

Abstract
Prochiral heteroaryl ketones containing furan, thiophene, chroman and thiochroman moieties were successfully reduced in the presence of 1 – 10 mol % of spiroaminoborate ester 1 with different borane sources to afford non-racemic alcohols in up to 99% ee. In addition, modest enantioselectivity, around 80% ee, was achieved in the reduction of linear α,β-unsaturated heteroaryl ketones.
1. Introduction
Heterocyclic fragments containing oxygen or sulfur in the ring are important cores found in natural and synthetic products used in the flavor, fragrance and pharmaceutical industry.1 Recently, enantiomerically pure (R)-2-furanyl-2-ethanol was used as a building block by O’Doherty and et al. to introduce chirality in the synthesis of the anthrax tetrasaccharide produced by Bacillus antharacis.2a Other furfuryl enantiopure alcohols were used by the same group to obtain iminosugar natural products.2b Benzofuran carbinols are important units found in chiral drugs such as bufuralols, potent nonselective β-blocker antagonists. 2d,e In addition, sulfur heterocycles are important intermediates in the synthesis of chiral biologically active compounds such as the antidepressant Duloxetine 2 (Cymbalta),3 and the carbonic anhydrase inhibitor, MK-0417 3,4 used for the treatment of glaucoma (Figure 1). Zileuton 4 from Abbott and other related furan, thiophene and benzofuran 5-lipoxygenase inhibitors have been developed for the treatment of asthma and other inflammatory deseases.5 In addition, chroman and thiochroman carbinols are found in many drugs, such as 4-hydroxy tertalolol 5, which is employed in the treatment of high blood pressure.
Figure 1.

Biologically active heterocyclic compounds containing sulfur.
Consequently, the synthesis of single isomeric heteroaryl carbinols is currently a vast area of research. Although conventional chemical resolutions of racemic drug precursors are still being used to obtain enantiopure products, more efficient bioenzymatic resolution methods are being developed.7 For example, the kinetic resolution of racemic heterocyclic alcohols by lipases has been demonstrated to be a useful tool since it provides high enantiopure heteroaryl ethanols.7a,b Nevertheless, the maximum yield is still 50% of the desired non-racemic alcohols. To overcome this limitation, the quantitative chemoenzymatic reduction of ketones has also been widely studied.8 Baker’s yeast reduction has been applied effectively to obtain (S)-1-heteroarylethanols in high enantiopurity, however, the opposite enantiomer required a chemical transformation.8c Other limitations of biocatalysts are the lack of generality and difficulties encountered in scaling-up the methods for pharmaceutical production.8 The Noyori asymmetric transfer hydrogenation method, which uses ruthenium diamino complexes, has been successfully applied to the synthesis of heterocyclic enantiopure secondary alcohols.2,9
Asymmetric addition of boron, aluminum and silicon hydrides to the carbonyl group of prochiral ketones is an area of major interest since these methods provide a less expensive, efficient and suitable access to a great variety of non-racemic alcohols with predictable stereochemistry.10 Oxazaborolidines have been the most widely used boron catalysts for the borane reduction of heteroaryl ketones.11,12 However, the oxazaborolidine reduction of ketones containing oxygen, sulfur and nitrogen, have been observed in some substrates, to provide inferior enantioselectivity, particularly at the 2-position, possibly due to a competitive intramolecular reduction by the heterocyclic-borane complex.12a
Recently, a series of novel stable aminoborate esters with oxazaborolidine-like enantioselectivity were synthesized and characterized by our group. The structure of some of these aminoborates esters was established by X-ray analysis.13c,e The highly crystalline and stable spiroaminoborate complex 1, derived from diphenylprolinol (Scheme 1) and ethylene glycol, was found to be highly enantioselective and a convenient catalysts for the asymmetric borane reduction of prochiral ketones.13a–e As part of our interest in the enantioselective synthesis of biological active precursors, we decided to apply our spiroaminoborate methodology for the asymmetric reduction of heteroaryl compounds containing oxygen or sulfur.
Scheme 1.

2. Results and Discussion
In previous work, we have studied the borane-mediated reduction of aryl and pyridyl ketones using different aminoborate catalysts, and the process was optimized using different solvents, borane sources, temperatures and amount of catalyts.13a–c To assess the catalytic performance of 1, a curve of catalytic load vs. enantiomeric excess was established using the acetophenone reduction with boranedimethyl sulfide in THF at 25 °C, as a model. The maximum enantioselectivity (99% ee) was attained with 10 molar % of aminoborate 1, and the catalytic cut-off was 0.5 mol % obtaining 98% ee.13c Additionally, we have carried out similar studies with the 3- and 4-acetyl pyridines observing excellent enantioselectivities, 98% ee and 99% ee, respectively, with 1 mol % of catalyst.13d
Derivatives of 2-acetylfuran are important synthons for the preparation of α-hydroxy and other α-functionalized acids by ozonolysis of the furan ring.14 The reduction of 2-acetylfuran in the presence of 10 mol % of 1 gave (+)-(R)-1-(furan-2-yl)ethanol in 95% ee. When 1% mol of catalyst was used, the selectivity was still high, 95% ee. The yield was moderate probably due to the high volatility of the alcohol causing product loss during distillation in the purification process (Table 1, entry 1 and 2). Other commercially available substrates with a furanyl fragment were studied (entry 3–6). Unexpectedly, 2-acetyl-5-methylfuran provided the corresponding alcohol with significantly lower selectivity with 1% of catalyst, but with 10 mol %, the enantiopurity of the corresponding alcohol increased to 91% ee (entry 3). The enantioselectivity for the 2-acetyl benzofuran reduction was 96% ee using 1% and 10% of 1, in good yield (entries 5 and 6).
