in Figure 2 show that miR-155 promotes Th1 differentiation, the results in Figure 1 show equal expression of miR-155 in Th1- and Th2-polarizing conditions. MiR-155, present at low basal levels in unstimulated CD4+ T cells, is up-regulated by a stimulus common to both Th1- and Th2-polarizing conditions, such as TCR ligation or IL-2, reaching peak levels by 24 hours after TCR ligation (Fig. S1A). To determine whether the observed Th1 suppression in the presence of miR-155 antagonism was secondary to an alteration of TCR mediated T cell activation, we measured the expression of activation markers and early IL-2 production in the presence or absence of miR-155 antagonism (Fig. S1B, C). We observed a mild decrease in the up-regulation of the activation markers CD25 and CD69 and the down-regulation of CD62L, while early IL-2 production was not significantly altered. While these results suggest that miR-155 may contribute to T cell activation after TCR ligation, the magnitude of the changes suggests that they are unlikely to account for the observed effects of miR-155 on Th1 differentiation.
The only confirmed targets of miR-155 in T cells are c-Maf, a transcription factor that promotes IL-4 production, and SOCS-1, a negative regulator of cytokine signaling [13, 18]. Regulation of SOCS-1 expression by miR-155 has been implicated in regulatory T cell development, and could contribute to the effects of miR-155 on Th1/Th2 differentiation [13]. However, in our system, SOCS1 protein levels in activated CD4+ T cells were unchanged in the presence of miR-155 over-expression (Fig S2). While miR-155 targeting of c-Maf may, in part, account for the effects of miR-155 on Th1/Th2 differentiation, two lines of evidence support the involvement of other targets. The first lies in the observation that of 53 transcripts up-regulated in bic deficient Th1 cells compared to wild type Th1 cells 46 were found to have computationally predicted miR-155 target sites [7]. Similarly, of 99 transcripts up-regulated in bic deficient Th2 cells compared to wild type Th2 cells, 53 were found to have computationally predicted miR-155 target sites [7]. The second is that the effects of miR-155 on Th1/Th2 differentiation shown in Figure 2 are all observed on day 4 after activation, while c-maf induction is not seen until day 8 [18]. To identify potential mechanisms by which miR-155 could alter Th1/Th2 differentiation, additional targets of miR-155 in CD4+ T cells were sought.
IFN-γRα is a target of miR-155 in CD4+ T cells
As cytokine signal transduction is known to be a key element of CD4+ T cell subset differentiation, computationally derived lists of putative miR-155 targets were examined for cytokine signal transduction components. Lists were obtained using the following searchable websites: PicTar (http://pictar.bio.nyu.edu), TargetScan (http://genes.mit.edu/targetscan), miRanda (http://www.microrna.org//miranda.html) and mirBase ((http://microrna.sanger.ac.uk/targets/v2/). The IFN-γRα chain was identified as a putative target, and was of particular interest as there is evidence that the regulation of IFN-γ signaling through the IFN-γR is important to the differentiation of Th1 cells [19-21]. During maturation, Th1 cells down-regulate the β chain of the IFN-γR and the blockade of this down-regulation through the expression of an IFN-γRβ transgene results in impaired Th1 differentiation [19-21]. Earlier in the course of CD4+ T cell activation, the IFN-γRα chain is transiently down-regulated in a manner that is independent of IFN-γ signaling [22]. The mechanism of this down-regulation of IFN-γRα is unknown. To determine whether IFN-γRα expression was modified by miR-155, CD4+ T cells were stimulated and cultured in Th1- and Th2-polarizing conditions in the presence or absence of antagomir directed against miR-155, as above. Antagonism of miR-155 led to increased expression of IFN-γRα in cells cultured under Th1 conditions, with no difference observed in cells cultured under Th2 inducing conditions (Fig. 3A). Conversely, IFN-γRα expression was diminished in CD4+ T cells over-expressing miR-155, an effect also seen in Th1-, but not Th2-polarizing conditions (Fig. 3B). The greater effect of miR-155 antagonism on IFN-γRα expression compared to miR-155 over-expression may be related to the observation that miR-155 is already significantly induced in activated CD4+ T cells (Fig. 1). CD4+ cells cultured in Th2 conditions express higher levels of the miR-155 target c-Maf in the presence of antagomir, providing evidence that the antagomir functionally antagonizes miR-155 in Th2 cells (Fig. S3). The absence of IFN-γRα mRNA targeting by miR-155 in Th2 inducing conditions could be explained by preferential binding of miR-155 to high affinity Th2 specific mRNA targets, such as c-Maf, in lieu of binding to IFN-γRα mRNA. Alternatively, miR-155 may interact with IFN-γRα message in Th2 cells, but the interaction may be insufficient for silencing, requiring additional factors, such as other miR, that are present in Th1 cells.
