

Abstract
Background and purpose:
Abnormal glutamatergic activity is implicated in neurologic and neuropsychiatric disorders. Selective glutamate receptor antagonists were highly effective in animal models of stroke and seizures but failed in further clinical development because of serious side effects, including an almost complete set of symptoms of schizophrenia. Therefore, the novel polyvalent glutamatergic agent 3,5-dibromo-L-phenylalanine (3,5-DBr-L-Phe) was studied in rat models of stroke, seizures and sensorimotor gating deficit.
Experimental approach:
3,5-DBr-L-Phe was administered intraperitoneally as three boluses after intracerebral injection of endothelin-1 (ET-1) adjacent to the middle cerebral artery to cause brain injury (a model of stroke). 3,5-DBr-L-Phe was also given as a single bolus prior to pentylenetetrazole (PTZ) injection to induce seizures or prior to the administration of the N-methyl-D-aspartate (NMDA) receptor antagonist dizocilpine (MK-801) to cause disruption of prepulse inhibition (PPI) of startle (sensorimotor gating deficit).
Key results:
Brain damage caused by ET-1 was reduced by 52%, which is comparable with the effects of MK-801 in this model as reported by others. 3,5-DBr-L-Phe significantly reduced seizures induced by PTZ without the significant effects on arterial blood pressure and heart rate normally caused by NMDA antagonists. 3,5-DBr-L-Phe prevented the disruption of PPI measured 3 days after the administration of ET-1. 3,5-DBr-L-Phe also eliminated sensorimotor gating deficit caused by MK-801.
Conclusion and implications:
The pharmacological profile of 3,5-DBr-L-Phe might be beneficial not only for developing a therapy for the neurological and cognitive symptoms of stroke and seizures but also for some neuropsychiatric disorders.
Keywords: stroke, seizures, prepulse inhibition, glutamate, sensorimotor gating
Introduction
Glutamate is a major excitatory neurotransmitter in the CNS and plays a crucial role in neuronal physiology by activating ionotropic glutamate receptors of the N-methyl-D-aspartate (NMDA), (RS)-amino-3-hydroxy-5-methyl-4-isoxazolepropioinic acid (AMPA), and kainate subtypes (receptor nomenclature follows Alexander et al., 2008). Disturbances in glutamatergic activity contribute to the pathophysiology of many neurological and cognitive disorders. Excessive release of glutamate and abnormal activation of ionotropic glutamate receptors is considered one of the primary events in stroke-induced brain damage and in the hyperexcitability that leads to epileptic seizures (During and Spencer, 1993; Mehta et al., 2007; Mattson, 2008). Despite the obvious promise of glutamate antagonists for the treatment of stroke and epilepsy, the clinical use of selective anti-glutamatergic agents, primarily NMDA receptor antagonists and to a lesser degree AMPA receptor antagonists, had to be abandoned because of serious side effects. NMDA channel blockers such as phencyclidine (PCP), ketamine and dizocilpine (MK-801) improve neurological function and reduce infarct volume in animal models of stroke, and depress epileptic seizures. Unfortunately, these compounds also cause symptoms of schizophrenia in both animals and healthy volunteers and exacerbate symptoms in schizophrenic patients (Kemp and McKernan, 2002; Rung et al., 2005; Eyjolfsson et al., 2006; Braun et al., 2007). Such side effects can be especially harmful when considering that both stroke and epilepsy may result in significant cognitive complications of their own (Jorge et al., 2003; Carreño et al., 2008).
The finding that the NMDA antagonists caused symptoms of schizophrenia was one of the bases for the hypothesis that reduced NMDA receptor activation contributes to the development of schizophrenia (Javitt and Zukin, 1991; Olney and Farber, 1995; Abekawa et al., 2006; Rasmussen et al., 2007). NMDA receptor antagonists such as PCP, ketamine and MK-801 have, among other effects, been shown to increase glutamate release in the prefrontal cortex of rats (Moghaddam et al., 1997; Idris et al., 2005; Large et al., 2005; Razoux et al., 2007). Both the anti-epileptic drug lamotrigine, which may reduce glutamate release, and AMPA/kainate receptor antagonists diminished the adverse effects of PCP and ketamine on cognitive function. This indicates that NMDA receptor antagonists may elicit unwanted side effects at least in part by increasing the release of glutamate and stimulating postsynaptic non-NMDA glutamate receptors (Moghaddam et al., 1997; Brody et al., 2003). Thus, reduced NMDA receptor activity in schizophrenia or selective targeting of NMDA receptors to depress excessive excitation during the acute phase of stroke or during epileptic seizures may in fact lead to increased glutamate release and further dysregulation of glutamatergic activity. A new polyvalent anti-glutamatergic agent, the halogenated derivative of the aromatic amino acid phenylalanine, 3,5-dibromo-L-phenylalanine (3,5-DBr-L-Phe), acts as a partial NMDA receptor agonist, reduces glutamate release and depresses the activity of AMPA/kainate receptors (Yarotskyy et al., 2005). Here, we present data that 3,5-DBr-L-Phe reduced neurological deficits and infarct volume in a rat model of stroke caused by intracerebral injection of endothelin-1 (ET-1) adjacent to the middle cerebral artery (MCA) and depressed seizures in rats induced by pentylenetetrazole (PTZ). In contrast to selective NMDA receptor antagonists, 3,5-DBr-LPhe did not cause disruption of prepulse inhibition (PPI) of the acoustic startle response. Furthermore, 3,5-DBr-L-Phe reduced the PPI deficit caused by stroke and prevented the disruption of PPI caused by the NMDA antagonist MK-801.
