

Abstract
Background and purpose:
The aim of this study was to investigate the effect of chronic treatment with antihypertensive and non-antihypertensive doses of zofenopril on cardiovascular changes in spontaneously hypertensive rats (SHR).
Experimental approach:
Male SHR were treated with 0.5 or 10 mg·kg−1 per day of zofenopril (Z0.5 and Z10) for 3 months. SHR and Wistar-Kyoto rats (WKY) receiving vehicle were used as controls. Systolic blood pressure was measured using the tail cuff method. Left ventricular weight/body weight ratio was calculated as cardiac hypertrophy index. Angiotensin converting enzyme (ACE) activity was determined in plasma and tissues by a fluorimetric method. Vascular reactivity was evaluated on aortic rings by acetylcholine and sodium nitroprusside relaxations. Effects on vascular structure were assessed by lumen diameter, wall thickness and medial cross-sectional area determination. Superoxide anion generation was quantified using lucigenin-amplified chemiluminescence in aorta.
Results:
Long-term daily administration of zofenopril (10 mg·kg−1) to SHR reduced blood pressure to WKY values, decreased cardiac hypertrophy, improved the acetylcholine-induced relaxant response and reversed the vascular remodelling. ACE inhibition and antioxidant activity were involved in these effects. 0.5 mg·kg−1 per day of zofenopril slightly modified blood pressure and the other effects were weaker.
Conclusions and implications:
Antihypertensive effects of chronic treatment with zofenopril were accompanied by recovery of endothelial function and improvement of cardiovascular structure. Low-dose zofenopril had little effect on blood pressure, with some benefits on cardiovascular structure and function. Inhibition of ACE and antioxidant activity were involved in these effects.
Keywords: hypertension, angiotensin converting enzyme, zofenopril, superoxide anion, vascular reactivity, remodelling
Introduction
Angiotensin converting enzyme (ACE) inhibitors are well-established drugs in the treatment of hypertension (Weir, 2007; Heran et al., 2008; Mancia et al., 2009) and protection of the cardiovascular system during congestive heart failure (Šimko et al., 2003). Although the pharmacological mechanisms of ACE inhibitors are not fully understood, various studies suggest that these agents may, besides inhibition of angiotensin II production, stimulate synthesis of prostaglandins and bradykinin, inhibit production of superoxide, scavenge free radicals, and increase expression of endothelial nitric oxide synthase (eNOS) (Buczko et al., 2006).
High blood pressure is the main risk factor for stroke, coronary heart disease and renal vascular disease. The renin–angiotensin system plays an important role in blood pressure regulation and fluid homeostasis. Within the enzyme cascade of this system, ACE generates angiotensin II from its inactive precursor, angiotensin I, by cleaving a dipeptide from its C-terminal (Erdös, 1976). Angiotensin II induces vasoconstriction, aldosterone release, production of reactive oxygen species and other physiological actions that act to raise the blood pressure. ACE also catalyses the degradation of bradykinin, a blood pressure lowering peptide in the kallikrein–kinin system (Bhoola et al., 1992). ACE is widely distributed, not only in the cardiovascular system, but also in other tissues. It has been localized in endothelial cells throughout the body, in the epithelial cells of various organs and in male germinal cells (Danser, 1996).
Angiotensin converting enzyme inhibitors are known to inhibit the conversion of angiotensin I to angiotensin II and to reduce blood pressure both clinically and in animal models of hypertension. ACE activity is easily detected in plasma, and the inhibition of ACE activity in plasma has been used as an indicator of ACE inhibition after treatment with ACE inhibitors. Zofenopril, a sulfhydryl ACE inhibitor derived from the amino acid proline, has been shown to have beneficial cardiovascular effects in patients with infarction or heart failure (Ambrosioni et al., 1995; Borghi et al., 1999) and anti-atherogenic activity (Napoli et al., 1999; de Nigris et al., 2001).
The endothelium plays a primary role in the modulation of vascular tone and structure. Several cardiovascular disease states, such as essential hypertension and congestive heart failure, are associated with impaired endothelium-dependent vasodilation (Kubo et al., 1991; Panza et al., 1995). In hypertension, endothelial dysfunction is believed to be associated with the production of reactive oxygen species. These react with nitric oxide (NO) in a setting of decreased antioxidant defences, culminating in attenuated endothelium-dependent vasodilation (Kerr et al., 1999; Lerman et al., 2001). The most characteristic pathophysiological feature of endothelial dysfunction is a diminished bioactivity of endothelium-derived NO due to reduced eNOS activity and/or increased metabolism through its interaction with superoxide anion (O2-) produced in the vascular wall by free-radical-generating enzymes, such as NADPH oxidase (Cai and Harrison, 2000).
The aim of this study was to investigate the effects of chronic treatment with zofenopril, on the prevention of hypertension, cardiac hypertrophy and vascular reactivity of aortic arteries and, if possible, to correlate those effects with its antioxidant properties.
Methods
Animals and experimental design
All animal care and experimental procedures were performed according to the European Union guidelines for the ethical care of animals. The study was conducted on 12-week-old male Wistar-Kyoto rats (WKY) (n= 7) and SHR (n= 36) that were purchased from Janvier (France) and maintained in the Animal Experimentation Service of Salamanca University. SHR were randomly divided into three groups: (i) SHR (n= 12); (ii) zofenopril 0.5 mg·kg−1·day−1 (Z0.5n= 12); and (iii) zofenopril 10 mg·kg−1·day−1 (Z10n= 12). Animals had free access to standard rat diet and tap water ad libitum. The drug was administered for 3 months in the drinking water and the concentration of zofenopril was adjusted weekly according to body weight and water intake. Untreated SHR and WKY rats received tap water during the time of study.
Experimental protocol
Body weight and blood pressure were determined periodically during the course of the experiment. Systolic blood pressure (SBP) was determined by the tail cuff method with a photoelectric sensor (Niprem 546, Cibertec S.A., Spain) using a MacLab software (AD Instruments, UK).
Animals were killed for further cardiovascular structural and functional analysis, immediately at the end of drug treatment. Rats were weighed and anaesthetized with sodium pentobarbital (60 mg·kg−1 i.p.). The left carotid artery was cannulated and heparinized physiological saline solution was infused. Rats were bled through the femoral artery. The kidney, heart and thoracic aorta were isolated and placed in Krebs solution of the following composition (in mM): NaCl, 118; KCl, 4.7; CaCl2, 2.5; KH2PO4, 1.2; MgSO4, 1.2; NaHCO3, 25; and glucose, 11. The pH of the solution after saturation with a 95% O2 and 5% CO2 mixture was 7.4.
Cardiac and renal hypertrophy
Hearts were removed and placed immediately in gassed Krebs solution at 37°C to wash out residual blood. Then, hearts were stopped in diastole by transferring them to another solution containing 150 mM KCl. Atria were removed and all the epicardial fat was scraped off. Right and left ventricles were separated from each other, regarding interventricular septum as an integral part of the left ventricle and this portion was weighed. The left ventricular hypertrophy index was calculated using the left ventricle weight/body weight ratio (LVW/BW). Right kidneys were isolated, dried and weighed to calculate the renal hypertrophy index as the kidney weight/body weight ratio (KW/BW).