Figure 3.
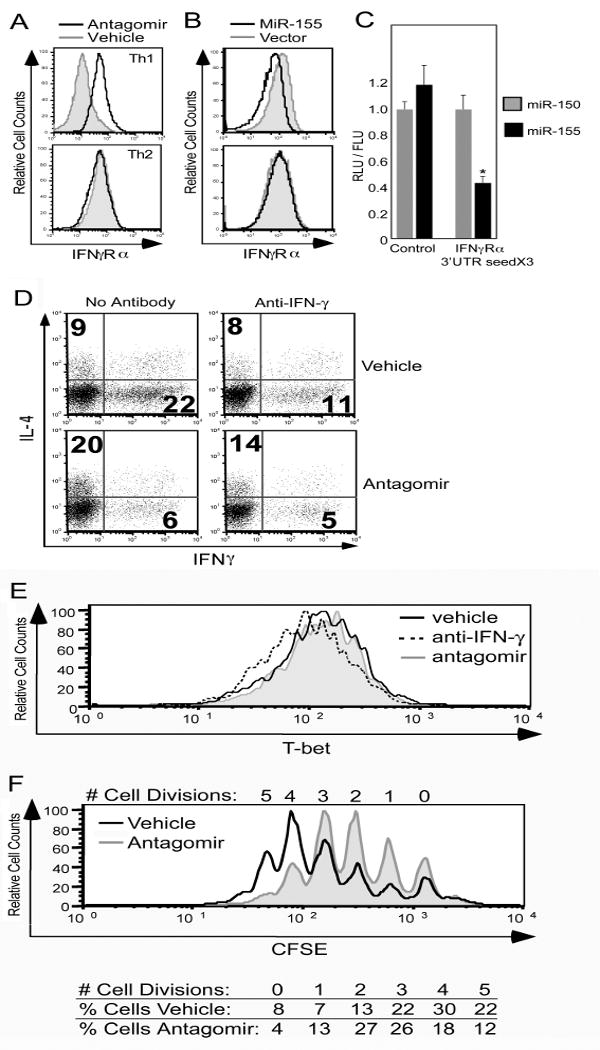
IFN-γRα is a target of miR-155 in Th1 cells. (A and B) CD4+ T cells from C57BL/6 mice were stimulated and cultured under Th1- or Th2-polarizing conditions with antagomir (black line) or vehicle control (gray filled line) (A) or transduced with retrovirus encoding GFP alone (vector, gray filled line) or with miR-155 (black line) (B). After four days, IFN-γRα surface expression was measured by flow cytometry. Plots in (B) are gated on GFP+ cells. (C) NIH 3T3 cells were transduced with retrovirus encoding miR-155 or miR-150 and then co-transfected with an unmodified firefly luciferase reporter and a renilla luciferase reporter either unmodified or modified to include three copies of the 27 base pair IFN-γRα 3′ UTR sequence containing the putative miR-155 target site. The ratio of renilla luciferase to firefly luciferase activity (RLU/FLU) is represented in the bar graph as mean ± SEM from three independent experiments. *p<0.01 versus miR-155 control; Student's two-tailed t-test. (D) CD4+ T cells from C57BL/6 mice were stimulated and cultured under unbiased conditions in the presence of antagomir or vehicle control, with or without anti-IFN-γ antibody. After four days cells were re-stimulated and production of IL-4 and IFN-γ was measured by flow cytometry. Data are representative of three independent experiments. (E and F) CD4+ T cells from C57BL/6 mice unlabeled (E) or CFSE labeled (F) were stimulated and cultured under unbiased conditions with anti-IFN-γ antibody (dashed line, E only), antagomir (gray filled line), or vehicle control (black line). (E) After 16 hours, CD4 and T-bet expression were analyzed by flow cytometry. (F) After three days cells were analyzed by flow cytometry for CFSE content. Histograms are gated on live CD4+ cells (E, F).