Table 1.
Reduction of ketones containing the furanyl ring and using spiroborate ester 1 in 10 and 1 mol %a
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Cat. 1 (%) |
Yieldb (%) |
Eec (%) |
| 1 |  |
10 | 63 | 95 |
| 2 | 1 | 65 | 95 | |
| 3 |  |
10 | 87 d | 91 |
| 4 | 1 | 91 | 85 | |
| 5 |  |
10 | 88d | 96 |
| 6 | 1 | 95 | 96 | |
| 7 | 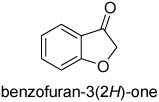 |
10 | 86d | 92 |
| 8 | 5 | 78d | 92 | |
| 9 |  |
10 | 82 | >99e |
The reaction was carried out using 0.7 equiv of borane DMS in THF at room temperature.
Yield after distillation in a Kugelrohr apparatus.
Determined by GC of the acetate on a CP-Chirasil-Dex CB column.
Yield after column chromatography.
Determined by 31P NMR of a phosphonate (CDA) derivative.15
The unstable and enolizable benzofuran-3(2H)-one afforded the corresponding alcohol with good selectivity (92%) using 10% and 5% of catalyst (entry 7 and 8 in Table 1). However, a lower level of asymmetric induction was observed with 1% of catalyst, obtaining the dihydrobenzofuranol with less than 50% ee and in low yield (33%). However, remarkable enantioselectivity was achieved for the 4-chromanone reduction with 10 mol % of catalyst 1, providing the chromanol in >99% ee (entry 9).
Our study was then extended to the synthesis of representative sulfur containing heteroaryl alcohols. Prochiral thiophenyl ketones and thiochromanones were, in general, successfully reduced to the corresponding alcohols with 10 and even 1 mol % of catalyst 1 (Table 2). Outstanding enantioselectivities (>96% ee) were attained in most instances even with a low catalytic load, and the reactions were completed in less than an hour. The products were easily isolated in high yield and purified, usually, by a simple distillation. In addition, we studied the reduction of some sulfur heterocyclic ketone using borane diethylaniline (DEA) as a more stable borane sources.15 In general, for the selected thiophene compounds, we observed high enantioselectivity, up to 99% ee with 10% catalyst 1 (as shown in parenthesis in Table 2). However the yields were modest, possibly due to the more laborious DEA extraction process during the work-up.
Table 2.
Reduction of heteroaryl ketones containing sulfur using 1and 10 mol % of spiroaminoborate ester 1.a
| Entry | Substrate | Cat. 1 (%) |
Yieldb (%) |
Eec (%) |
|---|---|---|---|---|
| 1 |  |
10 | 92 | 98 |
| 2 | 1 | 87 | 96 | |
| 3 | 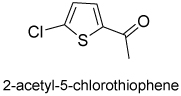 |
10 | 91(83)d | 98 (99)d |
| 4 | 1 | 91 | 98 | |
| 5 |  |
10 | 75 | 97 |
| 6 | 1 | 86 (71)d | 98 (82)d | |
| 7 |  |
10 | 90 (83)d | 95 (99)d |
| 8 | 1 | 89 | 96 | |
| 9 |  |
10 | 92 (76)d | 99(99)d |
| 10 | 1 | 92 | 99 | |
| 11 |  |
10 | 92(53)d | 94.5(94)d |
| 12 | 1 | 93 | 94 | |
| 13 |  |
10 | 80c,d | 93d |
| 14 | 1 | 92 | 99 (92)d | |
| 15 |  |
10 | 85c | 100 |
| 16 | 1 | 92 | 99 | |
The reaction was carried out using 0.7 equiv of borane DMS in THF at room temperature.
Isolated yield after distillation in a Kugelrohr apparatus.
Determined by GC of the acetate on chiral column.
The reaction was carried out using BH3-DEA
Chiral allylic alcohols are very important synthons in organic synthesis and can be prepared by asymmetric reduction of the corresponding unsaturated ketones.16 For reduction of heteroaryl α,β-unsaturated ketones, we investigated the reaction of (E)-4-phenylbut-3-en-2-one 6 using different amount of catalyst 1 and a variety of borane reagents (Table 3, entries 1–6). The optimal enantiomeric excess (80%) was achieved using borane-DMS with 10% of 1. The reaction was conducted in THF at 0 °C, and no reduction of the double bond was observed. The reduction of commercially available enones containing thiophenyl and furyl substituents 7 and 8 was studied with different catalytic loads of 1 using a borane-DMS complex under the previous conditions. Similarly, using 10% mol of catalyst 1 provided the optimal selectivity affording (R,Z)-4-(furan-3-yl)but-3-en-1-ol and (R,E)-4-(thiophen-3-yl)but-3-en-2-ol (entries 8 and 11) in 80 and 83% ee, respectively. The absolute configuration of the products was assigned based on the comparison with the literature specific rotation values and by the predicted stereochemistry outcome produced by catalyst 1.
Table 3.