Vascular functionality of thoracic aorta rings
The thoracic aorta was cleaned of adherent fat and rings 3 mm long were cut and placed between stainless-steel hooks for isometric tension recording in organ chambers. One of the hooks was fixed to the bath and the other connected to an isometric force transducer (UF1; Harvard Apparatus Inc., South Natick, MA, USA). The force was recorded on a PC computer using a Chart version 3.4 software and a PowerLab/800 data acquisition system (AD Instruments, Chalgrove, UK).
Rings were stretched to 2 g of tension and equilibrated for 1 h. After pre-contraction with phenylephrine (10−6 M) the presence of endothelium was verified by the ability of acetylcholine (ACh, 10−6 M) to induce relaxation. Concentration-response curves of aortic rings with endothelium to ACh (10−8–10−4 M) and sodium nitroprusside (SNP, 10−9–10−5 M) were performed with and without indomethacin (5 × 10−6 M; 30 min) and apocynin (10−3 M; 30 min).
To evaluate the formation of basal NO, the contraction induced by phenylephrine (10−6 M) was measured before and 30 min after aortic incubation with Nω-nitro-L-arginine methyl ester hydrochloride (L-NAME, 10−4 M), an inhibitor of endothelial NO synthase (eNOS).
In these experiments, the vasodilator responses to ACh and SNP are expressed as percentages of phenylephrine contraction.
Vascular histomorphometric study
We selected intrarenal arteries, with an external diameter between 25 and 50 µm, as resistance arteries and aorta as conductance arteries.
Kidney and aortic sections were stained with Sirius Red and examined under light microscopy with an attached video camera. Using an image analyser (Image J Software, http://rbs.info.nih.gov/ij), the internal and external perimeters of the media layer were measured and converted into internal and external radii (Ri and Re respectively) according to the formula: perimeter = 2ΠR, where 2 × Ri is the internal diameter (L) and Re– Ri is the medial thickness (Wm). Medial cross-sectional area (CSAm) was calculated as: CSAm=Π (Re2– Ri2).
Tissue preparation and measurement of ACE activity
Blood and tissue samples were collected for measuring ACE activity by a fluorimetric method adapted from Friedland and Silverstein (1976). Briefly, frozen samples of left ventricle and aorta were homogenized in 0.05 M potassium phosphate buffer (pH 7.5). The plasma and tissue samples were mixed with 12.5 nM of hippuryl-L-histidyl-L-leucine (H-H-L), and the mixture was incubated at 37°C for 10 min. After the reaction had been stopped by adding NaOH (0.28 M), o-phthalaldehyde 1% in methanol was added, the mixture was kept at room temperature for 10 min, and then HCl (3 M) was added. After that, the amount of liberated histidyl-L-leucine (H-L) was determined by measuring the fluorescent intensity of its adduct with o-phthalaldehyde (excitation at 364 nm and emission at 486 nm).
The protein contents of the plasma and tissue extracts were measured with Bradford assay (Bradford, 1976), using bovine serum albumin as a standard. ACE activity was expressed as nmol H-L formed per (mg protein) per min.
Detection of vascular O2− production
Production of O2- in intact aortic segments was quantified by lucigenin-enhanced chemiluminescence, as previously described by Ohara et al. (1993). Aortic rings from all experimental groups were incubated for 30 min at 37°C in HEPES-containing physiological salt solution (pH 7.4) of the following composition (in mM): NaCl, 119; HEPES, 20; KCl, 4.6; MgSO4, 1; Na2HPO4, 0.15; KH2PO4, 0.4; NaHCO3, 5; CaCl2, 1.2; and glucose, 5.5 and in presence of ammonium diethyldithiocarbamate (DDC, 0,5 M). Rings were then placed in tubes containing HEPES solution with lucigenin (5 × 10−6 M). Changes in O2- release were determined by measuring luminescence over 300 s, at 30 s intervals, in a luminometer (Lumat LB 9507, Berthold, Germany). Aortic production of O2- was stimulated by addition of NADPH (10−4 M). A cell-permeable, non-enzymatic, scavenger of O2-, tiron (4,5-dihydroxy-1,3-benzene disulphonic acid, 1 M), was added to quench the O2--dependent chemiluminiscence. Repeated measurements were taken again during 5 min. The difference between the initial set of readings and the readings after tiron was taken to calculate O2- production. O2- release is expressed as relative luminescence units (RLU) per min.
Statistical analysis
Data are expressed as mean ± SEM. Dose-response curves were analysed using the GraphPad Prism 4.0 software (GraphPad, San Diego, CA, USA) and adjusted to a logistic equation. Statistical calculations for significant differences were performed using Student's t-test in order to compare SHR and WKY or one-way anova when effect of treatment was analysed. Significance was accepted at P < 0.05.
Materials
Phenylephrine hydrochloride, acetylcholine chloride, sodium nitroprusside, L-NAME (Nω-nitro-L-arginine methyl ester hydrochloride), apocynin (4-hydroxy-3-methoxyacetophenone), indomethacin, DDC (ammonium diethyldithiocarbamate), tiron (4,5-dihydroxy-1,3-benzene disulphonic acid) and all the other compounds used for Krebs solution were obtained from Sigma-Aldrich (Química, S.A., Spain). Zofenopril was kindly supplied by the Menarini Laboratory (Menarini Ricerche SpA, Florence, Italy). Stock solutions of drugs were made up in ultrapure water and stored at −20°C, and appropriate dilutions were made on the day of the experiments. Drug and molecular target nomenclature follows Alexander et al. (2008).
Results
Systolic blood pressure
At the beginning of the study, SBP was higher in SHR (175 ± 2 mmHg) than in WKY rats (119 ± 3 mmHg, P < 0.05), and this difference persisted as time progressed. Treatment with zofenopril at 10 mg·kg−1 per day induced an acute and significant reduction in systolic blood pressure; this reached a maximum in the fourth week and persisted until the end of the study (134 ± 3 mmHg, P < 0.05). Blood pressure in SHR was maintained without increase during the treatment period with 0.5 mg·kg−1 per day of zofenopril (Figure 1).
Figure 1.
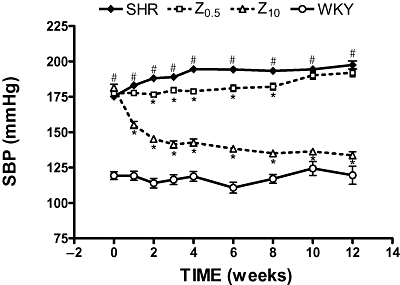
Effects of zofenopril on systolic blood pressure (SBP) during treatment. SHR, spontaneously hypertensive rats; WKY, Wistar-Kyoto rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day. *P < 0.05 versus SHR. #P < 0.05 versus WKY.
Cardiac and renal hypertrophy
At the beginning of the study, all 3-month-old SHR had similar body weights but this variable was, however, significantly higher in the WKY group (data not shown). Treatment with either dose of zofenopril did not modify the body weight in SHR rats.
The left ventricular hypertrophy index (LVW/BW) and renal hypertrophy index (KW/BW) in SHR were significantly greater than in WKY. These parameters were significantly reduced in zofenopril-treated SHR as compared with untreated SHR, without reaching the values seen in the WKY animals (Table 1).
Table 1.