To verify that IFN-γRα mRNA could be a direct target of miR-155, the computationally identified target site within the 3′ untranslated region (UTR) of IFN-γRα was tested for its ability to confer regulation by miR-155 to a heterologous reporter gene. An expression construct for renilla luciferase reporter was modified to include 3 copies of the 27 base pair region of the IFN-γRα 3′ UTR including the miR-155 target site. The modified construct was co-transfected with a control plasmid expressing firefly luciferase into NIH 3T3 cells over-expressing either miR-155 or miR-150. Expression of renilla luciferase relative to firefly luciferase was diminished in the presence of miR-155, but not miR-150, only in the presence of the IFN-γRα target site, indicating the ability of miR-155 to target this site (Fig. 3C). While we cannot entirely exclude the possibility that IFN-γRα expression is at least in part reduced by increased IFN-γ production by miR-155 expressing Th1 cells, our data support the direct targeting of IFN-γRα mRNA by miR-155 in CD4+ T cells in Th1 inducing conditions.
To investigate the functional significance of miR-155 disruption of IFN-γ signaling in Th1 differentiation, CD4+ T cells were activated and cultured in the presence or absence of antagomir, as in Figure 2D, with or without the addition of anti-IFN-γ antibody to the culture (Fig. 3D). Anti-IFN-γ inhibits Th1 differentiation in the absence of antagomir (Fig. 3D, top panels), but does not alter Th1 differentiation when antagomir is present (Fig. 3D, bottom panels). The degree of Th1 repression is greater with antagomir than with anti-IFN-γ (Fig 3D), suggesting that IFN-γRα is one of multiple targets of miR-155 in the inhibition of Th1 differentiation in CD4+ T cells.
IFN-γ has pleiotropic effects on CD4+ T cell differentiation. The induction of T-bet, a transcription factor critical to Th1 differentiation, has been shown to occur within 3 hours of CD4+ T cell activation and to be dependent on IFN-γ signaling through STAT1 [23, 24]. The inhibition of this activity by neutralizing anti-IFN-γ antibody would therefore inhibit Th1 differentiation by preventing early T-bet induction in activated CD4+ T cells. The early induction of T-bet would not be predicted to be impaired by miR-155, as IFN-γ signaling would likely have occurred by the time IFN-γRα protein levels were influenced by miR-155 induction and subsequent targeting of IFN-γRα mRNA. Consistent with this prediction, we observed that antagonism of miR-155 does not appear to interfere with T-bet expression at an early time point (Fig. 3E), showing that miR-155 does not influence IFN-γ mediated T-bet induction.
A second way in which IFN-γ is thought to promote Th1 differentiation is by inhibiting the proliferation of Th2, but not Th1 cells [25]. Subsequent studies have suggested a relationship between this finding and the previously mentioned finding that committed Th1 cells down-regulate the expression of IFN-γRβ, losing responsiveness to IFN-γ [19, 20]. MiR-155 targeting of IFN-γRα would be predicted to promote proliferation of CD4+ T cells by reducing the sensitivity of the cells to the anti-proliferative effects of IFN-γ. Consistent with this prediction, we observe reduced proliferation in CD4+ T cells cultured in the presence of miR-155 antagomir (Fig. 3F). Therefore, we propose a mechanism whereby miR-155 targeting of IFN-γRα enhances Th1 maturation by decreasing sensitivity of the cells to the antiproliferative effects of IFN-γ.
Concluding Remarks
We present evidence that miR-155 inhibits the expression of IFN-γRα early in the course of CD4+ T cell activation under Th1 inducing conditions and contributes to Th1 differentiation. IFN-γRβ is known to be down-regulated specifically in Th1 cells over the course of two weeks [19, 20]. A more rapid down-regulation of IFN-γRα induced by miR-155 in Th1 cells suggests a model whereby miR may serve as rapid and reversible gene regulators in cells that are undergoing differentiation prior to subsequent stabilization of gene expression during irreversible cell commitment [26]. Cytokine signal transduction is a pivotal element in the differentiation of CD4+ T cells into different effector subsets, and the data presented above represents the first evidence that miR play a role in inhibiting cytokine signal transduction in CD4+ T cells. Loss of miR-155 function is seen to promote Th2 differentiation (Fig 2D and [7, 8]), whereas the loss of all miR by Dicer ablation promotion leads to Th1 biased differentiation [3], providing evidence that miR other than miR-155 are likely to influence CD4+ effector differentiation as well.