Studies on the reduction of unsaturated ketones using spiroborate ester 1.a
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Cat. (%) |
Borane | Yieldb (%) |
Eec (%) |
| 1 |  |
25 | BH3- DMS | 87g | 79 |
| 2 | 10 | BH3- DMS | 73 | 80 | |
| 3 | 1 | BH3- DMS | 73 | 70 | |
| 4 | 10 | BH3- DEA | -d | 67 | |
| 5 | 10 | BH3- THFe | -d | 72 | |
| 6 | 10 | BH3- THFf | -d | 74 | |
| 7 |  |
25 | BH3- DMS | 55g | 72 |
| 8 | 10 | BH3- DMS | 83 | 80 | |
| 9 | 1 | BH3- DMS | 87 | 62 | |
| 10 |  |
25 | BH3- DMS | 73g | 81 |
| 11 | 10 | BH3- DMS | 83 | 83 | |
| 12 | 1 | BH3- DMS | 88 | 65 | |
The reaction was carried out using 0.6 equiv of borane reagent in THF at 0 °C.
Yield after distillation in a Kugelrohr apparatus.
Determined by GC of the acetate on a chiral column.
The product was not isolated.
Borane-THF complex was stabilized by < 0.005M of NaBH4.
Borane-THF complex was stabilized by < 0.005M of N-tert-butyl-N-isopropyl- methylamine.
Yield after column chromatography.
3. Conclusions
In general, the asymmetric borane reduction of prochiral furyl, chroman, thiochromanyl, and thiophenyl ketones was successfully achieved with high enantioselectivity using only 1% mol of spiroborate ester 1 as a catalyst and under very mild and convenient conditions, offering an efficient synthetic tool for the preparation of enantiopure secondary alcohols. Although only moderate selectivity was achieved in the reduction of linear α,β-unsaturated aromatic ketones, it is a facile method and further studies will be considered to improve the enantiomeric purity.
4. Experimental part
All reactions were preformed in oven-dried glassware under an N2 atmosphere. Air- and moisture sensitive reagents and solvents were transferred via syringe. All reagents were obtained commercially from Aldrich unless otherwise noted. Common solvents were dried and distilled by standard procedures.
Chromatographic purification of products was accomplished using flash chromatography on Merck silica gel, 200–400 mesh, as eluent, hexane-AcOEt 100:1 to 1:1 using HPLC grade solvents. Thin layer chromatography (TLC) was performed on Merck silica plates. The spots were visible with an UV lamp. Infrared analyses were performed in a Perkin Elmer Spectrum 100 FT-IR with ATR. 1H, 13C NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer with standard pulse sequences operating at 400.152 MHz and 100.627 MHz in CDCl3. Chiral gas chromatography analysis was processed on a Hewlett Packard GC 5890 equipped with a Chrompack Chiralsil-Dex-CB column (30 m × 0.25 mm × 0.25µm). GC-MS analysis was processed on a Finnegan Trace GC/Polaris Q Mass detector using a Restek RTX-5MS column. A Perking Elmer Polarimeter, Model 341, was used for optical rotation analysis.
4.1 General procedure for the preparation of racemic alcohols
To a solution of the ketone (1 mmol) in THF (3 mL) and MeOH (3 mL), solid sodium borohydride (37 mg, 1 mmol) was added at 0 °C. After the reaction mixture was stirred at 25 °C over 1 h, the solvents were evaporated. The residue was dissolved in dichloromethane (DCM) (10 mL) and added into water (15 mL) in a separatory funnel and extracted. The aqueous phase was extracted with DCM (2 × 10 mL) and dried over Na2SO4. The solvents were removed under vacuum. The crude product was used for the preparation of the O-acetyl derivative for the enantiopurity determination by GC-analysis.
4.2 General procedure for the preparation of O-acetyl derivatives for GC-analysis of alcohols
To a solution of the racemic or enantio-enriched alcohol (0.5 mmol) in dry ethyl ether (3 mL,) in a 10 mL vial, neat triethylamine (0.5 mL, 3.60 mmol), acetic anhydride (0.5 mL, 5.30 mmol ) and a small crystal of DMAP (2 mg, 0.02 mmol ) were added. The mixture was left at 25 °C for 5 min, then H2O (3 mL) and the mixture were shaken for 5 min. Solid Na2CO3 (3 g) was carefully added a small portions and left for 5 min, shaking from time to time. Extreme evolution of gas was observed. The O-acetyl derivative was extracted with additional diethyl ether (2 mL) and the organic phase was transferred with a Pasteur pipette to another vial. Chiral gas chromatography analysis was processed on a Hewlett Packard GC 5890 equipped with a Chrompack Chiralsil-Dex-CB column (30 m × 0.25 mm × 0.25µm).
4.3 General optimized method for the asymmetric reduction of ketones using 1 mol% of spiroborate 1
Borane-SMe2 complex (10 M, 0.7 mL, 7.0 mmol) was added to a solution of (S)-2-[(1,3,2-dioxaborolan-2-yloxy)diphenylmethyl]pyrrolidine 1 (32 mg, 0.10 mmol) in dry THF (5 mL) at 25 °C and the mixture was stirred for 1h. A solution of ketone (10.0 mmol) in THF (5 mL) was added for 1 h using an infusion pump. The reaction mixture was stirred at 25 °C over 1 h, then cooled at 0 °C and quenched with methanol (5 mL), concentrated. The residue was dissolved in methanol (10 mL) and refluxed over 3 h, concentrated, re-dissolved in methanol (5 mL) and concentrated again. The residue was distilled in a Kugelrohr apparatus under vacuum to give the final product.