Parameters of left ventricular and renal hypertrophy
| Parameter | SHR | Z0.5 | Z10 | WKY |
|---|---|---|---|---|
| BW (g) | 402 ± 3# | 414 ± 3 | 402 ± 6 | 538 ± 14 |
| LVW (mg) | 1092 ± 12# | 1080 ± 15 | 874 ± 15* | 941 ± 34 |
| LVW/BW | 2.82 ± 0.03# | 2.68 ± 0.04* | 2.22 ± 0.04* | 1.86 ± 0.03 |
| KW (mg) | 1241 ± 15# | 1221 ± 27 | 1213 ± 20 | 1421 ± 37 |
| KW/BW | 3.20 ± 0.05# | 3.06 ± 0.06* | 3.07 ± 0.05* | 2.81 ± 0.10 |
Values are means ± SEM.
P < 0.05 compared with SHR;
P < 0.05 compared with WKY.
SHR, spontaneously hypertensive rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day; WKY, Wistar-Kyoto rats; BW, body weight; LVW, left ventricular weight; KW, kidney weight.
Vascular functionality of thoracic aorta rings
Aortic rings from SHR and WKY rats presented similar responses to phenylephrine (10−6 M), with this concentration inducing around 80% of the maximum response. Aortas from untreated SHR rats showed decreased endothelium-dependent vasodilator responses to ACh in arteries stimulated by phenylephrine, compared with aortas from WKY rats (Emax= 51 ± 5% vs. 82 ± 4%, P < 0.05). Zofenopril produced a significant, dose-dependent, increase in the relaxation induced by ACh, compared with untreated SHR rats (Emax, Z0.5= 64 ± 3% and Z10= 82 ± 5%) (Figure 2A). The response to SNP was also improved by zofenopril treatment (Figure 2B).
Figure 2.
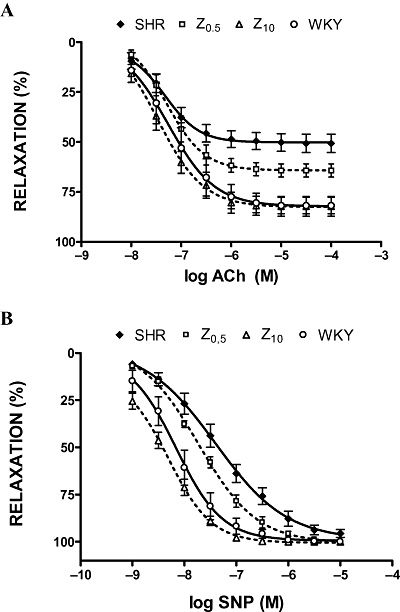
Acetylcholine (ACh, A) and sodium nitroprusside (SNP, B) induced relaxations of phenylephrine-preconstricted aortic rings. SHR, spontaneously hypertensive rats; WKY, Wistar-Kyoto rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day.
Exposure of aortic rings to indomethacin (5 × 10−6 M) significantly increased ACh-induced vasodilatation (in the untreated SHR and Z0.5 groups) (Figure 3A,B), without affecting rats treated with the high dose of zofenopril (Figure 3C). As shown in Figure 4A and B, concentration curves to SNP were similarly affected by pretreatment with indomethacin in untreated SHR and SHR treated with low-dose zofenopril, without affecting rats treated with the high dose (10 mg·kg−1 per day) of zofenopril (Figure 4C).
Figure 3.
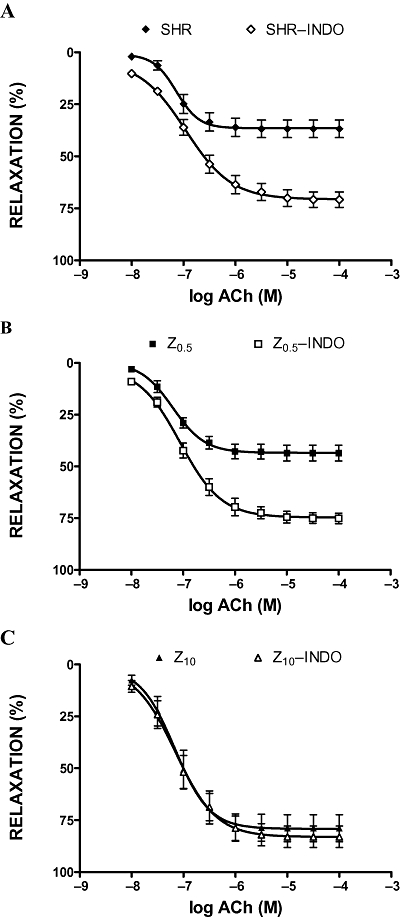
Acetylcholine (ACh) induced relaxations of phenylephrine-preconstricted aortic rings in the absence or presence of indomethacin (INDO, 5 × 10−6 M). SHR, spontaneously hypertensive rats (A); Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day (B); Z10, SHR treated with zofenopril 10 mg·kg−1 per day (C).
Figure 4.
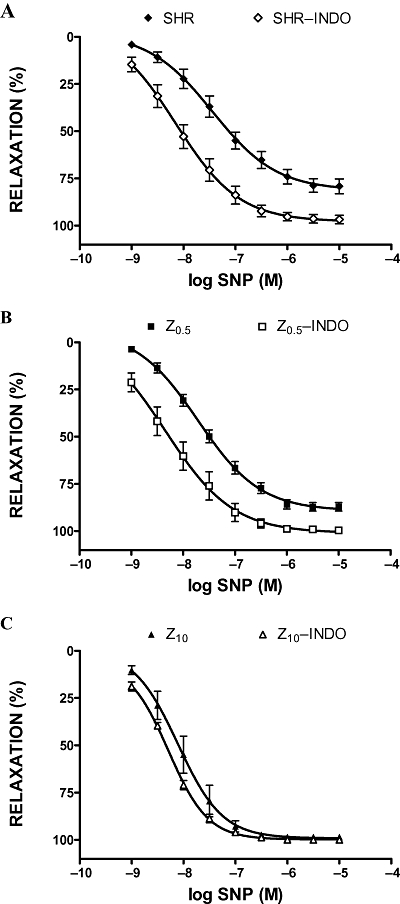
Sodium nitroprusside (SNP) induced relaxations of phenylephrine-preconstricted aortic rings in the absence or presence of indomethacin (INDO, 5 × 10−6 M). SHR, spontaneously hypertensive rats (A); Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day (B); Z10, SHR treated with zofenopril 10 mg·kg−1 per day (C).
Relaxation to ACh and SNP was less in the presence of apocynin in aortic rings from untreated SHR and SHR treated with low-dose zofenopril (Figures 5 and 6). Apocynin was ineffective in rings from SHR treated with the high dose of zofenopril (Figures 5C and 6C).
Figure 5.