Materials and Methods
Mice
All animal work was in accordance with Institutional Animal Care and Use Guidelines of the University of Pennsylvania (Philadelphia, PA). C57BL/6 and BALB/c mice were obtained from the Jackson Laboratory.
CD4+ T cell isolation and culture
CD4+ T cells were isolated from splenocytes by positive selection using CD4 MACS micro beads and MACS columns (Miltenyi Biotec). CD4+ T cells were activated with plate bound anti-CD3 antibody (BD Biosciences) and soluble anti-CD28 antibody (1 mg/ml, BD Biosciences) and cultured in the presence of 100 U/ml human recombinant IL-2 (unbiased condition). Th1- and Th2-polarizing conditions were as previously described [27]. Anti-IFN-γ antibody was purchased from Bio X Cell and used at a final concentration of 5 μg/ml.
Retroviral transduction
Retroviruses expressing GFP in conjunction with miR-155, miR-150 or miR-146a were constructed and prepared as described [28], using the previously described MSCV PIG vector (a generous gift of Scott Lowe) [29]. Retroviral transduction was performed after 36 hours of activation as previously described [27].
Flow Cytometry
Surface staining, intracellular staining and flow cytometry were done as previously described [27]. All antibodies used for flow cytometry were purchased from BD Biosciences, with the exception of APC conjugated T-bet antibody (eBioscience). Carboxyfluorescein diacetate succinimidyl ester (CFSE) (Molecular Probes) labeling was performed by incubating 1×107 cells/ml in PBS with 10 μM CFSE for 9 minutes at room temperature. Labeling was stopped with 1 volume fetal calf serum and cells were washed three times prior to stimulation.
Analysis of micro-RNA expression by Northern blot
Total RNA was isolated using Trizol (Invitrogen). For Northern blotting, 50 mg of total RNA per sample was separated by 15% Urea-PAGE and transferred to Nytran membrane (ISC Bioexpress) by semi-dry transfer. Probes were DNA oligonucleotides of complementary sequence to the miR of interest (miR-155 probe: 5′-CCCCTATCACAATTAGCATTAA-3′, miR-150 probe: 5′-CACTGGTACAAGGGTTGGGAGA-3′, miR-146a probe: 5′-AACCCATGGAATTCAGTTCTCA-3′) end-labeled with (γ-32P)ATP.
Antagonism of miR function
Antagomirs were designed as described and purchased from Dharmacon [17]. Sequence of antagomir used was 5′- cscsccuaucacaauuagcaususasas-Chol-3′, with subscripted “s” denoting phorphorothioate linkage and “Chol” representing a 3′ cholesterol moiety. Cells were cultured in the presence of 50 μg/ml antagomir.
Reporter Assays
Plasmids encoding renilla luciferase (pRL-TK, Promega) and firefly luciferase (pGL3, Promega) were transfected into NIH 3T3 cells by the calcium phosphate method. Luciferase activities were determined using a Dual Luciferase Reporter Assay System (Promega). 3 copies of the 27 bp sequence of the IFN-γRα 3′ untranslated region containing the miR-155 target site (5′-ACATACTTTTTTATGGAGCATTACTTA-3′) were cloned into the XbaI/NotI site of pRL-TK.
Acknowledgments
We are grateful to members of the Reiner laboratory for discussion. Supported by the National Institutes of Health (AI042370, S.L.R. and CA076931, A.B.) and the Abramson Family.