Methods
Animals
All experimental procedures were approved by the University of Florida Institutional Animal Care and Use Committee. In addition, the principles governing the care and treatment of animals, as stated in the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (publication No. 85-323, revised 1996) were followed at all times during this study. Male Sprague Dawley rats (250–300 g) were housed in groups of two at room temperature on a 12 h light/dark cycle.
Induction of stroke by intracerebral injection of ET-1
Ischaemic brain damage (stroke) was induced by intracerebral injection of ET-1 adjacent to the MCA (Sharkey et al., 1993; Mecca et al., 2009). This procedure was performed essentially as follows. Rats were anaesthetized with a mixture of O2 (1 L·min−1) and 4% isoflurane, placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA), and anaesthesia was maintained for the duration of the surgery using an O2/isoflurane (2%) mixture delivered through a nose cone attached to the frame. The skull was exposed and a small hole was drilled in the cranium dorsal to the right hemisphere using the following stereotaxic coordinates (1.6 mm anterior and 5.2 mm lateral to the bregma). A Hamilton syringe was stereotactically positioned into the hole and lowered to a depth of 8.7 mm DV, and 3 µL of saline solution containing 240 pmoles of ET-1 were injected at a rate of 1 µL·min−1. The syringe was left in place for 4 min before removal and wound closure. During surgery, body temperature was maintained between 37.5 and 38.0°C using a heating pad. Rats were treated in a randomized fashion with i.p. injections of 3,5-DBr-L-Phe or equal volumes of saline. Rats received 3 bolus injections of 3,5-DBr-L-Phe (30 mg·kg−1) or saline at 30, 120 and 240 min after intracerebral administration of ET-1, which was considered as the onset of induction of stroke. Previously, using a cranial window method to visualize the MCA branches we have demonstrated that injection of ET-1 into the brain parenchyma adjacent to the MCA elicits abrupt constriction of the proximal MCA branches to 0% baseline vessel diameter within minutes, followed by recanalizaton of the vessel (Mecca et al., 2009).
In order to assess the neurological function of each group of rats, three neurological tests were performed 72 h following the administration of ET-1. In addition, the animals were trained 3–5 days before testing by performing the same behavioural procedures without data recording.
Sunflower seed eating task (Gonzalez and Kolb, 2003): with the rat in a clear plastic cage the time to eat five sunflower seeds, as well as the number of shell pieces after consumption, was recorded.
Neurological examination reported by Bederson et al. (1986): each rat was evaluated for absence or degree of presence of abnormal forelimb flexion, symmetric resistance to a lateral push and circling behaviour, which were graded 0 (best), 1, 2 and 3 (worst) respectively.
Neurological examination reported by Garcia et al. (1995): six tests, which were scored individually and summed for a neurological score ranging from 3 (worst) to 18 (best) (Table 1).
Table 1.
Neurological examination (Garcia et al., 1995)
| Category | Assessment (range of scores) | Scoring |
|---|---|---|
| Spontaneous activity | Exploratory behaviour in a cage (0–3) | 3 approaches at least three cage walls |
| 2 hesitant, approaches at least one cage wall | ||
| 1 minimal attempts at movement or rising up | ||
| 0 no movement | ||
| Symmetry in extremity movement | Rat is held by base of tail one foot above lab bench (0–3) | 3 all four limbs extend |
| 2 limbs on one side extend less or more slowly | ||
| 1 minimal movement on one side | ||
| 0 forelimb immobile | ||
| Forepaw extension | Rat is held by base of tail and made to walk on forelimbs (0–3) | 3 symmetric walk |
| 2 asymmetric walk | ||
| 1 minimal movement on one side | ||
| 0 forelimb immobile | ||
| Climbing | Ability to climb wall of wire cage (1–3) | 3 rat climbs easily and holds on tightly |
| 2 asymmetry in climbing and holding onto wire | ||
| 1 unable to climb; circling | ||
| Body proprioception | Reaction to touch of both sides of the body (1–3) | 3 symmetric startle and turning to touched side |
| 2 asymmetry in promptness or extent of response | ||
| 1 no response on one side | ||
| Response to vibrissae touch | Reaction to brush against the vibrissae on each side (1–3) | 3 symmetric startle and turning to touched side |
| 2 slow response to stimulus on left side only | ||
| 1 no response on the left side |
Three days after the stroke induction procedure, the brain was removed and sectioned coronally into seven slices of 2 mm thickness, starting from the frontal pole. Slices were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) for 30 min at room temperature. Areas ipsilateral to the occlusion, which were not stained red with TTC, were defined as infarcted. Total infarct volume was computed by integrating infarct areas of sequential brain sections. The infarct volume in the cortex and subcortex was evaluated. After fixation with 10% formalin, brain sections were scanned on a flatbed scanner (Epson V500, Epson America Inc., Long Beach, CA, USA) and analysed using ImageJ software (NIH). To compensate for the effect of brain oedema, the corrected infarct volume was calculated as a percentage of the contralateral hemisphere.
Pentylenetetrazole-induced seizures
A rat model of PTZ-induced seizures was used to study the anti-epileptic effects of 3,5-DBr-L-Phe. 30 mg·kg−1 of 3,5-DBr-L-Phe (treatment group), or the equivalent volume of 0.9% NaCl (control group), were administered i.p. 15 min prior to PTZ injection (60 mg·kg−1, i.p.). The rats were observed and videotaped for 20 min after the administration of PTZ. In-house software was used for semi-automatic scoring of the duration (number of 1 s intervals) and the severity of seizures. The severity of seizures was scored using a seven-point behavioural seizure score: 0 – no response; 1 – ear and facial twitching; 2 – convulsive waves through the body; 3 – myoclonic jerks and/or rearing; 4 – clonic-tonic seizures; 5 – generalized clonic-tonic seizures, loss of postural control; 6 – lethal.
