

Abstract
Background and purpose:
Although both natural and synthetic cannabinoid compounds have been shown to exert an antinociceptive effect on acute and persistent pain, the anatomical locus of the target of cannabinoid-induced analgesia has not been fully elucidated. Here, we investigated the effects of the cannabinoid agonist WIN 55,212-2 on GABA-activated currents (IGABA) in rat primary sensory neurones.
Experimental approach:
In the present study, experiments were performed on neurones freshly isolated from rat trigeminal ganglion (TG) by using whole-cell patch clamp and repatch techniques.
Key results:
GABA-evoked inward currents were potentiated by pretreatment with WIN 55,212-2 in a concentration-dependent manner (10−10–10−8 M). WIN 55,212-2 shifted the GABA concentration–response curve upwards, with an increase of 30.3 ± 3.7% in the maximal current response but with no significant change in the EC50 (agonist concentration producing a half-maximal response) value. WIN 55,212-2 potentiated the responses to GABA in a manner independent of holding potential and in the absence of any change in the reversal potential of the current. This potentiation of IGABA induced by WIN 55,212-2 was almost completely blocked by AM 251 (3 × 10−8 M), a CB1 receptor antagonist, and, using the repatch technique, was found to be abolished after intracellular dialysis with the protein kinase A (PKA) activator cAMP or the PKA inhibitor H89.
Conclusions and implications:
The potentiation by WIN 55,212-2 of IGABA in primary sensory neurones may help to elucidate the mechanism underlying the modulation of analgesia by cannabinoids in the spinal dorsal horn.
Keywords: potentiation; WIN 55,212-2; GABA-activated current; trigeminal ganglion neurones; patch clamp technique; intracellular dialysis
Introduction
It is known that cannabis has been used for pain relief for many centuries (Walker and Huang, 2002). A growing body of evidence has suggested that cannabinoids are involved in the modulation of nociception and analgesia (Fox et al., 2001; Walker and Huang, 2002; Agarwal et al., 2007). The biological effects of cannabinoids are mediated via binding to type 1 and type 2 G-protein–coupled cannabinoid receptors (CB1 and CB2 respectively), which activate inhibitory Gi/o proteins (Freund et al., 2003; Piomelli, 2005). CB1 receptors are constitutionally active and abundantly expressed in peripheral and central terminals of primary sensory neurones including trigeminal ganglion (TG) neurones, second-order spinal dorsal-horn neurones, and brain regions involved in pain-modulating pathways (Tsou et al., 1998; Ahluwalia et al., 2000; Bridges et al., 2003; Price et al., 2003; Rea et al., 2007). Thus, the CB1 receptor has been implicated in cannabinoid-induced analgesia. Recently, Agarwal et al., by using specific deletion of CB1 in nociceptive neurones of primary sensory ganglia (the expression of CB1 in CNS remained unchanged), concluded that the contribution of CB1 type receptors expressed on the peripheral, rather than the central, terminals of nociceptors is paramount to cannabinoid-induced analgesia. In contrast, a number of other studies have indicated that CB1 receptors expressed on central terminals of primary sensory neurones are involved in cannabinergic modulation of pain. Cannabinoids are antihyperalgesic when administered to the spinal cord as well as when administered peripherally at the site of tissue injury (Calignano et al., 1998; Martin et al., 1999; Fox et al., 2001; Johanek et al., 2001). It has been reported that cannabinoids activate CB1 receptors at the nerve terminals of small-diameter primary afferent fibres and induce pre-synaptic inhibition (Liang et al., 2004). Moreover, cannabinoids have been shown to decrease the release of transmitters from terminals of dorsal root ganglion neurones in the spinal cord and skin by inhibiting the N-type voltage-gated calcium channels (Morisset and Urban, 2001; Khasabova et al., 2004).
In the present study, to further explore whether the analgesic effect of cannabinoids occurs in the central terminals of primary sensory neurones, the effects of cannabinoids on the GABAA receptor were investigated in rat TG neurones. We found that WIN 55,212-2 (a synthetic cannabinoid agonist of CB1 and CB2 receptors), when applied at nanomolar concentrations (10−11–10−8 M), enhanced the GABA-activated currents in the TG neurones. Our findings may throw light on the understanding of the mechanism underlying the modulation of analgesia by cannabinoids (Li et al., 2008).