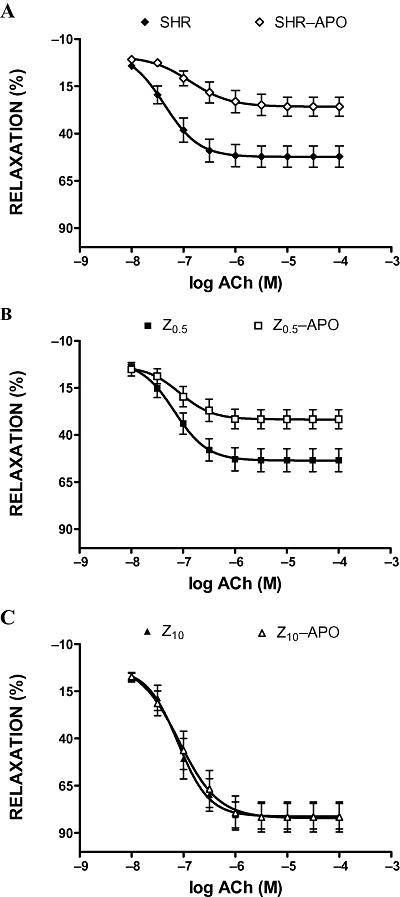
Acetylcholine (ACh) induced relaxations of phenylephrine-preconstricted aortic rings in the absence or presence of apocynin (APO, 10−3 M). SHR, spontaneously hypertensive rats (A); Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day (B); Z10, SHR treated with zofenopril 10 mg·kg−1 per day (C).
Figure 6.
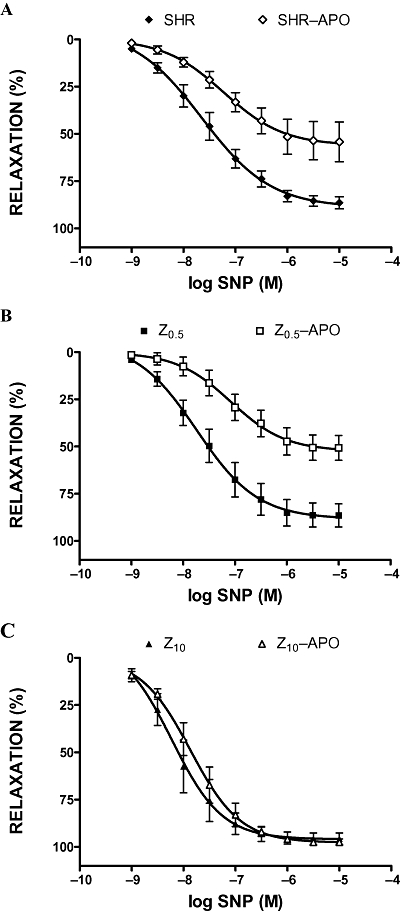
Sodium nitroprusside (SNP) induced relaxations of phenylephrine-preconstricted aortic rings in the absence or presence of apocynin (APO, 10−3 M). SHR, spontaneously hypertensive rats (A); Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day (B); Z10, SHR treated with zofenopril 10 mg·kg−1 per day (C).
Treatment of aortic rings for 30 min with L-NAME abolished ACh-induced relaxation in all experimental groups. After treatment with L-NAME, ACh evoked concentration-dependent contractions which were larger in rings from untreated SHR compared with the Z05 group, whereas it did not contract the rings from the Z10 group of SHR (Figure 7).
Figure 7.
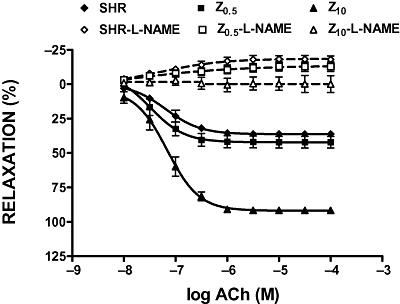
Acetylcholine (ACh) induced relaxations of phenylephrine-preconstricted aortic rings in the absence or presence of L-NAME (10−4 M). SHR, spontaneously hypertensive rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day.
The NOS inhibitor L-NAME induced a significantly higher contraction to phenylephrine in aortic rings from WKY rats than in rings from untreated SHR (Figure 8), indicating a reduced basal NO formation in SHR. Aortic rings from the SHR treated with the high dose of zofenopril showed values close to those from the WKY group.
Figure 8.
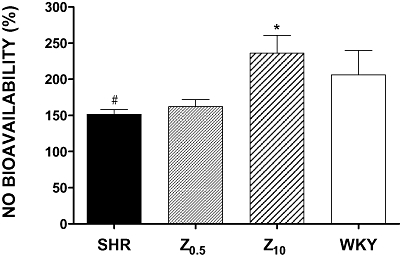
Ratio between contractions induced by phenylephrine before and after incubation of aortic rings with L-NAME (10−4 M). This ratio is shown as NO bioavailability (%) and contractions before L-NAME were set to 100%. NO, nitric oxide; SHR, spontaneously hypertensive rats; WKY, Wistar-Kyoto rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day. *P < 0.05 compared with SHR. #P < 0.05 compared with WKY.
Vascular histomorphometric study
A prolonged hypertensive state induces a range of structural modifications, depending of the vascular bed under study. In conductance arteries, hypertension leads to thickening of the arterial wall and increased lumen (internal diameter). This leads to greater values of the Wm/L ratio in SHR, with respect to the control normotensive rats. Zofenopril induced a dose-related reduction in aortic media thickness, lumen and medial cross-sectional area (Table 2).
Table 2.
Thoracic aortic wall geometry
| Parameter | SHR | Z0.5 | Z10 | WKY |
|---|---|---|---|---|
| L (µm) | 1702 ± 15 | 1583 ± 28* | 1530 ± 66* | 1646 ± 37 |
| Wm (µm) | 135.0 ± 2.5# | 116.7 ± 7.9* | 101.0 ± 3.7* | 105.5 ± 4.4 |
| Wm/L | 0.079 ± 0.002# | 0.074 ± 0.005 | 0.066 ± 0.003* | 0.064 ± 0.003 |
| CSAm (mm2) | 0.77 ± 0.02# | 0.62 ± 0.05* | 0.51 ± 0.03* | 0.58 ± 0.03 |
Values are means ± SEM.
P < 0.05 compared with SHR;
P < 0.05 compared with WKY.
SHR, spontaneously hypertensive rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day; WKY, Wistar-Kyoto rats; L, lumen; Wm, medial thickness; CSAm, medial cross-sectional area.
On small-sized intrarenal arteries of the SHR, the vascular changes consisted of luminal narrowing accompanied by increased arterial wall thickness compared with those of WKY. Both low and high doses (0.5 and 10 mg·kg−1 per day) of zofenopril increased the lumen but Wm only was decreased by the high dose of zofenopril (Table 3).
Table 3.
Wall geometry of small intrarenal resistance arteries
| Parameter | SHR | Z0.5 | Z10 | WKY |
|---|---|---|---|---|
| L (µm) | 11.8 ± 0.7# | 14.1 ± 1.0* | 14.5 ± 0.9* | 21.1 ± 2.6 |
| Wm (µm) | 11.6 ± 0.5# | 11.4 ± 0.5 | 10.1 ± 0.4* | 8.9 ± 0.5 |
| Wm/L | 1.06 ± 0.06# | 0.91 ± 0.07 | 0.77 ± 0.06* | 0.47 ± 0.05 |
| CSAm (µm2) | 793 ± 60 | 893 ± 67 | 761 ± 59 | 664 ± 98 |
Values are means ± SEM.
P < 0.05 compared with SHR;
P < 0.05 compared with WKY.
SHR, spontaneously hypertensive rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day; WKY, Wistar-Kyoto rats; L, lumen; Wm, medial thickness; CSAm, medial cross-sectional area.