Measurements of cardiovascular parameters and locomotor activity in conscious rats
Rat telemetry transducers (DSI, St. Paul, MN) were implanted into the abdominal aorta (Li et al., 2006). Rats were left to recover for 10–14 days before drug administration. The doses, volumes and timing for drug administrations were the same as described above for the ET-1-induced stroke experiments. Heart rate, mean arterial blood pressure, pulse pressure and locomotor activity were recorded from 2 h before until 12 h after drug administration.
Measurements of acoustic startle response and PPI of startle
The PPI test was performed in two groups of animals. The first group of rats was given the stroke induction procedure via cerebral injection of ET-1, and animals were randomly treated with either 3,5-DBr-L-Phe or 0.9% saline as described above. The PPI test was performed 3 days after the cerebral administration of ET-1.
Rats in the second group were used to study the effect of 3,5-DBr-L-Phe on the disruption of PPI caused by MK-801. Rats were handled within 2 days of shipment arrival. Approximately 6 days after arrival, the rats were exposed to a ‘matching’ startle session, which was essentially the same as one that was used for the test (see below). Based on data for PPI from the matching session the rats were assigned to treatment groups to ensure similar baseline PPI levels between groups. The effect of 3,5-DBr-L-Phe on the disruption of PPI caused by MK-801 was studied 3 days after the matching session. The rats received either a single bolus of 3,5-DBr-L-Phe (30 mg·kg−1, i.p.) or equal volumes of saline 15 min before the PPI test. All animals were injected with MK-801 (0.15 mg·kg−1, i.p.) or saline 5 min after administration of 3,5-DBr-L-Phe or saline. Testing occurred during the light phase of the dark–light cycle. The SR-LAB apparatus and accompanying software (San Diego Instruments, San Diego, CA) were used to perform the tests. At the beginning of every testing session, each animal was placed into the cylindrical animal enclosure and then was exposed to a 75 dB white noise (background) for a 5 min acclimation period. The acclimation period was then followed by a test session consisting of five different types of trials: (i) 120 dB pulse-only of 40 ms duration; (ii–iv) a 120 dB pulse of 40 ms duration preceded by a prepulse of 20 ms duration at 5 dB, 10 dB and 15 dB above background; and (v) a no-stimulus trial of background noise. The delay between the onset of the prepulse and the onset of the pulse was 100 ms. The trials were presented in pseudorandom order with variable inter-trial intervals with an average duration of 15 s. The first four trials and last three trials consisted of 120 dB pulse-only trials. All five types of trials were presented eight times each in pseudorandom order after the first four and last three pulse-only trials. The %PPI for each prepulse intensity was calculated using the following formula: %PPI = 100 ×[(pulse alone) − (prepulse + pulse)]/pulse alone (Geyer and Dulawa, 2003). The responses to the first four and last three pulse-only stimuli were not included in the calculations. The startle and PPI of startle were compared between three groups of animals. Two groups of rats received two i.p. injections, a first injection 15 min and a second injection 10 min prior to the test, which altogether delivered the following combinations of treatments: (i) saline – MK-801 (0.15 mg·kg−1) and (ii) 3,5-DBr-L-Phe (30 mg·kg−1) – MK-801 (0.15 mg·kg−1). The control group (group 3) consisted of all animals from groups 1 and 2 tested 3 days before any injections.
Statistical analysis
For parametric data, single comparisons were tested using the t-test, whereas multiple comparisons among groups were analysed using one-way or two-way repeated measures anova followed by Bonferonni t-test. For non-parametric data, the Mann–Whitney Rank Sum test was used. P < 0.05 was considered significant. SigmaStat 3.11 software (Systat Software, Inc, Point Richmond, CA) was used for statistical analysis.
Materials
3,5-DBr-L-Phe was obtained from Showa Denko K.K. (Kawasaki, Japan). All other compounds were purchased from Sigma-Aldrich (St. Louis, MO) and Tocris Cookson Inc. (Ellisville, MO).
Results
Neuroprotective effects of 3,5-DBr-L-Phe in a rat model of stroke caused by cerebral injection of ET-1 adjacent to the MCA
Compared to the baseline values, prior to induction of stroke, rats treated with 0.9% saline exhibited remarkable neurological deficits 3 days after intracerebral administration of ET-1 into the area adjacent to the MCA. They produced significantly more shell pieces during opening of the sunflower seeds [F(3, 42) = 10.65, P < 0.05 overall and post hoc]. The saline-treated rats spent significantly longer time to consume five seeds [F(3, 42) = 22.28, P < 0.05 overall and post hoc; Figure 1A]. In contrast, rats treated with 3,5-DBr-L-Phe produced similar numbers of shell pieces before and 3 days after administration of ET-1 (P= 0.7, but P < 0.05 relative to saline-treated animals 3 days after administration of ET-1). The time spent by the 3,5-DBr-L-Phe-treated rats to eat five sunflower seeds was significantly greater after the ET-1 administration in comparison with the time spent by the same rats before this procedure (P < 0.05), but it was nonetheless significantly shorter than that of saline-treated animals 3 days after they received ET-1 (P < 0.05; Figure 1B). Results from both of the neurological examinations described by Bederson et al. (1986) and by Garcia et al. (1995) revealed significant preservation of neurological function in the 3,5-DBr-L-Phe-treated animals compared with rats receiving 0.9% saline as the treatment (Figure 1C,D). Histological examination of brain tissue in these rats using TTC staining revealed pronounced infarcts in the right hemisphere in both the cortex and the subcortical region of rats treated with 3,5-DBr-L-Phe and those that received 0.9% saline. However, the proportion of infarcted brain tissue was significantly less in the 3,5-DBr-L-Phe-treated animals (Figure 2). Brain damage was reduced by approximately 52% after i.p. administration of 3,5-DBr-L-Phe relative to the 0.9% saline-treated controls (t= 2.1, d.f. = 21, P < 0.05), which is comparable with the effects of MK-801 in this model (Pringle et al., 2003; Moyanova et al., 2007). The neuroprotective effects of 3,5-DBr-L-Phe were more evident in the cortex (t= 2.28, d.f. = 21, P < 0.05), with a non-significant effect in the subcortex (t= 1.75, d.f. = 21, P= 0.10).