Methods
Isolation of TG neurones
Two- to three-week old Sprague-Dawley rats were anaesthetized with ether and then decapitated. The TGs were taken out and transferred immediately into Dulbecco's modified Eagle's medium (DMEM) at pH 7.4. After the removal of the surrounding connective tissues, the TGs were minced with fine scissors, and the ganglion fragments were placed in a flask containing 5 mL of DMEM in which trypsin (type II-S) 0.5 mg·mL−1, collagenase (type I-A) 1.0 mg·mL−1 and DNase (type IV) 0.1 mg·mL−1 had been dissolved, and incubated at 35°C in a shaking water bath for 30–35 min. Soybean trypsin inhibitor (type II-S) 1.25 mg·mL−1 was then added to stop trypsin digestion. Dissociated neurones were placed into a 35 mm Petri dish and kept for at least another 30 min before electrophysiological recording. The neurones selected for patch clamp experiments were 15–45 µm in diameter.
Electrophysiological recordings
Whole-cell patch clamp recordings were carried out at room temperature (22–24°C) by using a whole-cell patch clamp amplifier (CEZ-2400, Nihkon Kohden, Japan). The micropipettes were filled with internal solution containing (mM): KCl 140, MgCl2 2.5, HEPES 10, EGTA 11 and ATP 5. pH was adjusted to 7.2 with KOH, and osmolarity was adjusted to 310 mOsm·L−1 with sucrose. Cells were bathed in an external solution containing (mM): NaCl 150, KCl 5, CaCl2 2.5, MgCl2 2, HEPES 10, d-glucose 10. osmolarity was adjusted to 330 mOsm·L−1 with sucrose, and pH was adjusted to 7.4 with NaOH. The resistance of the recording pipette was in the range of 2–5 MΩ. A small patch of membrane underneath the tip of the pipette was aspirated to form a gigaseal, and then a negative pressure was applied to rupture it, thus establishing a whole-cell configuration. The adjustment of capacitance compensation and series resistance compensation was carried out before the membrane currents were recorded. The holding potential was set at −60 mV, except when indicated otherwise. Membrane currents were filtered at 10 kHz (−3 dB), and the data were registered by a pen recorder (Nihon Kohden).
Intracellular dialysis using the repatch technique
For the repatch experiment, the first patch is used as the control, with the pipette filled with normal internal solution. After the current had been recorded, the pipette was discarded. On the same neuronee, the second patch was performed by using another pipette that was filled with internal solution containing guanosine 5 =−O-(2-thiodiphosphate) (GDP)-β-S, cAMP or another test substance. After 30 min, the membrane current is recorded again and compared with that of the control.
Drug application
Drugs used in the experiments were purchased from Sigma Chemical Co. and include GABA, WIN 55,212-2, AM 251, DMEM, trypsin, collagenase, Dnase and soybean trypsin inhibitor. All drugs were dissolved daily in the external solution just before use and held in a linear array of fused silica tubes (o.d./i.d. = 500 µm/200 µm) connected to a series of independent reservoirs. The distance from the tube mouth to the cell examined was around 100 µm. The application of each drug was driven by gravity and controlled by the corresponding valve, and rapid solution exchange could be achieved within about 100 ms by shifting the tubes horizontally with a micromanipulator. Cells were constantly bathed in normal external solution flowing from one tube connected to a larger reservoir between drug applications. In the experiments where GDP-β-S and cAMP were applied intracellularly, they were dissolved in the internal solution before use.
Data analysis
Data were statistically compared by using Student's t-test or anova. Statistical analysis of concentration–response data was performed by using the non-linear, curve-fitting program ALLFIT. The values for current are expressed as mean ± SEM.
Results
Effects of WIN 55,212-2 on the GABA-activated current in rat TG neurones
Freshly isolated neurones from rat TG in the range of 15–45 µm were used in the present study. In the majority of the neurones examined (95.2%, 79/83), GABA induced a concentration-dependent (10−6–10−3 M) inward current. This GABA-activated current (IGABA, 10−4 M) could be blocked reversibly by bicuculline (3 × 10−5 M), a selective antagonist of GABAA receptors, indicating this current was mediated by the GABAA receptor (Figure 1A). When GABA was applied regularly for durations of 3 s with 4 min intervals, the IGABA was stable for at least 90 min, and the change in amplitude was within 7.0% (data not shown). Thus, we used this pattern of GABA applications in the following experiments.
Figure 1.