ACE activity in plasma and tissues
The effects of zofenopril on ACE activity are shown in Table 4. ACE activity in plasma and tissues of SHR was significantly increased with respect to the WKY group. Only treatment with 10 mg·kg−1 per day of zofenopril inhibited activity (by 83%) of plasma ACE. Treatment with zofenopril also produced a dose-dependent lowering of ACE activity in the aorta and in the heart to levels well below those in the untreated SHR.
Table 4.
Effects of zofenopril on ACE activity in plasma, aorta and heart
| ACE activity [nmol HL·(mg protein)−1·min−1] | SHR | Z0.5 | Z10 | WKY |
|---|---|---|---|---|
| PLASMA | 2.44 ± 0.25# | 1.86 ± 0.12 | 0.41 ± 0.09* | 1.09 ± 0.23 |
| AORTA | 5.05 ± 0.58# | 2.49 ± 0.35* | 1.45 ± 0.42* | 1.97 ± 0.64 |
| HEART | 0.052 ± 0.01# | 0.022 ± 0.01* | 0.015 ± 0.00* | 0.012 ± 0.00 |
Values are means ± SEM.
P < 0.05 compared with SHR;
P < 0.05 compared with WKY.
SHR, spontaneously hypertensive rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day; WKY, Wistar-Kyoto rats.
Detection of vascular O2− production
Chronic treatment of SHR with 10 mg·kg−1 per day of zofenopril induced a significant decrease of basal superoxide anion production by aortic rings, relative to values from untreated SHR (Figure 9).
Figure 9.
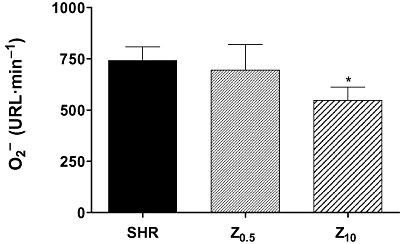
Superoxide anion (O2-) release quantified by lucigenin-enhanced chemiluminescence, from intact aortic segments. SHR, spontaneously hypertensive rats; Z0.5, SHR treated with zofenopril 0.5 mg·kg−1 per day; Z10, SHR treated with zofenopril 10 mg·kg−1 per day. *P < 0.05 compared with SHR.
Incubation of aortic rings with 10−4 M NADPH enhanced O2- production in all experimental groups. Chronic treatment with zofenopril did not modify this NADPH-stimulated O2- production.
Discussion
In this study, we have treated SHR for 3 months, with two doses (0.5 and 10 mg·kg−1 per day) of zofenopril, an ACE inhibitor. With the high dose, we observed a marked decrease of systolic blood pressure (SBP) accompanied by a recovery of endothelial function and improvement of cardiovascular morphology. Although the effect of low-dose zofenopril on SBP was minimal, this dose did provide clearer benefits for cardiovascular structure and function.
Treatment of SHR with 10 mg·kg−1 of zofenopril daily gradually reduced the SBP. Most of the reduction occurred over the first 4 weeks of treatment and then this reduction was maintained over the following 8 weeks. There was a time-dependent increase of SBP in untreated SHR which was suppressed by the low dose of zofenopril for the two first months and in the last month, this low-dose treatment became ineffective in controlling the hypertension. While some authors report, in the chronic stage of hypertension, an increased ACE activity in vessels but a normal activity in plasma (Okunishi et al., 1991; Shiota et al., 1992) others have found more activity in WKY than in SHR (Webb et al., 1997). We observed significant differences between normotensive and hypertensive animals and a good correlation between plasma ACE activity and SBP in treated animals. Nevertheless, both doses inhibited ACE activity in aorta and heart.
In hypertension, the increase in cardiac mass is essentially considered to be an adaptation of the heart in response to this pathological situation, in which the myocardium must work harder and for a longer time than normal to supply the organism's requirements and to decrease the tension in the walls of the ventricles. However, this adaptive process cannot compensate this pathology indefinitely, and the ventricles acquire a lower degree of compliance and cardiac output decreases (Agabiti-Rosei et al., 2007). Although ACE inhibitors have been reported to regress left ventricular hypertrophy (LVH) in hypertensive humans and various experimental models, the mechanisms underlying this process have not yet been fully elucidated. It is known that ACE activity in the heart may be an important factor in the development of cardiac hypertrophy (Raasch et al., 2002). Consistent with that finding, the hearts of SHR, in our study, showed higher enzyme activity and hypertrophy than hearts from WKY rats and the antihypertensive effect of zofenopril was accompanied by a greater reduction in the left ventricular hypertrophy index and cardiac ACE activity. On the other hand, a decrease in cardiac hypertrophy was also observed with low-dose zofenopril where treatment only delayed the blood pressure increase observed in the untreated SHR group. Our results are in agreement with those of Linz et al. (1995) who have reported that ramipril, at a dose that did not decrease blood pressure, reversed LVH in aortic-banded rats. Moreover, Mori et al. (1995) have observed that short-term treatment with lisinopril, 3 mg·kg−1 per day for 2 weeks, inhibited the progression of hypertension and suppressed the development of LVH in SHR, whereas 0.5 mg·kg−1·day−1 of lisinopril suppressed the development of LVH without reducing blood pressure. In order to clarify this point, the correlations between tissue ACE activities, blood pressure and cardiac hypertrophy were analysed by Takai et al. (2004), using several ACE inhibitors. The authors established a significant correlation between LVH and ACE activity in the heart but not between plasma ACE activity and SBP. They concluded that the inhibition of ACE activity in vascular tissues may be the most important mechanism for the antihypertensive effects of ACE inhibitors, and inhibition of ACE activity in the heart may contribute to the suppression of cardiac hypertrophy.
The potential of sulphydryl angiotensin-converting enzyme inhibitors in scavenging free radical oxygen species has been proposed as a contributing factor to the cardioprotection exerted by this class of compounds (Gagnon et al., 2004). Zofenopril has antioxidant properties, acting as a scavenger of various reactive oxygen species in vitro (Chopra et al., 1992; Cominacini et al., 2002; Evangelista and Manzini, 2005) as well as in vivo (Buikema et al., 2000; de Nigris et al., 2001; Sacco et al., 2001). These properties, together with its high lipophilicity, could provide an enhanced penetration into cardiac tissue (Subissi et al., 1999) and be involved in its beneficial effect in patients with myocardial infarction (Ambrosioni et al., 1995) or chronic heart failure (Borghi et al., 1996). Recently, experimental studies in SHR (Liu et al., 2007) and in essential hypertensive patients (Pasini et al., 2007) indicate that the presence of the sulphydryl group may confer properties other than ACE inhibition, such as improvement of endothelial dysfunction.
Hypertrophic remodelling of the vascular wall may result from hyperplasia, hypertrophy, architectural rearrangement of existing cells, increased deposition of fibrillar or nonfibrillar intercellular matrix, or from a combination of these events (Schiffrin and Hayoz, 1997). According to Mulvany et al. (1996), the most common change found in large arteries in hypertension is an ‘outward hypertrophic remodelling’, increased lumen diameter and increased media cross-sectional area with increased wall thickness. In hypertensive large arteries, these changes appear to be the consequence of cellular hypertrophy and redisposition of the wall mass around a larger internal diameter. Histological examination of the vessels from our SHR controls revealed medial thickening and a cross sectional area larger than in WKY. According to other authors (Marque et al., 1999), the lumen was increased as compared with WKY but without significant differences. Three months of treatment with zofenopril produced a marked improvement in all parameters. We observed a complete recovery of remodelling when the treatment achieved SBP values close to the normotensive animals.