Reduction of 2-acetylfuran using 1 mol% of spiroborate
A solution of freshly redistilled 1-(furan-2-yl)ethanone (1.10 g, 10.0 mmol) in THF (5 mL) was added to the reaction flask. After following the general procedure, (+)-(R)-1-(furan-2-yl)ethanol was obtained as colorless oil (730 mg, 65%). Bp: 150 °C/1 mmHg. IR (cm−1): 3335, 2981, 1505, 1148, 1066, 1008. MS m/z (relative intensity, ion): 112.0 (28, M), 97.1 (46, M-CH3), 95.1 (100, M-OH, - H), 69.2 (32, M-CHOHCH3). 1H NMR (400 MHz, CDCl3): δ̣ 1.48 (d, J = 6.6 Hz, 3H, Me), 3.08 (br. s, 1H, OH), 4.81 (q, J = 6.6 Hz, CHMe), 6.19 (dt, J = 3.3, 0.7 Hz, 1H, C3’H), 6.29 (dd, J = 3.2, 1.8 Hz, 1H, C4’H), 7.33 (dd, J = 1.8, 0.8 Hz, 1H, C5’H); 13C NMR (100 MHz, CDCl3):δ̣ 21.3, 66.4, 105.1, 110.1, 141.8, 157.8. Chiral GC of O-acetyl derivative at T1 = 70 °C (10 min), gradient = 4 °C/min, T2 = 190 °C (35 min): RtR 19.2 min; RtS 17.4 min, indicated 95% ee. [α]22d = +21 (c 1.45, CHCl3). Lit7d (R): [α]d = +22 (c 2.7, CHCl3), 95% ee.
Reduction of 1-(5-methylfuran-2-yl)ethanone using 10 mol % of spiroborate
A solution of freshly redistilled 1-(5-methylfuran-2-yl)ethanone (0.62 g, 5.00 mmol) in THF (5 mL) was added to the reducing mixture for 1 h using an infusion pump. After the usual work-up, the crude product was distilled in a Kugelrohr apparatus under vacuum to give the final product, (+)-(R)-1-(5-methylfuran-2-yl)ethanol, as a colorless oil (468 mg, 74%). Bp: 150 °C/1 mmHg. The reaction was repeated to improve the yield and the crude product was purified by column chromatography with silica gel 60 Å and hexane/EtOAc gradients to give 0.546 g (87%). 1H NMR (400 MHz, CDCl3): δ̣ 1.49 (d, J = 6.6 Hz, 3H, CHMe), 2.26 (d, J = 0.8 Hz, 3H, C5’Me), 2.45 (br. s, 1H, OH), 4.79 (q, J = 6.6 Hz, CHMe), 5.87–5.88 (m, 1H, C4’, H), 6.07 (d, J = 3.0 Hz, 1H, C3’H); 13C NMR (100 MHz, CDCl3): δ̣ 13.5, 21.2, 63.5, 105.9, 106.0, 151.6, 155.9. GC/MS m/z (relative intensity) 126 M+ (16%), 110 (32%), 109 (100%), 95 (52%), 81 (12%). Chiral GC of O-acetyl derivative at T1 = 70 °C (10 min), gradient = 4 °C/min, T2 = 190 °C (35 min): RtR 21.3 min, RtS 16.9 min, indicated 91% ee, determined from crude derivative. [α]23 d = +7.2 (c 1.25 , CHCl3). Lit7d (R): [α]d = +8.5 (c 2.2, CHCl3), 95% ee.
Reduction of 2-acetylbenzofuran using 1 mol% of spiroborate 1
A solution of freshly redistilled 2-acetylbenzofuran (1.60 g, 10.0 mmol) in THF (5 mL) was added for 1 h using an infusion pump. After usual work-up, the crude residue was distilled in a Kugelrohr apparatus under vacuum to give the final (+)-(R)-1-(benzofuran-2-yl)ethanol as a colorless oil (1.54 g, 95%). Bp: 220 °C/0.5 mmHg 1H NMR (400 MHz, CDCl3): δ̣ 1.57 (d, J = 6.6 Hz, 3H, Me), 2.88 (br. s, 1H, OH), 4.94 (q, J = 6.4 Hz, CHMe), 6.53 (s, 1H, C3’H), 7.18–7.27 (m, 2H, C5’H and C6’H), 7.44 (dd, J = 8.0, 0.6 Hz, C4’H), 7.51 (d, J = 6.0 Hz, 1H, C7’H); 13C NMR (100 MHz, CDCl3): δ̣ 21.4, 66.0, 101.7, 111.2, 121.0, 122.7, 124.1, 128.2, 154.7, 160.3. Chiral GC of O-acetyl derivative at T1 = 90 °C (10 min), gradient = 1°C/min, T2 = 150 °C (35 min): RtR 47.2 min; RtS 46.2 min, indicated 96% ee. [α]23 d = +18 (c 3, CHCl3). Lit7a (S) [α]20 d = −16.6 (c 1.0, CHCl3), 98.6% ee.