Abbreviations
- miR
micro-RNA
- UTR
untranslated region
- IFN-γRα
IFN-γ receptor alpha chain
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Reiner SL. Development in motion: helper T cells at work. Cell. 2007;129:33–36. doi: 10.1016/j.cell.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 2.Cobb BS, Nesterova TB, Thompson E, Hertweck A, O'Connor E, Godwin J, Wilson CB, Brockdorff N, Fisher AG, Smale ST, Merkenschlager M. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, Braich R, Manoharan M, Soutschek J, Skare P, Klein LO, Davis MM, Chen CZ. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–159. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 6.Zhou B, Wang S, Mayr C, Bartel DP, Lodish HF. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proc Natl Acad Sci U S A. 2007;104:7080–7085. doi: 10.1073/pnas.0702409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 9.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 10.Monticelli S, Ansel KM, Xiao C, Socci ND, Krichevsky AM, Thai TH, Rajewsky N, Marks DS, Sander C, Rajewsky K, Rao A, Kosik KS. MicroRNA profiling of the murine hematopoietic system. Genome Biol. 2005;6:R71. doi: 10.1186/gb-2005-6-8-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, Manjunath N. miRNA profiling of naive, effector and memory CD8 T cells. PLoS ONE. 2007;2:e1020. doi: 10.1371/journal.pone.0001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol. 2009;182:2578–2582. doi: 10.4049/jimmunol.0803162. [DOI] [PubMed] [Google Scholar]
- 13.Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, Loeb GB, Lee H, Yoshimura A, Rajewsky K, Rudensky AY. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30:80–91. doi: 10.1016/j.immuni.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A. 2005;102:3627–3632. doi: 10.1073/pnas.0500613102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haasch D, Chen YW, Reilly RM, Chiou XG, Koterski S, Smith ML, Kroeger P, McWeeny K, Halbert DN, Mollison KW, Djuric SW, Trevillyan JM. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol. 2002;217:78–86. doi: 10.1016/s0008-8749(02)00506-3. [DOI] [PubMed] [Google Scholar]
- 16.van den Berg A, Kroesen BJ, Kooistra K, de Jong D, Briggs J, Blokzijl T, Jacobs S, Kluiver J, Diepstra A, Maggio E, Poppema S. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer. 2003;37:20–28. doi: 10.1002/gcc.10186. [DOI] [PubMed] [Google Scholar]
- 17.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 18.Ho IC, Hodge MR, Rooney JW, Glimcher LH. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996;85:973–983. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 19.Bach EA, Szabo SJ, Dighe AS, Ashkenazi A, Aguet M, Murphy KM, Schreiber RD. Ligand-induced autoregulation of IFN-gamma receptor beta chain expression in T helper cell subsets. Science. 1995;270:1215–1218. doi: 10.1126/science.270.5239.1215. [DOI] [PubMed] [Google Scholar]
- 20.Pernis A, Gupta S, Gollob KJ, Garfein E, Coffman RL, Schindler C, Rothman P. Lack of interferon gamma receptor beta chain and the prevention of interferon gamma signaling in TH1 cells. Science. 1995;269:245–247. doi: 10.1126/science.7618088. [DOI] [PubMed] [Google Scholar]
- 21.Tau GZ, von der Weid T, Lu B, Cowan S, Kvatyuk M, Pernis A, Cattoretti G, Braunstein NS, Coffman RL, Rothman PB. Interferon gamma signaling alters the function of T helper type 1 cells. J Exp Med. 2000;192:977–986. doi: 10.1084/jem.192.7.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skrenta H, Yang Y, Pestka S, Fathman CG. Ligand-independent down-regulation of IFN-gamma receptor 1 following TCR engagement. J Immunol. 2000;164:3506–3511. doi: 10.4049/jimmunol.164.7.3506. [DOI] [PubMed] [Google Scholar]
- 23.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 24.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O'Shea JJ. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gajewski TF, Fitch FW. Anti-proliferative effect of IFN-gamma in immune regulation. I. IFN-gamma inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J Immunol. 1988;140:4245–4252. [PubMed] [Google Scholar]
- 26.Hobert O. Gene regulation by transcription factors and microRNAs. Science. 2008;319:1785–1786. doi: 10.1126/science.1151651. [DOI] [PubMed] [Google Scholar]
- 27.Mullen AC, Hutchins AS, Villarino AV, Lee HW, High FA, Cereb N, Yang SY, Hua X, Reiner SL. Cell cycle controlling the silencing and functioning of mammalian activators. Curr Biol. 2001;11:1695–1699. doi: 10.1016/s0960-9822(01)00533-4. [DOI] [PubMed] [Google Scholar]
- 28.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 29.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]