Vascular remodelling of the small resistance arteries can involve a reduction in internal lumen (eutrophic remodelling) or a lumen increase (outward remodelling) (Izzard et al., 2005). We also examined small intrarrenal arteries of SHR and WKY and, our data confirmed an eutrophic remodelling which was partially reversed with the high dose (10 mg·kg−1 per day) of zofenopril. Also zofenopril at the low dose increased the lumen to a similar extent as the antihypertensive dose. Our results agree with those of other authors that ACE inhibitors are effective in controlling and even reversing vascular remodelling, independently of their blood pressure reducing effect (Galderisi and de Divitiis, 2008).
Another objective of our study was to investigate whether long-term treatment with zofenopril might improve the endothelial function of conductance arteries. It is known that structural and/or functional alterations of the vascular endothelium occur in many pathological conditions, including hypertension (Lüscher and Vanhoutte, 1986). Regression of these alterations is an important target of antihypertensive therapy. It has been proposed that impaired relaxation to ACh in the large conductance arteries of SHR may result from decreased NO synthesis (Hayakawa et al., 1993; Panza et al., 1995) and perhaps from an increase in superoxide anions or other contracting factors, released from a dysfunctional endothelium.
Increased vasodilator capacity to ACh after zofenopril treatment of SHR was observed in our study and is consistent with the findings of Buikema et al. (2000), who reported in rats with heart failure following myocardial infarction, an improvement of endothelial dysfunction through increased activity of NO released from the endothelium into the vessel wall. In our experiments, indomethacin enhanced ACh-induced relaxations and this effect was greater in untreated SHR than in the Z0.5 group, whereas this enhancement was not observed in the Z10 group. The same pattern of effect was observed with SNP. In different models of hypertension, an increased level of vascular basal cyclooxygenase-2 (COX-2) protein expression and prostanoid production from COX-2 (Garcia-Cohen et al., 2000; Adeagbo et al., 2005; Alvarez et al., 2005) has been reported. Our results with indomethacin suggest that zofenopril could modify the expression or activity of this enzyme, although this point should be corroborated by further studies.
The involvement of reactive oxygen species in ACh-induced endothelium-dependent contraction in the SHR aorta has been noted earlier (Yang et al., 2002; 2003; Tang and Vanhoutte, 2009). These authors indicate that superoxide anions or secondary reactive oxygen species, such as hydrogen peroxide and hydroxyl radicals, could activate COX-1 in smooth muscle and produce prostanoids that in turn stimulate thromboxane (TP) receptors. We observed that the contractile response to ACh in aortic rings incubated with L-NAME was greater in untreated SHR than in the low-dose zofenopril (Z0.5) group and was absent in the high-dose zofenopril (Z10) group. This effect of zofenopril could be mediated by decreasing COX expression or superoxide production.
Apocynin has been used in many laboratories as a potent and selective inhibitor of the O2- anion-generating NADPH oxidase of activated neutrophils (Van den Worm et al., 2001). In order to achieve a complete inhibition of NADPH oxidase, apocynin has been used under a wide variety of experimental conditions. In our experiments, it was surprising that there was less relaxation to ACh and SNP in presence of apocynin in aortic rings from untreated SHR and the low-dose Z0.5 groups, but not in rings from treated rats with 10 mg·kg−1 per day. Earlier work has suggested that high concentrations of apocynin can induce superoxide anion generation in a mouse glial cell line (Riganti et al., 2006) and in endothelial cells (Vejrazka et al., 2005). We used 1 mM of apocynin and this concentration may exert a pro-oxidant effect and thus cause the impairment of ACh response. In our opinion, the higher dose (10 mg·kg−1 per day) of zofenopril prevented the apocynin pro-oxidant effect, as this treatment decreased the O2- generation measured by chemiluminescence in aortic rings.
In conclusion, both antihypertensive and non-antihypertensive doses of zofenopril produced a reduction of heart hypertrophy and vascular remodelling as well as an improvement of endothelial function, although these effects were more marked when zofenopril was used at an antihypertensive dose. Inhibition of ACE and antioxidant activity were involved in these effects of zofenopril.
Acknowledgments
We thank Cristina Novoa of Departamento de Medicina y Cirugía Animal of Universidad Complutense de Madrid for the excellent technical assistance in histological study. This work has been supported by a collaborative project (art 83, LOU) between the University of Salamanca and Menarini IFR. M Gómez-Roso was supported by a FPI fellowship from Ministerio de Educación y Ciencia, Spain.
Glossary
Abbreviations:
- ACE
angiotensin converting enzyme
- BW
body weight
- COX
cyclooxygenase
- CSAm
medial cross sectional area
- DDC
ammonium diethyldithiocarbamate
- HHL
Hippuryl-L-Histidyl-L-Leucine
- HL
Histidyl-L-Leucine
- KH
kidney hypertrophy
- KW
kidney weight
- L
lumen
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- LVH
left ventricular hypertrophy
- LVW
left ventricle weight
- NOS
nitric oxide synthase
- O2-
superoxide anion
- Re
external radii
- Ri
internal radii
- RLU
relative luminescence units
- SBP
systolic blood pressure
- SHR
spontaneously hypertensive rat
- SNP
sodium nitroprusside
- WKY
Wistar Kyoto rat
- Wm
medial wall thickness
- Z0.5 and Z10
SHR treated with 0.5 or 10 mg·kg−1 per day of zofenopril
Conflict of interest
The authors have declared that no conflict of interest exists.