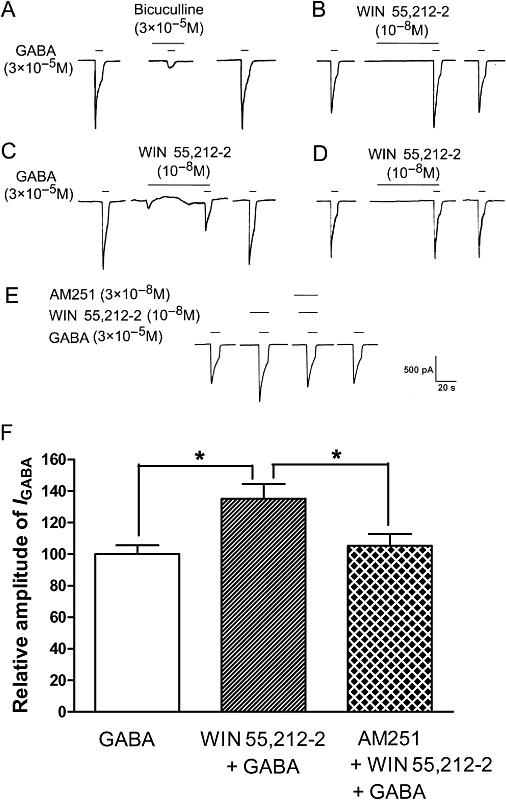
Modulatory effects of WIN 55,212-2 on GABA-activated currents in rat trigeminal ganglion (TG) neurones. (A) The inward current evoked by 3 × 10−5 M GABA could be blocked by the GABAA receptor antagonist bicuculline. Pre-application of WIN 55,212-2 (10−8 M) exerts an enhancing (B), inhibitory (C) and no effect (D) on GABA-activated currents in rat TG neurones. (E) Current traces demonstrating that the CB1 receptor antagonist AM 251 blocked the potentiation by WIN 55,212-2 of GABA-activated currents. (F) Shows the inhibitory effect of AM 251 (3 × 10−8 M) on WIN 55,212-2 (10−8 M)-induced potentiation of GABA (3 × 10−5 M)-activated currents. *P < 0.05, one way analysis of variance followed by Bonferroni's post hoc test, n= 7.
Of the 79 TG neurones sensitive to GABA, 73 cells were pretreated for 30 s with the CB1 receptor agonist WIN 55,212-2. We observed that the majority (69.9%, 51 out of 73) of IGABA (3 × 10−5 M) recorded were obviously potentiated by the pre-application of WIN 55,212-2 (10−8 M); IGABA increased by 36.3 ± 2.6% in 51 neurones (P < 0.01, paired t-test); the amplitude of IGABA decreased by 56.2 ± 7.9% in 3 neurones examined (4.1%, 3/73); while, in the remainder (26.0%, 19/73), WIN 55,212-2 had no effect on the IGABA (3.9 ± 1.2%, P > 0.05, paired t-test) (Figure 1B–D). In the present study, we established a cut-off value for the effect of WIN 55,212-2, which was a change in the amplitude of the IGABA exceeding 12.0%. After pooling all the data from the 73 neurones examined, pre-application of WIN 55,212-2 was found to increase the IGABA by 22.0 ± 3.9% (P < 0.05, paired t-test). The potentiating effect of WIN 55,212-2 on IGABA disappeared after a 4–8 min washout and was reproducible in the same TG neurone. WIN 55,212-2 itself did not induce a current in any of the neurones examined, with the exception of three neurones where it had an inhibitory effect on IGABA induced by an apparent inward current (Figure 1C).
Blockade of WIN 55,212-2-induced potentiation of IGABA by AM 251
To verify whether the potentiation of IGABA by WIN 55,212-2 was mediated by the CB receptor, we examined the effect of the pre-application of WIN 55,212-2 (10−8 M) with AM 251(3 × 10−8 M), a selective CB1 receptor antagonist, on IGABA. The potentiation of IGABA (3 × 10−5 M) by pretreatment with WIN 55,212-2 was almost completely reversed by the addition of AM 251 (Figure 1E,F, P < 0.01, one-way anova following by Bonferroni's post hoc test, n= 7). Moreover, AM 251(3 × 10−8 M) itself had no effect on IGABA (data not shown).