Reduction of benzofuran-3(2H)-one using 10 mol % of borate ester 1
To a dry 25 mL round flask under a nitrogen flow, spiroborate 1 (0.097 g, 0.3 mmol), was added and then, dry THF (5 mL) was added to make a clear solution. Borane dimethyl sulfide complex (0.21 mL, 10.0 M, 2.1 mmol) was added to the solution and the mixture was stirred during 1 h. The initial cloudy mixture changed to a colorless solution. Benzofuran-3(2H)-one (0.402 g, 3.0 mmol) in dry THF (5.0 mL) was added to the solution over 1 h using an infusion pump, and the mixture was allowed to stir for an additional 1 h. The reaction was then monitored by TLC using a silica plate and hexane as eluent to confirm the substrate consumption. The reaction was quenched with MeOH (2 mL) and the mixture was stirred overnight. The solvents were removed in the rotovaporator at 60 °C at approximately 20 mmHg and finally concentrated to afford a yellow oil. The crude product was treated with H2O (10 mL), extracted with methylene chloride (3 × 10 mL), dried over sodium sulfate, filtered and concentrated in the rotovaporator at 60 °C under vacuum. The crude product was purified by column chromatography using silica gel 60 Å Mesh 70–130 and hexane/EtOAc 1:1 as eluent to give (+)-(S)-2,3-dihydrobenzofuran-3-ol as a yellow solid, m.p. 48–50. (0.349 g, 86% yield). 1H-NMR (400 MHz, CDCl3) δ ppm: 2.24 (s, 1H, OH); 4.45 (dd, J1 = 2.6 Hz, J2 = 10.6 Hz, 1H, CH2); 4.56 (m, 1H, CH2); 5.36 (d, J = 4.8 Hz, 1H, HO-C-H); 6.99 (td, J1 = 0.8 Hz, J2 = 7.4 Hz , 1H, Ar); 7.31 (td, J1 = 0.8 Hz, J2 = 8.0 Hz, 1H, Ar); 13C-NMR (100 MHz, CDCl3) δ ppm: 72.2, 78.2, 110.6, 121.1, 125.5, 128.3, 130.8, 160.2. Chiral GC of O-acetyl derivative at T1 = 70 °C (10 min), gradient = 4 °C/min, T2 = 190 °C (35 min): RtR 21.3 min; RtS 19.9 min, indicated 92% ee. [α] 22 d = +13 (c 2.2, CHCl3). With 5% it was afforded 78% chemical yield, 92% ee and similar specific rotation: [α]22 d = +61 (c 1.8, CHCl3). Lit.7e (R): [α]d = +67 (c 0.63, CHCl3), >98% ee.
Reduction of chroman-4-one using 10 mol% of spiroborate 1
To a 100 mL, one neck round bottom flask, previously dried and under nitrogen was placed (S)-2-[(1,3,2-dioxaborolan-2-yloxy)diphenylmethyl]pyrrolidine (1) (0.323 g, 1.0 mmol) and dissolved in dry THF (40 mL). Borane-SMe2 complex (10 M, 2.0 mL, 20 mmol) was added via an infusion pump during 30 min, and the mixture was stirred at 25 °C for 1 h. A solution of chroman-4-one (1.48 g, 10 mmol) in THF (5 mL) was added for 1 h using an infusion pump. The reaction mixture was stirred at 25 °C over 3 h, then cooled at 0 °C and quenched with methanol (5 mL) and left stirring overnight at room temperature. After the mixture was concentrated in the rotoevaporator at 70 °C, the residue was dissolved in methanol (10 mL) and concentrated again. The residue was treated with NH4Cl (20 mL), and extracted with diethyl ether (4 × 15 mL) and water (3 × 10 mL). The organic phases were collected and the organic phase was dried with K2CO3. After concentration at 30 °C in the rotoevaporator, the crude product was purified by flash column chromatography on silica gel (45 g): ethyl acetate: hexane 1:5, obtaining the (+)-(R)-4-chromanol as a white solid (1.23 g, 82 % yield). 1H-NMR (400 MHz, CDCl3): δ 1.8 (s, 1H, OH), 2.5 (m, 2H, CH2), 4.3 (m, 2H, CH2), 4.83 (m, 1H, CH-OH), 6.86 (d, J = 6.40, 1H, Ar), 6.95 (t, J = 7.60, 1H, Ar), 7.24 (m, 1H, Ar), 7.35 (m, 1H, Ar); 13C-NMR (100 MHz, CDCl3-d): δ 63.5, 30.5, 62, 154.5, 116.8, 129, 120.5, 130.1, 124; FT-IR ν (cm−1): 3434 (O-H), 3054, 2987, 1062(C–O, OH). GC Rt 6.97 min/MS m/z: 150 M+ (50%), 131 (100%), 95 (20%), 121 (80%). The alcohol was derivatized with the CDA Alexakis reagent15 for 31P-NMR (161.992 MHz, CDCl3): δ 146 provide >99.8% ee. [α]20 d = +61 ( c 0.045, CHCl3). Lit.9a (S): [α]25 d = −60.5 (c 2.55, EtOH), 95% ee.
Reduction of 2- acetylthiophene using 1 mol% of borate ester 1
A solution of freshly redistilled 2-acetylthiophene (1.26 g, 10.0 mmol) in THF (5 mL) was added for 1 h using an infusion pump. After the usual work-up, the crude product was distilled in a Kugelrohr apparatus under vacuum to give the final product (+)-(R)-1-(thiophen-2-yl)ethanol as a colorless oil (1.109 g, 87%). Bp: 200 °C/1 mmHg; IR (cm−1) 3342 (OH), 2974 (CH3), 1435 (CH), 1370 (CH), 1066 (C-O), 849 (C-S); 1H NMR (400 MHz, CDCl3): δ̣ 1.53 (d, J = 6.4 Hz, 3H, Me), 2.91 (br. s, 1H, OH), 5.04 (q, J = 6.4 Hz, Me), 6.91–6.93 (m, 2H, C3’H and C4’H), 7.17–7.19 (m, 1H, C5’H); 13C NMR (100 MHz, CDCl3): δ̣ 25.2, 66.0, 123.1, 124.3, 126.6, 150.0. GC/MS m/z128 M+ (10%), 113 (32%), 95 (20%), 85 (100%). Chiral GC of O-acetyl derivative at T1 = 90 °C (10 min), gradient = 1 °C/min, T2 = 150 °C (35 min): RtR 20.2 min; RtS 17.4 min, indicated 96% ee. [α]23 d = +27 (c 2.5, CHCl3). With 10% of 1, it was obtained 92% yield and 98% ee [α]23 d = +26 (c 2.0, CHCl3). Lit.7d: [α]d = +24.2 (c 5, CHCl3), 100% ee.