Figure 1.
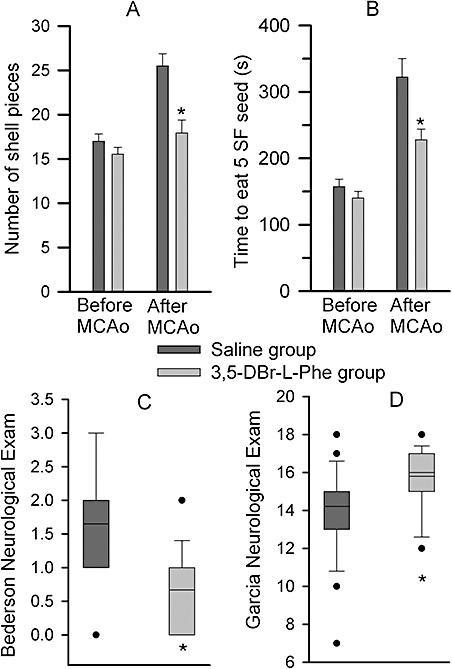
3,5-dibromo-L-phenylalanine (3,5-DBr-L-Phe) decreases neurological deficits caused by stroke after intracerebral injection of endothelin-1 (ET-1). Rats each received three bolus injections of 3,5-DBr-L-Phe (30 mg·kg−1; i.p.) or equal volumes of 0.9% saline at 30, 120 and 240 min after injection of ET-1. The neurological evaluations were performed 3 days later. (A and B) The results of the sunflower seed test in the same groups of animals before and 3 days after administration of ET-1. (C and D) Respective results from the Bederson and Garcia neurological examinations. Data are presented as a box and whiskers plot. The boundary of the box closest to zero indicates the 25th percentile, lines within the box mark the median and the mean, and the boundary of the box farthest from zero the 75th percentile. Whiskers (error bars) above and below the box indicate the 90th and 10th percentiles. Outliers are represented as individual data points. *P < 0.05 relative to the saline-treated animals, n= 15 (3,5-DBr-L-Phe) and n= 8 (saline). MCAo, middle cerebral artery occlusion.
Figure 2.
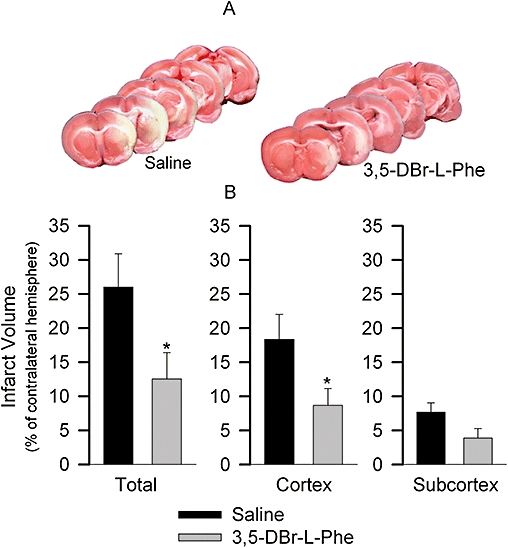
3,5-dibromo-L-phenylalanine (3,5-DBr-L-Phe) decreases the volume of infarcted brain tissue caused by intracerebral injection of endothelin-1 (ET-1). Results obtained from the same rats as in Figure 1. Histopathological analysis of the infarcted brain tissue was performed 3 days after administration of ET-1 using 2,3,5-triphenyltetrazolium chloride (TTC) staining. (A) TTC-stained sections of brain at five coronal levels from representative rats that received either saline or 3,5-DBr-L-Phe. (B) total volume of infarcted brain tissue as well as volume of infarcted brain tissue in the cortex and in the subcortex presented as a percent of the contralateral hemisphere. *P < 0.05 versus 0.9% Saline.
Anti-seizure effects of 3,5-DBr-L-Phe in a rat model of PTZ-induced seizures
Administration of PTZ caused changes in rat behaviour that ranged from no response to generalized clonic-tonic seizures (see Methods for behavioural response grading). The response to PTZ in the saline-treated rats began within times ranging from 5 to 241 s (median was 38 s, 25 and 75% percentile were 13 and 68 s, respectively, n= 10) as ear and facial twitching (stage 1) and convulsive waves through the body (stage 2). This could be followed by seizures of different degrees of severity. The pro-convulsive effects of PTZ were significantly diminished in the 3,5-DBr-L-Phe-treated rats (Figure 3). In two animals from the 3,5-DBr-L-Phe-treated group, there was no visible response observed after PTZ injection. In animals treated with 3,5-DBr-L-Phe that had seizures, the latency period before the appearance of the seizure response was significantly increased. The seizures began within times ranging from 9 to 270 s (median was 107 s, 25 and 75% percentile were 69 s and 196 s, respectively, n= 8, P < 0.05 vs. saline). Two animals treated with 3,5-DBr-L-Phe did not have seizures and therefore were not included in the analysis of the latent period before seizures. 3,5-DBr-L-Phe decreased both the intensity and the duration of seizures (Figure 3).
Figure 3.