Concentration-dependent potentiation of IGABA by WIN 55,212-2
The IGABA was potentiated by the pre-application of WIN 55,212-2 (10−10–10−8 M) in a concentration-dependent manner. Figure 2 shows that increasing the concentration of WIN 55,212-2 used to pretreat the TG from 10−10 to 10−8 M potentiated the amplitudes of IGABA (3 × 10−5 M) in a stepwise, concentration-dependent manner. This effect peaked with 10−8 M WIN 55,212-2, and with doses higher than this, the potentiation of IGABA gradually decreased (Figure 2A,B).
Figure 2.
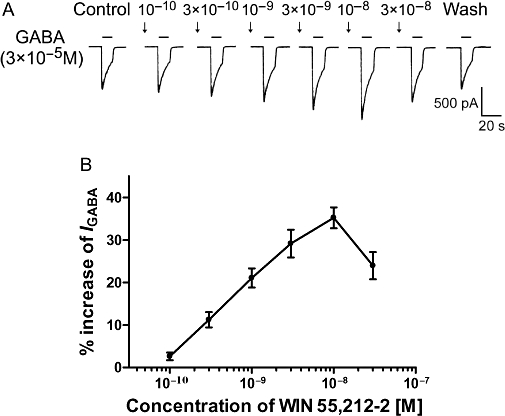
Concentration-dependent potentiation of GABA-activated currents by WIN 55,212-2. (A) Sequential current-traces illustrating the potentiation of IGABA induced by different concentrations of WIN 55,212-2(10−10–3 × 10−8 M) on a single TG neurone. (B) Shows that IGABA increased step by step on increasing the concentration of WIN 55,212-2 from 10−10 to 10−8 M; at the higher concentration, the increase of IGABA reached its peak value; beyond that, IGABA gradually decreased. The pretreatment time for WIN 55,212-2 was 60 s. Each point represents the mean ± SEM of 7–9 neurones.
Effect of pretreatment time of WIN 55,212-2 on its potentiation of IGABA
It can be seen from Figure 3 that, when WIN 55,212-2 was applied to the TG with GABA (i.e. no pretreatment), it did not potentiate the GABA-activated currents. However, after a pre-application of WIN 55,512-2 of at least 15 s, the potentiation of IGABA by WIN 55,212-2 emerged. This suggests that the potentiation by WIN 55,212-2 of IGABA is a time-dependent process and that an intracellular signal transduction pathway may be involved. Hence, an experiment was designed to explore the effect of the duration of WIN 55,512-2 pre-application on its potentiation of GABA-activated currents. The results depicted in Figure 3 demonstrate that increasing the pre-application time of WIN 55,212-2 from 15 s increased its potentiation of the amplitute of IGABA, step by step, until a peak value was reached at 75 s, thereafter, the IGABA was not increased anymore.
Figure 3.
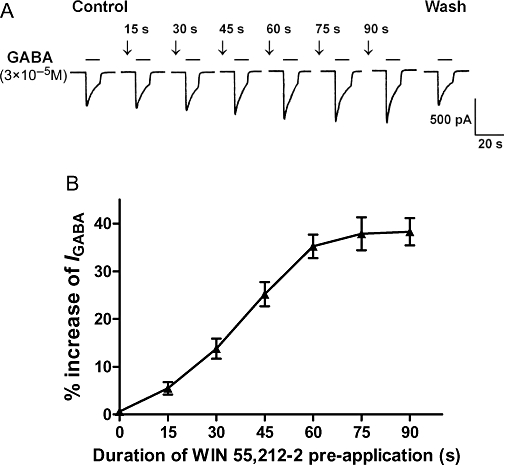
Effect of the duration of the pre-application of WIN 55,212-2 on its potentiation of GABA-activated currents. (A) Current traces demonstrating that the increase of IGABA induced by WIN 55,212-2 (10−8 M) is related to the duration of its pre-application from 15 to 90 s. (B) Shows that the potentiation of IGABA (3 × 10−5 M) by WIN 55,212-2 (10−8 M) was enhanced when the duration of the pre-application was increased from 15 to 90 s. Note that, at a pre-application duration of between 75 and 90 s, the potentiation of the GABA-activated current reached its maximum value. Each point represents the mean ± SEM of 6–9 neurones.