Reduction of 1-(5-chlorothiophen-2-yl)ethanone using 1 mol% of spiroborate 1
A solution of freshly redistilled 1-(5-chlorothiophen-2-yl)ethanone (1.61 g, 10.0 mmol) in THF (5 mL) was added for 1h using an infusion pump. The crude product was distilled to give the (+)-(R)-1-(5-chlorothiophen-2-yl)ethanol as a white solid (1.49 g, 91%). Bp: 250 °C/1 mmHg. 1H NMR (400 MHz, CDCl3): δ̣ 1.53 (d, J = 6.4 Hz, 3H, Me), 2.46 (br. s, 1H, OH), 4.97 (q, J = 6.4 Hz, Me), 6.70 (dd, J = 3.8, 0.8 Hz, 1H, C3’H), 6.74 (d, J = 3.8 Hz, C4’H); 13C NMR (100 MHz, CDCl3): δ̣ 25.0, 66.4, 122.4, 125.6, 129.0, 148.6. Chiral GC of O-acetyl derivative at T1 = 70 °C (10 min), gradient = 4 °C/min, T2 = 190 °C (35 min): RtR 28.8 min; RtS 28.0 min, indicated 98% ee. [α]23 d = −27.4 (c 1.8, CHCl3). Lit.10e: [α]25 d = −29.2 (c 2.1, CHCl3), 54% ee.
Reduction of 3-acetylthiophene using 1 mol% of spiroborate 1
A solution of freshly redistilled 3-acetylthiophene (630 mg, 5.00 mmol) in THF (5 mL) was added for 1h using an infusion pump. After the usual work-up, the crude was distilled to give (+)-(R)-1-(thiophen-3-yl)ethanol as a colorless oil (550 mg, 86%). Bp: 200 °C/1 mmHg. 1H NMR (400 MHz, CDCl3): δ̣ 1.45 (d, J = 6.5 Hz, 3H, Me), 2.82 (br. s, 1H, OH), 4.86 (q, J = 6.4 Hz, CHMe), 7.04 (dd, J = 5.0, 1.2 Hz, 1H, C4’H), 7.10–7.12 (m, 1H, C2’H), 7.24 (dd, J = 5.0, 3.0 Hz, 1H, C5’H); 13C NMR (100 MHz, CDCl3): δ̣ 24.4, 66.3, 120.1, 125.7, 126.0, 147.3. Chiral GC of O-acetyl derivative at T1 = 120 °C (10 min), gradient = 5 °C/min, T2 = 185 °C (3 min): RtR 5.4 min; RtS 5.15 min, indicated 98.4 % ee. [α]23 d = +27° (c 1.5, CHCl3). Lit.9b [α]24 d = +33.8 (c 0.43, EtOH), 91% ee and Lit.9c [α]25 d = +44.7 (c 1.00, EtOH), 99.8% ee
Reduction of 2,5-dimethyl-3-acetylthiophene using 1 mol% of spiroborate 1.7d
A solution of freshly redistilled 2,5-dimethyl-3-acetylthiophen (1.54 g, 10.00 mmol) in THF (5 mL) was added to the reducing solution for 1h using an infusion pump. After the work-up, the crude was distilled to give (+)-(R)-1-2,5-dimethyl-3-yl)ethanol as a white solid (1.395 g, 89%). Bp: 152 °C/9 mmHg; mp 83–84 °C. 1H NMR (400 MHz, CDCl3): δ̣ 1.41 (d, J = 6.4 Hz, 3H, CHMe), 2.02 (br. s, 1H, OH), 2.33 (s, 3H, Me), 2.38 (s, 3H, Me), 4.86 (q, J = 6.6 Hz, 1H, CHMe), 6.66 (s 1H, C4’H); 13C NMR (100 MHz, CDCl3): δ̣ 12.6, 15.1, 23.9, 64.4, 123.6, 131.5, 136.0, 141.3. Chiral GC of O-acetyl derivative at T1 = 70 °C (10 min), gradient = 4 °C/min, T2 = 190 °C (35 min): RtR 27.4 min; RtS 27.2 min, indicated 95.6% ee. [α]23 d = +18 (c 3.5, CHCl3). Lit.9c: [α]21 d = +7.0 (c 0.45, CHCl3), 97% ee.
Reduction of 2,5-dichloro-3-acetylthiophene using 1 mol% of spiroborate 1
A solution of freshly redistilled 2,5-dichloro-3-acetylthiophene (1.95 g, 10.00 mmol) in THF (5 mL) was added for 1h using an infusion pump. After the work-up, the crude was distilled to give (+)-(R)-1-(5-chlorothiophen-2-yl)ethanol as a white solid (1.795 g, 91%). Bp: 143 °C/8 mmHg; mp 66–68 °C. 1H NMR (400 MHz, CDCl3): δ̣ 1.42 (d, J = 6.5 Hz, 3H, CHMe), 2.25 (br. s, 1H, OH), (q, J = 6.5 Hz, 1H, CHMe), 6.87 (s 1H, C4’H); 13C NMR (100 MHz, CDCl3): δ̣ 23.2, 64.1, 121.4, 124.4, 126.9, 142.5. Chiral GC of O-acetyl derivative at T1 = 90 °C (10 min), gradient = 4 °C/min, T2 = 190 °C (35 min): RtR 12.6 min; RtS 12.4 min, indicated 98.6% ee. [α]23 d = +24° (c 3.8, CHCl3). For 99.8% ee: [α]20 D = +30° (c 0.011, CHCl3). Lit.9b [α]21 d = +6.75 (c 0.2, CHCl3), 92.1% ee.