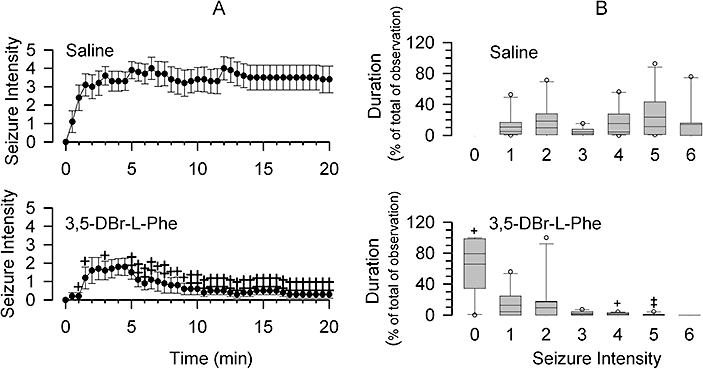
3,5-dibromo-L-phenylalanine (3,5-DBr-L-Phe) depresses pentylenetetrazole (PTZ)-induced seizures. Each group of animals received an injection of 3,5-DBr-L-Phe (30 mg·kg−1, i.p.) or equal volumes of saline 15 min prior to PTZ administration (60 mg·kg−1, i.p.). (A) seizure intensity at a given time (bin interval 30 s) during the 20 min observation period. See Methods for seizure intensity scoring. Data are presented as mean ± SE. (B) Box plots showing the duration of seizures of a given intensity (score) during the experiment. +P < 0.05 and ‡P < 0.01 versus saline (Mann–Whitney Rank Sum Test) for both (A) and (B), n= 10 per group.
3,5-DBr-L-Phe decreases deficits of PPI of acoustic startle response caused by stroke or MK-801
Some of the animals that were used to study the neuroprotective effects of 3,5-DBr-L-Phe following induction of ischaemic stroke (Figures 1 and 2) were also tested for the acoustic startle response and PPI of startle as part of their behavioural evaluation. The acoustic startle response and PPI of startle were measured 1 day before and 3 days after induction of stroke with ET-1. The startle responses were similar in all groups, in naïve animals tested prior to administration of ET-1, in those that were treated with 3,5-DBr-L-Phe and in those treated with saline [F(2, 29) = 0.59, P= 0.56]. PPI of the startle response, on the other hand, was significantly disrupted in animals that received only saline after stroke induction relative to control [F(2, 81) = 6.258, P < 0.05 overall and post hoc; Figure 4A). The 3,5-DBr-L-Phe-treated group had PPI responses similar to controls (P= 1.0 vs. control and P < 0.05 vs. saline group). There was no statistically significant treatment by prepulse interaction.
Figure 4.
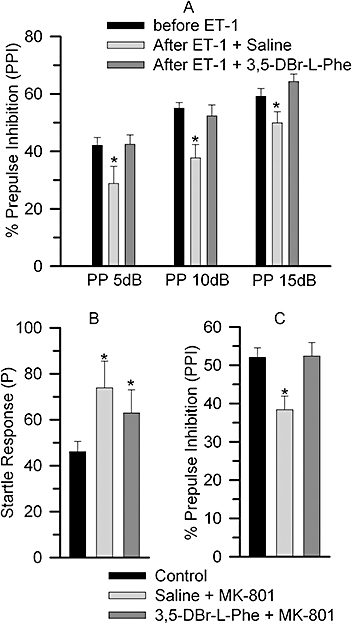
Effect of 3,5-dibromo-L-phenylalanine (3,5-DBr-L-Phe) on endothelin-1 (ET-1)- and dizocilpine (MK-801)-induced disruption of prepulse inhibition (PPI) of the acoustic startle response. (A) The PPI test was performed 1 day prior and 3 days after the administration of ET-1. The animals were treated with 3,5-DBr-L-Phe (n= 15) or saline (n= 8) as described in Figure 1. (B and C) Startle response and PPI were measured in all rats before treatment (n= 34) and after administration of (i) Saline and MK-801 (0.15 mg·kg−1) (n= 17) or (ii) 3,5-DBr-L-Phe (30 mg·kg−1) and MK-801 (0.15 mg·kg−1) (n= 17) (see text for details). *P < 0.05 relative to the control group. The average %PPI data for all three prepulse intensities for each treatment group is shown in Figure 4C.
The finding that 3,5-DBr-L-Phe alleviates the disruption of PPI caused by stroke prompted us to assess whether 3,5-DBr-L-Phe may counteract the disruption of PPI induced by MK-801, which is considered a gold standard for neuroprotection but is known to cause a sensorimotor gating deficit. Treatment with MK-801 increased the startle response from control values [F(2, 65) = 3.51, P < 0.05], whereas treatment with both 3,5-DBr-L-Phe and MK-801 did not (Figure 4B). There was, however, no statistically significant difference in startle responses between the saline-MK-801 and 3,5-DBr-L-Phe-MK-801 groups. MK-801 diminished PPI [F(2, 130) = 5.73, P < 0.05 overall and post hoc]. 3,5-DBr-L-Phe treatment reversed the MK-801-induced PPI deficit (P= 1.0 versus control and P < 0.05 versus MK-801). There was no statistically significant interaction between groups and prepulse intensities. Figure 4C demonstrates the average %PPI data for all three prepulse intensities.