Concentration-response and current-voltage relationships for GABA with and without pretreatment with WIN 55,212-2
Figure 4A shows the concentration–response curves for GABA in the absence and presence of WIN 55,212-2. From the graph in Figure 4A, it can be seen that (i) the dose–response curve for GABA after pretreatment with WIN 55,212-2 is shifted upwards compared with the control; (ii) the EC50 value in both curves are very close (41.4 ± 2.4 × 10−5 M after WIN 55,212-2 pretreatment vs. 43.3 ± 2.6 × 10−5 M without WIN 55,212-2 pretreatment); (iii) the maximum amplitude of IGABA after pretreatment with WIN 55,212-2 increased by 30.3 ± 3.7% as compared with the control; and (iv) the threshold values of both the curves are basically the same. The current–voltage (I-V) curves for GABA with and without pretreatment with WIN 55,212-2 are demonstrated in Figure 4B. WIN 55,212-2 enhanced IGABA at all holding potentials between −80 and 40 mV as shown by the increase in the slope of the I-V curve, and the reversal potentials of two curves are the same at 0 mV. Figure 4C shows that the WIN 55,212-2-induced enhancement of IGABA did not differ significantly at membrane holding potentials between −80 and 40 mV (n= 7, P > 0.1, ANOVA).
Figure 4.
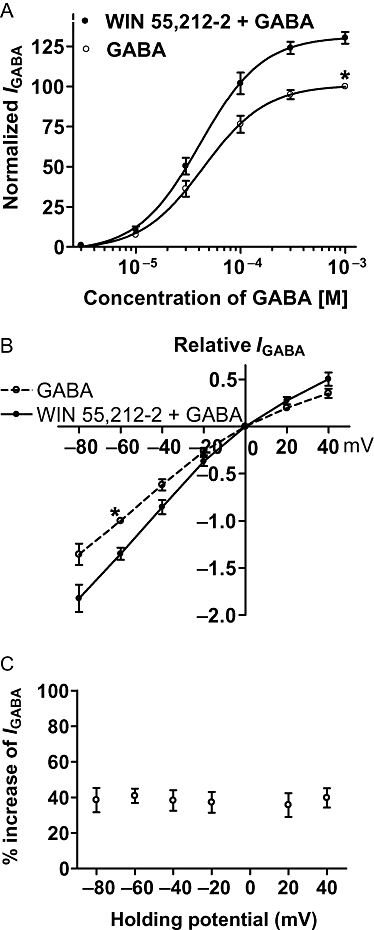
Concentration–response and current–voltage (I-V) relationships for GABA with or without the pre-application of WIN 55,212-2. (A) The concentration–response curves for GABA with or without WIN 55,212-2 (10−8 M) pre-application. Each point represents the mean ± SEM of 8–10 neurones. All GABA-induced currents were normalized to the response induced by 10−3 M GABA applied alone (marked with asterisk). (B) The I-V curves for GABA (3 × 10−5 M)-activated current with and without WIN 55,212-2 (10−8 M) pre-appplication. All current values from the same cell were normalized to the current response induced by 10−3 M GABA applied alone (marked with asterisk). Each point represents the mean ± SEM of 6–8 neurones. The experiment was carried out by using recording pipettes filled with CsCl containing internal solution. (C) Increase of IGABA (%) evoked by WIN 55,212-2 (10−8 M) at different holding potentials. Each point represents the mean ± SEM of 6–8 neurones.
Analysis of the intracellular signal transduction route involved in the potentiation of IGABA by WIN 55,212-2
As identified in Figure 1E,F, the WIN 55,212-2-induced potentiation of IGABA was blocked by the CB1 receptor antagonist AM 251. CB1 receptors belong to Gi/o protein-coupled receptor family, and the activation of the receptors leads to a cascade of events that decrease the intracellular cAMP level and inhibit the PKA system. To further explore whether the WIN 55,212-2-induced potentiation of IGABA is mediated through the G-protein-cAMP-PKA signalling pathway, GDP-β-S (a non-hydrolysable GDP analogue), cAMP (an activator of PKA) and H89 (a PKA inhibitor) were included in the recording pipette for intracellular dialysis using the repatch technique. As seen in Figure 5A, the IGABA relative to control (before repatch) was 98.2 ± 5.1% (n= 7) 30 min after repatch when the pipette was filled with normal internal solution, but this decreased to 68.4 ± 6.3% (n= 7, P < 0.05, post hoc Bonferroni's test) when the pipette was filled with internal solution containing 10−7 M cAMP, and increased to 141.9 ± 14.5% (n= 7, P < 0.05, post hoc Bonferroni's test) when the pipette was filled with internal solution containing H89 (10−8 M). However, the IGABA did not significantly change 30 min after intracellular dialysis with 2 × 10−7 M GDP-β-S (86.9 ± 9.0%, n= 7, P > 0.05, post hoc Bonferroni's test). In the control experiment with a pipette filled with normal internal solution, WIN 55,212-2-induced potentiation of IGABA was 35.2 ± 2.2% (n= 7). In contrast, the potentiation of IGABA induced by WIN 55,212-2 was almost completely abolished 30 min after intracellular dialysis with 10−7 M cAMP (4.1 ± 1.5%, n= 7, P < 0.01, post hoc Bonferroni's test), 10−8 M H89 (6.3 ± 2.5%, n= 7, P < 0.01, post hoc Bonferroni's test) and 2 × 10−7 M GDP-β-S (7.7 ± 1.8%, n= 7, P < 0.01, post hoc Bonferroni's test) (Figure 5B).