References
- Adeagbo AS, Zhang X, Patel D, Joshua IG, Wang Y, Sun X, et al. Cyclo-oxygenase-2, endothelium and aortic reactivity during deoxycorticosterone acetate salt-induced hypertension. J Hypertens. 2005;23:1025–1036. doi: 10.1097/01.hjh.0000166844.42227.5c. [DOI] [PubMed] [Google Scholar]
- Agabiti-Rosei E, Muiesan ML, Salvetti M. New approaches to the assessment of left ventricular hypertrophy. Ther Adv Cardiovasc Dis. 2007;1:119–128. doi: 10.1177/1753944707086350. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Y, Briones AM, Balfagón G, Alonso MJ, Salaices M. Hypertension increases the participation of vasoconstrictor prostanoids from cyclooxygenase-2 in phenylephrine responses. J Hypertens. 2005;23:767–777. doi: 10.1097/01.hjh.0000163145.12707.63. [DOI] [PubMed] [Google Scholar]
- Ambrosioni E, Borghi C, Magnani B. The effect of the angiotensin-converting-enzyme inhibitor zofenopril on mortality and morbidity after anterior myocardial infarction. The Survival of Myocardial Infarction Long-Term Evaluation (SMILE) Study Investigators. N Engl J Med. 1995;332:80–85. doi: 10.1056/NEJM199501123320203. [DOI] [PubMed] [Google Scholar]
- Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- Borghi C, Ambrosioni E, Magnani B. Effects of the early administration of zofenopril on onset and progression of congestive heart failure in patients with anterior wall acute myocardial infarction. The SMILE Study Investigators. Survival of Myocardial Infarction Long-term Evaluation. Am J Cardiol. 1996;78:317–322. doi: 10.1016/s0002-9149(96)00285-8. [DOI] [PubMed] [Google Scholar]
- Borghi C, Bacchelli S, Esposti DD, Bignamini A, Magnani B, Ambrosioni E. Effects of the administration of an angiotensin-converting enzyme inhibitor during the acute phase of myocardial infarction in patients with arterial hypertension. SMILE Study Investigators. Survival of Myocardial Infarction Long-term Evaluation. Am J Hypertens. 1999;12:665–672. doi: 10.1016/s0895-7061(99)00042-4. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye-binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Buczko W, Kramkowski K, Mogielnicki A. Are the endothelial mechanisms of ACE-Is already established? Pharmacol Rep. 2006;58:126–131. [PubMed] [Google Scholar]
- Buikema H, Monnink SH, Tio RA, Crijns HJ, de Zeeuw D, van Gilst WH. Comparison of Zofenopril and lisinopril to study the role of the sulfhydryl-group in improvement of endothelial dysfunction with ACE-inhibitors in experimental heart failure. Br J Pharmacol. 2000;130:1999–2007. doi: 10.1038/sj.bjp.0703498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Chopra M, Beswick H, Clapperton M, Dargie HJ, Smith WE, McMurray J. Antioxidant effects of angiotensin-converting enzyme (ACE) inhibitors: free radical and oxidant scavenging are sulfhydryl dependent, but lipid peroxidation is inhibited by both sulfhydryl- and nonsulfhydryl-containing ACE inhibitors. J Cardiovasc Pharmacol. 1992;19:330–340. doi: 10.1097/00005344-199203000-00005. [DOI] [PubMed] [Google Scholar]
- Cominacini L, Pasini A, Garbin U, Evangelista S, Crea AE, Tagliacozzi D, et al. Zofenopril inhibits the expression of adhesion molecules on endothelial cells by reducing reactive oxygen species. Am J Hypertens. 2002;15:891–895. doi: 10.1016/s0895-7061(02)02995-3. [DOI] [PubMed] [Google Scholar]
- Danser AH. Local renin-angiotensin systems. Mol Cel Biochem. 1996;157:211–216. doi: 10.1007/BF00227900. [DOI] [PubMed] [Google Scholar]
- Erdös EG. Conversion of angiotensin I to angiotensin II. Am J Med. 1976;60:749–759. doi: 10.1016/0002-9343(76)90889-5. [DOI] [PubMed] [Google Scholar]
- Evangelista S, Manzini S. Antioxidant and cardioprotective properties of the sulphydryl angiotensin-converting enzyme inhibitor zofenopril. J Int Med Res. 2005;33:42–54. doi: 10.1177/147323000503300103. [DOI] [PubMed] [Google Scholar]
- Friedland J, Silverstein M. A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am J Clin Pathol. 1976;66:416–424. doi: 10.1093/ajcp/66.2.416. [DOI] [PubMed] [Google Scholar]
- Gagnon C, Legault F, Geraldes P, Tanguay JF, Lambert C. Diverse effects of Ace inhibitors and angiotensin II receptor antagonists on prevention of cardiac hypertrophy and collagen distribution in spontaneously hypertensive rats. Int J Cardiol. 2004;97:373–381. doi: 10.1016/j.ijcard.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Galderisi M, de Divitiis O. Risk factor-induced cardiovascular remodelling and the effects of angiotensin-converting enzyme inhibitors. J Cardiovasc Pharmacol. 2008;51:523–531. doi: 10.1097/FJC.0b013e31817751a7. [DOI] [PubMed] [Google Scholar]
- Garcia-Cohen EC, Marin J, Diez-Picazo LD, Baena AB, Salaices M, Rodriguez-Martinez MA. Oxidative stress induced by tert-butyl hydroperoxide causes vasoconstriction in the aorta from hypertensive and aged rats: role of cyclooxygenase-2 isoform. J Pharmacol Exp Ther. 2000;293:75–81. [PubMed] [Google Scholar]
- Hayakawa H, Hirata Y, Suzuki E, Sugimoto T, Matsuoka H, Kikuchi K, et al. Mechanisms for altered endothelium-dependent vasorelaxation in isolated kidneys from experimental hypertensive rats. Am J Physiol. 1993;264:H1535–H1541. doi: 10.1152/ajpheart.1993.264.5.H1535. [DOI] [PubMed] [Google Scholar]
- Heran BS, Wong MM, Heran IK, Wright JM. Blood pressure lowering efficacy of angiotensin converting enzyme (ACE) inhibitors for primary hypertension. Cochrane Database Syst Rev. 2008;4:1–358. doi: 10.1002/14651858.CD003823.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzard AS, Rizzoni D, Agabiti-Rosei E, Heagerty AM. Small artery structure and hypertension: adaptive changes and target organ damage. J Hypertens. 2005;23:247–250. doi: 10.1097/00004872-200502000-00002. [DOI] [PubMed] [Google Scholar]
- Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. Superoxide anion production is increased in a model of genetic hypertension: role of the endothelium. Hypertension. 1999;33:1353–1358. doi: 10.1161/01.hyp.33.6.1353. [DOI] [PubMed] [Google Scholar]
- Kubo SH, Rector TS, Bank AJ, Williams RE, Heifetz SM. Endothelium-dependent vasodilation is attenuated in patients with heart failure. Circulation. 1991;84:1589–1596. doi: 10.1161/01.cir.84.4.1589. [DOI] [PubMed] [Google Scholar]
- Lerman LO, Nath KA, Rodriguez-Porcel M, Krier JD, Schwartz RS, Napoli C, et al. Increased oxidative stress in experimental renovascular hypertension. Hypertension. 2001;37:541–546. doi: 10.1161/01.hyp.37.2.541. [DOI] [PubMed] [Google Scholar]
- Linz W, Wiemer G, Schaper J, Zimmermann R, Nagasawa K, Gohlke P, et al. Angiotensin converting enzyme inhibitors, left ventricular hypertrophy and fibrosis. Mol Cell Biochem. 1995;147:89–97. doi: 10.1007/BF00944788. [DOI] [PubMed] [Google Scholar]