3,5-DBr-L-Phe does not affect blood pressure and heart rate
Further, we addressed the question of whether 3,5-DBr-L-Phe, at doses that produced neuroprotection following stroke, caused adverse effects that were previously reported for MK-801 and other selective NMDA receptor antagonists, such as increases in blood pressure and heart rate. In order to investigate the cardiovascular effects of 3,5-DBr-L-Phe, rats (which did not undergo ET-1-induced stroke) received 3,5-DBr-L-Phe using the same dose and timing protocol for drug administration as in the ET-1-induced stroke experiments (see above). Recordings of mean arterial pressure, heart rate, and activity started 2 h prior to 3,5-DBr-L-Phe administration and continued for 13 h. Therefore, each animal served as its own control. All measured parameters fluctuated during the recording period, but these fluctuations were not related to 3,5-DBr-L-Phe administration (Figure 5). Injections of 3,5-DBr-L-Phe caused short lasting increases in all measured parameters. These changes were largely caused by procedural manipulations during injections, as they were also readily apparent in the control group injected with saline. Each subsequent injection had a markedly smaller effect (Figure 5).
Figure 5.
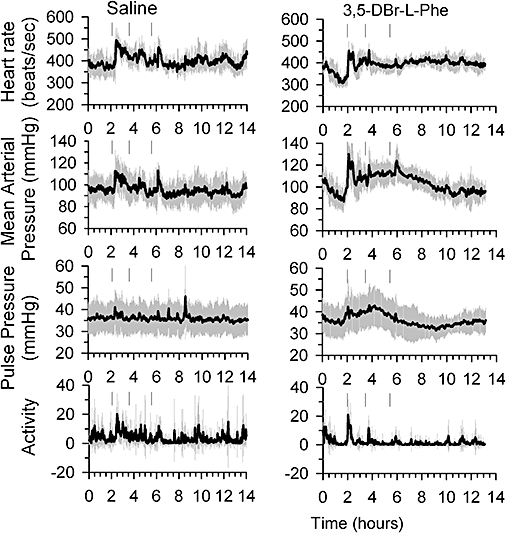
Effects of 3,5-dibromo-L-phenylalanine (3,5-DBr-L-Phe) on cardiovascular parameters and locomotor activity in the rats. Adult male SD rats, fitted with telemetry pressure transducers, received three bolus i.p. injections of 3,5-DBr-L-Phe (right panel) 30 mg·kg−1 or equal volume of saline (left panel) at 120, 210 and 330 min after start of recordings. Arrows indicate time points at which drugs were administered. Data are means ± SE from six rats for each treatment group.
Discussion
The main finding of this study is that 3,5-DBr-L-Phe, which modifies glutamatergic activity by targeting multiple pre- and postsynaptic sites, produced effective neuroprotection in a rat model of stroke, reduced acute seizures in rats, and did not cause clinically significant changes in arterial blood pressure and heart rate. 3,5-DBr-L-Phe decreased disruption of PPI caused by stroke. This is in striking contrast to MK-801, which is considered a standard for new therapeutic agents in animal models of stroke and epilepsy due to its high efficacy but causes a deficit in PPI by itself. In fact, our results indicate that 3,5-DBr-L-Phe decreased the PPI deficit caused by MK-801.
A decreased response to a startle stimulus when it is presented shortly after a weak prepulse stimulus, that is, PPI, is a measure of an organism's ability to filter non-essential information. PPI is deficient in schizophrenia and in some other neuropsychiatric disorders (Braff et al., 2001). For this reason, and because of the relative simplicity of experimental setting to measure PPI, PPI has become a major experimental paradigm in neuropsychiatric research to study the neurobiology of these disorders and to test new antipsychotics (Geyer et al., 2001). Many patients with epilepsy display PPI deficits and psychotic symptoms similar to those observed in schizophrenic patients (Takeda et al., 2001; Tremolizzo et al., 2005). The deficiency of PPI can be alleviated by antipsychotic drugs or by some anti-epileptic agents (Brody et al., 2003; Tremolizzo et al., 2005). A possibility that schizophrenia may increase the risk of epilepsy has also been suggested (Cascella et al., 2009). Cognitive complications after an acute stroke are well documented and are considered a poor prognostic factor for functional recovery. Tejkalováet al. (2007) demonstrated that rats that experienced an experimental stroke during the neonatal period displayed disrupted PPI after they achieved adulthood. Altogether, these data indicate a tight link between neurological and neuropsychiatric abnormalities in pathophysiological conditions that involve dysregulation of the glutamatergic system. Ideally, these should be all treated at the same time. These data also suggest the danger of exacerbating one group of symptoms while treating another one. Our data indicate that 3,5-DBr-L-Phe exhbits neuroprotective, anti-seizure and antipsychotic activities. The complex, unique action of 3,5-DBr-L-Phe at glutamatergic synapses may provide the basis for its polyvalent action in vivo. By simultaneously targeting the presynaptic glutamate release and activity of all subtypes of ionotropic glutamate receptors 3,5-DBr-L-Phe is more likely to normalize glutamatergic activity without unbalancing it, as found with selective NMDA receptor antagonists.
3,5-DBr-L-Phe acts as a partial NMDA receptor agonist. In addition, 3,5-DBr-L-Phe depresses the presynaptic glutamate release and activity of postsynaptic AMPA/kainate receptors (Yarotskyy et al., 2005). The partial agonism of 3,5-DBr-L-Phe at NMDA receptors should be efficient in the depression of excessive NMDA receptor activity during the acute phase of stroke or during epileptic seizures. The action of 3,5-DBr-L-Phe as a partial agonist of NMDA receptors may explain the observation that 3,5-DBr-L-Phe diminished the PPI deficit in the MK-801-treated animals.