Figure 5.
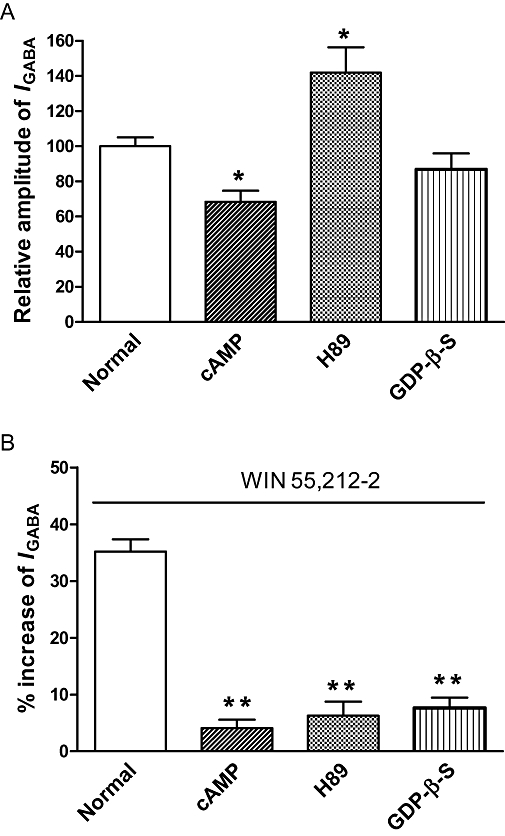
G-protein coupling and intracellular phosphorylation are shown to be involved in the potentiation of GABA-activated currents by WIN 55,212-2. (A) Intracellular dialysis of cAMP, an activator of PKA, significantly decreased IGABA. H89, a PKA inhibitor, significantly increased IGABA, while dialysis of guanosine 5=-O-(2-thiodiphosphate)(GDP)-β-S, a non-hydrolysable GDP analogue, had no effect on IGABA. *P < 0.05, post hoc Bonferroni's test, compared with normal internal solution, n= 7/each column. (B) Intracellular dialysis of cAMP, H89 and GDP-β-S abolished the enhancing effect of WIN 55,212-2 (10−8 M) on IGABA. The duration of the pre-application of WIN 55,512-2 was 30 s. **P < 0.01, post hoc Bonferroni's test, compared with normal internal solution, n= 7/each column.
Taken together, these results suggest that binding of WIN 55,212-2 to the CB1 receptor reduces the activity of adenylyl cyclase via the intermediacy of the Gi/oprotein. The resulting fall in the intracellular concentration of cAMP may decrease the stimulation of PKA that exerts an inhibitory effect upon the GABAA receptor. Hence, the potentiation by WIN 55,212-2 of IGABA may be due to a reduction of cAMP inhibition of GABA-activated currents.
Discussion
The most striking finding of this work is that WIN 55,212-2 potentiated GABA-activated currents via the activation of the CB1 receptor in primary sensory neurones.
Both CB1 and CB2 receptors belong to the G-protein coupled receptor family (Gi/o), which negatively couples to the effector enzyme, adenylyl cyclase, the later in turn catalyses ATP to produce cAMP and thus leads to less activation of PKA, a protein kinase that phosphorylates numerous intracellular proteins (Howlett, 2005). In addition, Gi/o protein can act on voltage-gated N and P/Q type calcium channels (VACC) negatively and on voltage-gated potassium channels positively (Morisset and Urban, 2001; Khasabova et al., 2004; Lovinger, 2008). In the present study, we found that the potentiation of GABA-activated currents by WIN 55,212-2 was blocked by AM 251, GDP-β-S, cAMP and H89. These results suggest that WIN 55,212-2 activates the signal pathway of CB1 receptors-Gi/o protein-cAMP in TG neurones.