Reduction of 6,7-dihydrobenzo[b]thiophen-4(5H)-one using 1 mol% of spiroborate 1
A solution of freshly redistilled 3-acetylthiophen (760 mg, 5.00 mmol) in THF (5 mL) was added for 1 h using an infusion pump. After the work-up, the residue was under vacuum to give (+)-(R)-4,5,6,7-tetrahydrobenzo[b]thiophen-4-ol as a white solid (716 mg, 93%). Bp: 152 °C/6 mmHg; mp 57–59 °C. 1H NMR (400 MHz, CDCl3): δ̣ 1.1.75–2.10 (m, 5H, C5H, C6H and OH), 2.67–2.84 (m, 2H, C7H), 4.76 (br s, 1H, C4H), 7.01 (d, J = 5.2 Hz, C3H), 7.08 (d, J = 5.2 Hz, C2H); 13C NMR (100 MHz, CDCl3): δ̣ 19.9, 25.0, 32.3, 65.4, 122.7, 122.6, 138.0, 138.7. Chiral GC of O-acetyl derivative at T1 = 110 °C (10 min), gradient1 = 1 °C/min, T2 = 120 °C (10 min), gradient2 = 0.5 °C/min, T3 = 130 °C (20 min), gradient3 = 10 °C/min, T4 = 150 °C/min (20 min): RtR 43.4; RtS 43.8 min, indicated 94% ee. [α]23 d = −7° (c 1, CHCl3).
Reduction of thiochroman-4-one using 1 mol% of spiroborate 1
A solution of freshly redistilled thiochroman-4-one (1.64 g, 10.0 mmol) in THF (5 mL) was added for 1 h using an infusion pump. After the usual work-up, the residue was distilled under vacuum to give (+)-(R)-thiochroman-4-ol as a white solid (1.53 g, 92%). Bp: 155 °C/4 mmHg; mp 78–80 °C. 1H NMR (400 MHz, CDCl3): δ̣ 1.96–2.04 (m, 1H, C3H), 2,21 (br s, 1H, OH), 2.24–2.31 (m, 1H, C3H), 2.79–2.84 (m, 1H, C2H), 3.22–3.30 (m, 1H, C2H), 4.72 (br tr, J = 3.9 Hz, 1H, C4H), 7.01–7.05 (m, 1H, C8H), 7.07–7.16 (m, 2H, C7H and C6H), 7.24–7.28 (m, 1H, C5H); 13C NMR (100 MHz, CDCl3): δ̣ 21.5, 30.0, 66.5, 124.2, 126.7,128.4, 129.7, 120.4, 133.2, 134.6; Chiral GC of O-acetyl derivative at T1 = 150 °C gradient = 1 °C/min, T2 = 185 °C: RtR 14.97 min; RtS 15.5 min, indicated 98.7% ee. [α]23 d = +134 (c 1.9, CHCl3). Lit.9a (S): [α]25 d = − 113.1 (c 0.46, CHCl3), 90% ee.
4.4.12 Reduction of 6-chlorothiochroman-4-one using 1 mol% of spiroborate 1
A solution of freshly redistilled 6-chlorothiochroman-4-one (995 mg, 5.00 mmol) in THF (5 mL) was added for 1 h using an infusion pump. After the work-up, the residue was distilled under vacuum to give (+)-(R)-6-chlorothiochroman-4-ol as a white solid (920 g, 92%). Mp 102–104 °C. 1H NMR (400 MHz, CDCl3): δ̣ 2.02–2.10 (m, 1H, C3H), 2.24–2.30 (m, 1H, C3H), 2.59 (br. s, 1H, OH), 2.87–2.93 (m, 1H, C2H), 3.22–3.28 (m, 1H, C2H), 4.71 (dd, J = 4.9, 3.1 Hz, 1H, C4H), 7.07 (d, J = 8.4 Hz, 1H, C8H), 7.15 (dd, J = 8.4, 2.1 Hz, 1H, C7H), 7.35 (d, J = 1.8 Hz, 1H, C5H); 13C NMR (100 MHz, CDCl3): δ̣ 21.8, 30.0, 66.4, 127.9, 128.5, 129.7, 129.9, 131.8, 136.3; Chiral GC of O-acetyl derivative at T1 = 150 °C gradient = 1 °C/min, T2 = 185 °C: RtR 25.6 min; RtS 26.7 min, indicated 98.7% ee. [α]23 d = +79 (c 2.2, CHCl3).
Reduction of (E)-4-phenylbut-3-en-2-one using 10% of spiroborate 1
A solution of (E)-4-phenylbut-3-en-2-one (730 g, 5.0 mmol) in THF (5 mL) was added at 0 °C for 1 h using an infusion pump. The reaction mixture was stirred at 25 °C over 30 min, then cooled at 0 °C and quenched with methanol (5 mL), concentrated. The residue was dissolved in methanol (10 mL) and refluxed over 5 h, concentrated, re-dissolved in methanol (10 mL) and concentrated again. The residue was distilled in a Kugelrohr apparatus under vacuum to give (+)-(R,E)-4-phenylbut-3-en-2-ol as a pale yellow oil (543 mg, 73%). Chiral GC of O-acetyl derivative at T1 = 90 °C gradient = 4 °C/min, T2 = 190 °C (35 min: RtR 37.4 min; RtS 36.5 min, indicated 80% ee. [α]23 d = +25.6 (c 4, CHCl3). Lit.10g [α]d = +27 (c 0.5, CHCl3) 84% ee, Lit.10f (S) [α]d = −32.2 (c 5, CHCl3) 81% ee.