It has been postulated that reduction in glutamate release by the anti-epileptic drug lamotrigine mediates its therapeutic effect on the perceptual abnormalities produced by ketamine in humans and the disruption of PPI caused by ketamine in mice (Anand et al., 2000; Brody et al., 2003). Similarly, in the current study the depressant effect of 3,5-DBr-L-Phe on glutamate release may contribute to the reduction in PPI deficit caused by stroke or MK-801 and to the depression of seizures caused by PTZ in rats. The ability of 3,5-DBr-L-Phe to depress presynaptic glutamate release and epileptic seizures may be important properties of these agents as neuroprotectors, allowing neuroprotection at later stages of stroke by depressing the ‘secondary or delayed’ glutamatergic activity associated with peri-infarct ischaemic depolarization and post-stroke epileptogenesis (Ferro and Pinto, 2004; Umegaki et al., 2005). By depressing glutamate release, 3,5-DBr-L-Phe may alleviate cognitive abnormalities associated with stroke, epileptic seizures, and schizophrenia as it is evident from its effect on PPI disrupted by stroke and MK-801.
3,5-DBr-L-Phe may produce its effects in vivo via additional mechanisms not readily detectable in electrophysiological experiments in vitro. Thus, aromatic amino acids, by trapping hydroxyl radicals (•OH), decrease toxicity induced by reactive oxygen species (ROS) and by preventing the •OH-induced inhibition of glutamate uptake. D-phenylalanine has been reported to restore the glutamate uptake impaired by ROS in the rat ventral tegmental area (Wolf et al., 2000). 3,5-DBr-L-Phe may increase glutamate uptake by a similar mechanism. Indeed, using the hydrogen peroxide-driven Fenton reaction and mass spectrometry, we have obtained evidence that 3,5-DBr-L-Phe is hydroxylated by trapping •OH (unpublished observation).
Another important property of 3,5-DBr-L-Phe that sets it apart from highly selective NMDA receptor antagonists is the absence of clinically significant effects on cardiovascular parameters in rats. Administration of 3,5-DBr-L-Phe at doses that produced effective neuroprotection after the ET-1-induced stroke caused only brief changes in heart rate and arterial blood pressure. These increases were probably due to the stress of the injection procedure rather than the drug as they were also seen during 0.9% saline injections, and their magnitude decreased with each successive administration. Hypotension is of major concern in stroke patients. Importantly, 3,5-DBr-L-Phe caused an increase (<15%), but not a decrease, in arterial blood pressure (Figure 5). Similar increases in arterial blood pressure can be observed during the administration of intravenous isotonic fluids to stroke patients as part of therapeutic procedures (Finley Caulfield and Wijman, 2008).
In summary, 3,5-DBr-L-Phe produced effective neuroprotection in a rat model of stroke caused by intracerebral injection of ET-1 adjacent to the MCA, reduced seizures induced by PTZ, and decreased the PPI deficit caused by both stroke and MK-801. Importantly, significant neuroprotection with 3,5-DBr-L-Phe could be achieved by administering this agent after the onset of stroke. The type of action that 3,5-DBr-L-Phe produces could be useful either alone or in combination with already known agents, for the treatment of certain neurological and neuropsychiatric disorders.
Acknowledgments
This work was supported by Grants NS060862 from the NIH, 08KB02 from the Florida Biomedical Research Program, by the University of Florida McKnight Brain Institute, by JS Gravenstein MD Endowment, and by I. Heermann Anesthesia Foundation, Inc.
We would like to thank Laura Bohatch and Loel Warsch for technical assistance.
Glossary
Abbreviations:
- AMPA
(RS)-amino-3-hydroxy-5-methyl-4-isoxazolepropioinic acid
- 3,5-DBr-L-Phe
3,5-dibromo-L-phenylalanine
- ET-1
endothelin-1
- MCA
middle cerebral artery
- MK-801
dizocilpine
- NMDA
N-methyl-D-aspartic acid
- PCP
phencyclidine
- PPI
prepulse inhibition
- PTZ
pentylenetetrazole
- TTC
2,3,5-triphenyltetrazolium chloride
Conflict of interest
None.
References
- Abekawa T, Ito K, Koyama T. Role of the simultaneous enhancement of NMDA and dopamine D(1) receptor-mediated neurotransmission in the effects of clozapine on phencyclidine-induced acute increases in glutamate levels in the rat medial prefrontal cortex. Naunyn Schmiedebergs Arch Pharmacol. 2006;374:177–193. doi: 10.1007/s00210-006-0115-9. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57:270–276. doi: 10.1001/archpsyc.57.3.270. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Braun I, Genius J, Grunze H, Bender A, Möller HJ, Rujescu D. Alterations of hippocampal and prefrontal GABAergic interneurons in an animal model of psychosis induced by NMDA receptor antagonism. Schizophr Res. 2007;97:254–263. doi: 10.1016/j.schres.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Brody SA, Geyer MA, Large CH. Lamotrigine prevents ketamine but not amphetamine-induced deficits in prepulse inhibition in mice. Psychopharmacology (Berl) 2003;169:240–246. doi: 10.1007/s00213-003-1421-2. [DOI] [PubMed] [Google Scholar]
- Carreño M, Donaire A, Sánchez-Carpintero R. Cognitive disorders associated with epilepsy: diagnosis and treatment. Neurologist. 2008;14:S26–34. doi: 10.1097/01.nrl.0000340789.15295.8f. [DOI] [PubMed] [Google Scholar]