It has been suggested that phosphorylation and dephosphorylation of the GABAA receptor-chloride channel complex are involved in the modulation of the GABA response (Chen et al., 1990). Biochemical studies have suggested that phosphorylation of the GABAA receptor by various protein kinases inhibits GABAA receptor function (Gyenes et al., 1994). In agreement with the present results, many studies have also shown that cyclic AMP-dependent protein kinase directly phosphorylates GABAA receptor and decreases GABA-activated currents in dorsal root ganglion and spinal neurones (Porter et al., 1990; Moss et al., 1992; Huang et al., 1996). We think that the function of the GABAA receptor may be tonically inhibited by cAMP-dependent protein kinase in primary sensory neurones. Thus, the GABAA receptor-mediated current increases when these kinases are inhibited by the activation of the CB1 receptor.
Although Agarwal et al. (2007) concluded that CB1 receptors expressed on the peripheral rather than the central terminals of nociceptors are mainly responsible for cannabinoid-induced analgesia, a number of studies have demonstrated that CB1 receptors expressed on central terminals of primary sensory neurones are involved in cannabinergic modulation on pain. Several reports have provided evidence for the presence of GABAA receptors in the primary sensory neurones of adult rats. For example, Toulméet al. (2007) and Kondo et al. (1994) have used immunocytochemistry to demonstrate the expression of GABAA receptors in the soma and central processes of nociceptive DRG and TG neurones in the adult rat. Our results revealed that the function of the GABAA receptor is potentiated by the activation of CB1 receptors in primary sensory neurones, indicating that the analgesic effect of cannabinoids may originate in the central terminals of primary sensory neurones via the activation of GABAA receptors expressed on the central terminals of the TG neurones.
GABA is an established inhibitory neurotransmitter that acts through the GABAA receptor. It opens the Cl- channel and is involved in primary afferent depolarization, an effect known as ‘pre-synaptic inhibition’ (Eccles, 1964). This action of GABA results in a decrease in the amount of neurotransmitter, including substance P and glutamate, released from primary afferent terminals. Under normal conditions, GABA exerts tonic modulation of nociceptive neurotransmission between primary afferents and second-order, spino-thalamic tract neurones (Malcangio and Bowery, 1996). In the present study, we used the cell body of TG neurones as a simple and accessible model to examine the characteristics of the membrane of central terminals. If WIN 55,212-2 enhances the GABA response at the central terminal of primary afferent neurones by activating the CB1 receptor, as it does at the soma membrane, potentiation of the ‘pre-synaptic inhibition’ would directly result in the inhibition of nociception in the spinal cord. Therefore, WIN 55,212-2 might be directly associated with the modulation of primary sensory information (including pain) at the central terminals of primary afferent neurones. This provides a reasonable explanation of cannabinoid-induced antinociception in the spinal dorsal horn.
Glossary
Abbreviations:
- IGABA
GABA-activated current
- I-V
current–voltage
- TG
trigeminal ganglion
Conflicts of interest
None.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahluwalia J, Urban L, Capogna M, Bevan S, Nagy I. Cannabinoid 1 receptors are expressed in nociceptive primary sensory neurons. Neuroscience. 2000;100:685–688. doi: 10.1016/s0306-4522(00)00389-4. [DOI] [PubMed] [Google Scholar]
- Bridges D, Rice AS, Egertová M, Elphick MR, Winter J, Michael GJ. Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience. 2003;119:803–812. doi: 10.1016/s0306-4522(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Chen QX, Stelzer A, Kay AR, Wong RK. GABAA receptor function is regulated by phosphorylation in acutely dissociated guinea-pig hippocampal neurons. J Physiol. 1990;420:207–221. doi: 10.1113/jphysiol.1990.sp017908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC. Presynaptic inhibition in the spinal cord. Prog Brain Res. 1964;12:65–91. doi: 10.1016/s0079-6123(08)60618-4. [DOI] [PubMed] [Google Scholar]