- Liu YH, You Y, Song T, Wu SJ, Liu LY. Impairment of endothelium-dependent relaxation of rat aortas by homocysteine thiolactone and attenuation by captopril. J Cardiovasc Pharmacol. 2007;50:155–161. doi: 10.1097/FJC.0b013e31805c9410. [DOI] [PubMed] [Google Scholar]
- Lüscher TF, Vanhoutte PM. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension. 1986;8:344–348. doi: 10.1161/01.hyp.8.4.344. [DOI] [PubMed] [Google Scholar]
- Mancia G, Giannattasio C, Seravalle G, Quarti-Trevano F, Grassi G. Protective effects of renin-angiotensin blockade beyond blood pressure control. J Hum Hypertens. 2009;23:570–577. doi: 10.1038/jhh.2008.171. [DOI] [PubMed] [Google Scholar]
- Marque V, Kieffer P, Atkinson J, Lartaud-Idjouadiene I. Elastic properties and composition of the aortic wall in old spontaneously hypertensive rats. Hypertension. 1999;34:415–422. doi: 10.1161/01.hyp.34.3.415. [DOI] [PubMed] [Google Scholar]
- Mori T, Nishimura H, Ueyama M, Kubota J, Kawamura K. Comparable effects of angiotensin II and converting enzyme blockade on hemodynamics and cardiac hypertrophy in spontaneously hypertensive rats. Jpn Circ J. 1995;59:624–630. doi: 10.1253/jcj.59.624. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Baumbach GL, Aalkjaer C, Heagerty AM, Korsgaard N, Schiffrin EL, et al. Vascular remodelling. Hypertension. 1996;28:505–506. [PubMed] [Google Scholar]
- Napoli C, Cicala C, D'Armiento FP, Roviezzo F, Somma P, de Nigris F, et al. Beneficial effects of ACE-inhibition with zofenopril on plaque formation and low-density lipoprotein oxidation in watanabe heritable hyperlipidemic rabbits. Gen Pharmacol. 1999;33:467–477. doi: 10.1016/s0306-3623(99)00043-9. [DOI] [PubMed] [Google Scholar]
- de Nigris F, D'Armiento FP, Somma P, Casini A, Andreini I, Sarlo F, et al. Chronic treatment with sulfhydryl angiotensin-converting enzyme inhibitors reduce susceptibility of plasma LDL to in vitro oxidation, formation of oxidation-specific epitopes in the arterial wall, and atherogenesis in apolipoprotein E knockout mice. Int J Cardiol. 2001;81:107–115. doi: 10.1016/s0167-5273(01)00542-3. [DOI] [PubMed] [Google Scholar]
- Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okunishi H, Kawamoto T, Kurobe Y, Oka Y, Ishii K, Tanaka T, et al. Pathogenetic role of vascular angiotensin-converting enzyme in the spontaneously hypertensive rat. Clin Exp Pharmacol Physiol. 1991;18:649–659. doi: 10.1111/j.1440-1681.1991.tb01639.x. [DOI] [PubMed] [Google Scholar]
- Panza JA, García CE, Kilcoyne CM, Quyyumi AA, Cannon RO., 3rd Impaired endothelium-dependent vasodilation in patients with essential hypertension. Evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation. 1995;91:1732–1738. doi: 10.1161/01.cir.91.6.1732. [DOI] [PubMed] [Google Scholar]
- Pasini AF, Garbin U, Nava MC, Stranieri C, Pellegrini M, Boccioletti V, et al. Effect of sulfhydryl and non-sulfhydryl angiotensin-converting enzyme inhibitors on endothelial function in essential hypertensive patients. Am J Hypertens. 2007;20:443–450. doi: 10.1016/j.amjhyper.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Raasch W, Bartels T, Schwartz C, Häuser W, Rütten H, Dominiak P. Regression of ventricular and vascular hypertrophy: are there differences between structurally different angiotensin-converting enzyme inhibitors? J Hypertens. 2002;20:2495–2504. doi: 10.1097/01.hjh.0000042885.24999.e9. [DOI] [PubMed] [Google Scholar]
- Riganti C, Costamagna C, Bosia A, Ghigo D. The NADPH oxidase inhibitor apocynin (acetovanillone) induces oxidative stress. Toxicol Appl Pharmacol. 2006;212:179–187. doi: 10.1016/j.taap.2005.07.011. [DOI] [PubMed] [Google Scholar]
- Sacco G, Bigioni M, Evangelista S, Goso C, Manzini S, Maggi CA. Cardioprotective effects of zofenopril, a new angiotensin-converting enzyme inhibitor, on doxorubicin- induced cardiotoxicity in the rat. Eur J Pharmacol. 2001;414:71–78. doi: 10.1016/s0014-2999(01)00782-8. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL, Hayoz D. How to assess vascular remodelling in small and medium-sized muscular arteries in humans. J Hypertens. 1997;15:571–584. doi: 10.1097/00004872-199715060-00002. [DOI] [PubMed] [Google Scholar]
- Shiota N, Miyazaki M, Okunishi H. Increase of angiotensin converting enzyme gene expression in the hypertensive aorta. Hypertension. 1992;20:168–174. doi: 10.1161/01.hyp.20.2.168. [DOI] [PubMed] [Google Scholar]
- Šimko F, Šimko J, Fábryová M. ACE-inhibition and angiotensin II receptor blockers in chronic heart failure: pathophysiological consideration of the unresolved battle. Cardiovasc Drugs Ther. 2003;17:287–290. doi: 10.1023/a:1026215712983. [DOI] [PubMed] [Google Scholar]
- Subissi A, Evangelista S, Giachetti A. Preclinical profile of Zofenopril: An angiotensin converting enzyme inhibitor with peculiar cardioprotective properties. Cardiovasc Drug Rev. 1999;17:115–133. [Google Scholar]
- Takai S, Jin D, Sakaguchi M, Miyazaki M. Significant target organs for hypertension and cardiac hypertrophy by angiotensin-converting enzyme inhibitors. Hypertens Res. 2004;27:213–219. doi: 10.1291/hypres.27.213. [DOI] [PubMed] [Google Scholar]
- Tang EH, Vanhoutte PM. Prostanoids and reactive oxygen species: team players in endothelium-dependent contractions. Pharmacol Ther. 2009;122:140–149. doi: 10.1016/j.pharmthera.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Van den Worm E, Beukelman CJ, Van den Berg AJ, Kroes BH, Labadie RP, Van Dijk H. Effects of methoxylation of apocynin and analogs on the inhibition of reactive oxygen species production by stimulated human neutrophils. Eur J Pharmacol. 2001;433:225–230. doi: 10.1016/s0014-2999(01)01516-3. [DOI] [PubMed] [Google Scholar]
- Vejrazka M, Mícek R, Stípek S. Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim Biophys Acta. 2005;1722:143–147. doi: 10.1016/j.bbagen.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Webb RL, Abramson ML, Beil ME, Odorico LM, Chatelain RE. Effects of the novel dual inhibitor of neutral endopeptidase and angiotensin-converting enzyme, CGS 30440, on blood pressure and cardiac hypertrophy in spontaneously hypertensive rats. J Cardiovasc Pharmacol. 1997;30:632–642. doi: 10.1097/00005344-199711000-00014. [DOI] [PubMed] [Google Scholar]
- Weir MR. Effects of renin-angiotensin system inhibition on end-organ protection: can we do better? Clin Ther. 2007;29:1803–1824. doi: 10.1016/j.clinthera.2007.09.019. [DOI] [PubMed] [Google Scholar]
- Yang D, Félétou M, Boulanger CM, Wu HF, Levens N, Zhang JN, et al. Oxygen-derived free radicals mediate endothelium-dependent contractions to acetylcholine in aortas from spontaneously hypertensive rats. Br J Pharmacol. 2002;136:104–110. doi: 10.1038/sj.bjp.0704669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Félétou M, Levens N, Zhang JN, Vanhoutte PM. A diffusible substance(s) mediates endothelium-dependent contractions in the aorta of SHR. Hypertension. 2003;41:143–148. doi: 10.1161/01.hyp.0000047651.45322.16. [DOI] [PubMed] [Google Scholar]