- Cascella NG, Schretlen DJ, Sawa A. Schizophrenia and epilepsy: is there a shared susceptibility? Neurosci Res. 2009;63:227–235. doi: 10.1016/j.neures.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet. 1993;341:1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Eyjolfsson EM, Brenner E, Kondziella D, Sonnewald U. Repeated injection of MK-801: an animal model of schizophrenia? Neurochem Int. 2006;48:541–546. doi: 10.1016/j.neuint.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Ferro JM, Pinto F. Poststroke epilepsy: epidemiology, pathophysiology and management. Drugs Aging. 2004;21:639–653. doi: 10.2165/00002512-200421100-00003. [DOI] [PubMed] [Google Scholar]
- Finley Caulfield A, Wijman CA. Management of acute ischemic stroke. Neurol Clin. 2008;26:345–371. doi: 10.1016/j.ncl.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Dulawa SC. Assessment of murine startle reactivity, prepulse inhibition, and habituation. Curr Protoc Neurosci. 2003 doi: 10.1002/0471142301.ns0817s24. Nov; Chapter 8: Unit 8.17. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Gonzalez CL, Kolb B. A comparison of different models of stroke on behaviour and brain morphology. Eur J Neurosci. 2003;18:1950–1962. doi: 10.1046/j.1460-9568.2003.02928.x. [DOI] [PubMed] [Google Scholar]
- Idris NF, Repeto P, Neill JC, Large CH. Investigation of the effects of lamotrigine and clozapine in improving reversal-learning impairments induced by acute phencyclidine and D-amphetamine in the rat. Psychopharmacology (Berl) 2005;179:336–348. doi: 10.1007/s00213-004-2058-5. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Jorge RE, Robinson RG, Arndt S, Starkstein S. Mortality and post-stroke depression: a placebo controlled trial of antidepressants. Am J Psychiatry. 2003;160:1823–1829. doi: 10.1176/appi.ajp.160.10.1823. [DOI] [PubMed] [Google Scholar]
- Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5:1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- Large CH, Webster EL, Goff DC. The potential role of lamotrigine in schizophrenia. Psychopharmacology (Berl) 2005;181:415–436. doi: 10.1007/s00213-005-0020-9. [DOI] [PubMed] [Google Scholar]
- Li H, Gao Y, Freire CD, Raizada MK, Toney GM, Sumners C. Macrophage migration inhibitory factor in the PVN attenuates the central pressor and dipsogenic actions of angiotensin II. FASEB J. 2006;20:1748–1750. doi: 10.1096/fj.06-5836fje. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann NY Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca AP, O'Connor TE, Katovich MJ, Sumners C. Candesartan pre-treatment is cerebroprotective in a rat model of endothelin-1 induced middle cerebral artery occlusion. Exp Physiol. 2009;94:937–946. doi: 10.1113/expphysiol.2009.047936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. Neuroscience. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyanova SG, Kortenska LV, Mitreva RG, Pashova VD, Ngomba RT, Nicoletti F. Multimodal assessment of neuroprotection applied to the use of MK-801 in the endothelin-1 model of transient focal brain ischemia. Brain Res. 2007;1153:58–67. doi: 10.1016/j.brainres.2007.03.070. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- Pringle AK, Schmidt W, Deans JK, Wulfert E, Reymann KG, Sundstrom LE. 7-Hydroxylated epiandrosterone (7-OH-EPIA) reduces ischaemia-induced neuronal damage both in vivo and in vitro. Eur J Neurosci. 2003;18:117–124. doi: 10.1046/j.1460-9568.2003.02734.x. [DOI] [PubMed] [Google Scholar]
- Rasmussen BA, O'Neil J, Manaye KF, Perry DC, Tizabi Y. Long-term effects of developmental PCP administration on sensorimotor gating in male and female rats. Psychopharmacology (Berl) 2007;190:43–49. doi: 10.1007/s00213-006-0584-z. [DOI] [PubMed] [Google Scholar]
- Razoux F, Garcia R, Lena I. Ketamine, at a dose that disrupts motor behavior and latent inhibition, enhances prefrontal cortex synaptic efficacy and glutamate release in the nucleus accumbens. Neuropsychopharmacology. 2007;32:719–727. doi: 10.1038/sj.npp.1301057. [DOI] [PubMed] [Google Scholar]
- Rung JP, Carlsson A, Ryden Markinhuhta K, Carlsson ML. (+)-MK-801 induced social withdrawal in rats; a model for negative symptoms of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:827–832. doi: 10.1016/j.pnpbp.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Sharkey J, Ritchie IM, Kelly PA. Perivascular microapplication of endothelin-1: a new model of focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1993;13:865–871. doi: 10.1038/jcbfm.1993.108. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Inoue Y, Tottori T, Mihara T. Acute psychosis during intracranial EEG monitoring: close relationship between psychotic symptoms and discharges in amygdala. Epilepsia. 2001;42:719–724. doi: 10.1046/j.1528-1157.2001.08700.x. [DOI] [PubMed] [Google Scholar]
- Tejkalová H, Kaiser M, Klaschka J, Stastný F. Does neonatal brain ischemia induce schizophrenia-like behavior in young adult rats? Physiol Res. 2007;56:815–823. doi: 10.33549/physiolres.931056. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Doueiri MS, Dong E, Grayson DR, Davis J, Pinna G, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Umegaki M, Sanada Y, Waerzeggers Y, Rosner G, Yoshimine T, Heiss WD, et al. Peri-infarct depolarizations reveal penumbra-like conditions in striatum. J Neurosci. 2005;25:1387–1394. doi: 10.1523/JNEUROSCI.4182-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Xue CJ, Li Y, Wavak D. Amphetamine increases glutamate efflux in the rat ventral tegmental area by a mechanism involving glutamate transporters and reactive oxygen species. J Neurochem. 2000;75:1634–1644. doi: 10.1046/j.1471-4159.2000.0751634.x. [DOI] [PubMed] [Google Scholar]
- Yarotskyy V, Glushakov AV, Sumners C, Gravenstein N, Dennis DM, Seubert CN, et al. Differential modulation of glutamatergic transmission by 3,5-dibromo-L-phenylalanine. Mol Pharmacol. 2005;67:1648–1654. doi: 10.1124/mol.104.005983. [DOI] [PubMed] [Google Scholar]