- Fox A, Kesingland A, Gentry C, McNair K, Patel S, Urban L, et al. The role of central and peripheral cannabinoid1 receptors in the antihyperalgesic activity of cannabinoids in a model of neuropathic pain. Pain. 2001;92:91–100. doi: 10.1016/s0304-3959(00)00474-7. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gyenes M, Wang Q, Gibbs TT, Farb DH. Phosphorylation factors control neurotransmitter and neuromodulator actions at the gamma-aminobutyric acid type A receptor. Mol Pharmacol. 1994;46:542–549. [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005;168:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- Huang CS, Ma JY, Marszalec W, Narahashi T. Effects of the nootropic drug nefiracetam on the GABAA receptor-channel complex in dorsal root ganglion neurons. Neuropharmacology. 1996;35:1251–1261. doi: 10.1016/s0028-3908(96)00074-3. [DOI] [PubMed] [Google Scholar]
- Johanek LM, Heitmiller DR, Turner M, Nader N, Hodges J, Simone DA. Cannabinoids attenuate capsaicin-evoked hyperalgesia through spinal and peripheral mechanisms. Pain. 2001;93:303–315. doi: 10.1016/S0304-3959(01)00336-0. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Harding-Rose C, Simone DA, Seybold VS. Differential effects of CB1 and opioid agonists on two populations of adult rat dorsal root ganglion neurons. J Neurosci. 2004;24:1744–1753. doi: 10.1523/JNEUROSCI.4298-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo E, Kiyama H, Araki T, Shida T, Ueda Y, Tohyama M. Coexpression of GABAA receptor gamma 1 and gamma 2 subunits in the rat trigeminal ganglion. Brain Res Mol Brain Res. 1994;21:363–367. doi: 10.1016/0169-328x(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Li ZW, Zhang J, Hu WP, Liu YW, Ai YX, Li CY. Potentiation by WIN55,212-2 of GABA-activated currents in rat trigeminal neurons. Neuroscience Meeting Abstract Nov 15–19, 2008, Washington DC.
- Liang Y-C, Huang C-C, Hsu K-S, Takahashi T. Cannabinoid-induced presynaptic inhibition at the primary afferent trigeminal synapse of juvenile rat brainstem slices. J Physiol. 2004;555:85–96. doi: 10.1113/jphysiol.2003.056986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008;184:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Bowery NG. GABA and its receptors in the spinal cord. Trends Pharmacol Sci. 1996;17:457–462. doi: 10.1016/s0165-6147(96)01013-9. [DOI] [PubMed] [Google Scholar]
- Martin BR, Mechoulam R, Razdan RK. Discovery and characterization of endogenous cannabinoids. Life Sci. 1999;65:573–595. doi: 10.1016/s0024-3205(99)00281-7. [DOI] [PubMed] [Google Scholar]
- Morisset V, Urban L. Cannabinoid-induced presynaptic inhibition of glutamatergic EPSCs in substantia gelatinosa neurons of the rat spinal cord. J Neurophysiol. 2001;86:40–48. doi: 10.1152/jn.2001.86.1.40. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG, Blackstone CD, Huganir RL. Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science. 1992;257:661–665. doi: 10.1126/science.1323140. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The endocannabinoid system: a drug discovery perspective. Curr Opin Investig Drugs. 2005;6:672–679. [PubMed] [Google Scholar]
- Porter NM, Twyman RE, Uhler MD, Macdonald RL. Cyclic AMP-dependent protein kinase decreases GABAA receptor current in mouse spinal neurons. Neuron. 1990;5:789–796. doi: 10.1016/0896-6273(90)90338-g. [DOI] [PubMed] [Google Scholar]
- Price TJ, Helesic G, Parghi D, Hargreaves KM, Flores CM. The neuronal distribution of cannabinoid receptor type 1 in the trigeminal ganglion of the rat. Neuroscience. 2003;120:155–162. doi: 10.1016/S0306-4522(03)00333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea K, Roche M, Finn DP. Supraspinal modulation of pain by cannabinoids: the role of GABA and glutamate. Br J Pharmacol. 2007;152:633–648. doi: 10.1038/sj.bjp.0707440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulmé E, Blais D, Léger C, Landry M, Garret M, Séguéla P, et al. An intracellular motif of P2X(3) receptors is required for functional cross-talk with GABA(A) receptors in nociceptive DRG neurons. J Neurochem. 2007;102:1357–1368. doi: 10.1111/j.1471-4159.2007.04640.x. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Walker JM, Huang SM. Cannabinoid anagesia. Pharmacol Ther. 2002;95:127–135. doi: 10.1016/s0163-7258(02)00252-8. [DOI] [PubMed] [Google Scholar]