Reduction of (Z)-4-(furan-3-yl)but-3-en-2-one using 10% of spiroborate 1
A solution of (Z)-4-(furan-2-yl)but-3-en-2-one (1.36 g, 10.0 mmol) in THF (5 mL) was added to the reducing solution at 0 °C for 1 h using an infusion pump. After the usual work-up, the crude was distilled in a Kugelrohr apparatus under vacuum to give (R,Z)-4-(furan-2-yl)but-3-en-2-ol as a yellow oil (1.14 g, 83%). 1H NMR (400 MHz, CDCl3): δ̣ 1.32 (d, J = 6.4 Hz, 1H), 2.26 (br. s, 1H, OH), 4.39–4.45 (m, 1H), 6.15–6.23 (m, 2H), 6.32–6.41 (m, 2H), 7.32 (d, J = 1.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ̣ 23.4, 68.3, 107.9, 111.3, 117.6, 132.4, 141.9, 152.5; Chiral GC of O-acetyl derivative at T1 = 90 °C gradient = 4 °C/min, T2 = 190 °C: RtR 8.5 min; RtS 7.7 min, indicated 83% ee. [α]23 d = +24 (c 2.5, CHCl3).
Reduction of (E)-4-(thiophen-3-yl)but-3-en-2-one using 10% of spiroborate 1
A solution of (Z)-4-(thiophen-2-yl)but-3-en-2-one (1.52 g, 10.0 mmol) in THF (5 mL) was added at 0 °C for 1h using an infusion pump. After the work-up, the residue was distilled in a Kugelrohr apparatus under vacuum to give the (R,E)-4-(thiophen-2-yl)but-3-en-2-ol as a yellow oil (1.14 g, 83%). 1H NMR (400 MHz, CDCl3): δ̣ 1.32 (d, J = 6.4 Hz, 1H), 2.26 (br. s, 1H, OH), 4.36–4.43 (m, 1H), 6.07 (dd, J = 15.8, 6.4 Hz), 6.65 (d, J = 15.8 Hz, 1H), 6.90–6.94 (m, 2H), 7.11–7.12 (m, 1H); 13C NMR (100 MHz, CDCl3): δ̣ 23.3, 68.4, 122.5, 124.2, 125.7, 127.3, 141.8; Chiral GC of O-acetyl derivative at T1 = 110 °C gradient = 1 °C/min, T2 = 140 °C: RtR 25.3 min; RtS 24.2 min, indicated 80% ee. [α]23 d = +18.2 (c 2.3, CHCl3). Lit.9c [α]25 d = +41.3 (c 0.49, CHCl3), 91% ee.
Acknowledgments
Financial support by the National Institute of Health through their MBRS (GM 08216) and AABRE (NC P20 RR-016470) grants is greatly appreciated. We express our special gratitude to the NSF-MRI (01–07) and NIH-MBRS programs to have made possible the acquisition of a 400 MHz NMR spectrometer. The NSF-PREM (DMR-03537730), NIH-INBRE, NIH-RISE, NIH-MARC and NSF-AMP undergraduate student’s support is also gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Bauer K, Garbe D, Horst S. Common Fragrance and Flavor Materials. 3th Ed. New York: Wiley-VCH Verlag; 1997. [Google Scholar]; (b) Macor JE, editor. Annual Reports In Medicinal Chemistry. Volume 42. Amsterdam: Elsevier Academic Press; 2007. [Google Scholar]
- 2.(a) Abrams JN, Babu RS, Guo H, Le D, Le J, Osbourn JM, O'Doherty GA. J. Org. Chem. 2008;73:1935. doi: 10.1021/jo702476q. [DOI] [PubMed] [Google Scholar]; (b) Guo H, O'Doherty GA. Org. Lett. 2006;8:1609. doi: 10.1021/ol0602811. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Noyori R, Hashiguchi S. Acc. Chem. Res. 1997;30:97. [Google Scholar]; (d) Hwang S-K, Juhasz A, Yoon S-H, Bodor N. J. Med. Chem. 2000;43:1525. doi: 10.1021/jm9904654. [DOI] [PubMed] [Google Scholar]; (e) Weerawarna SA, Geisshusler SM, Murthy SS, Nelson WL. J. Med. Chem. 1991;34:3091. doi: 10.1021/jm00114a019. [DOI] [PubMed] [Google Scholar]
- 3.Bymaste FP, Beedle EE, J. F, Findlay J, Gallagher PT, Krushinski JH, Mitchell S, Robertson DW, Thompson DC, Wallace L, Wong DT. Bioorg. Med. Chem. Lett. 2003;13:4477. doi: 10.1016/j.bmcl.2003.08.079. [DOI] [PubMed] [Google Scholar]
- 4.Jones TK, Mohan JJ, Xavier LC, Blacklock TJ, Mathre DJ, Sohar PE, Turner Jones T, Reamer RA, Roberts FE, Grabowski EJJ. J. Org. Chem. 1991;56:763. [Google Scholar]
- 5.(a) Ohemeng KA, Appollina MA, Nguyen VN, Schwender CF, Singer M, Steber M, Ansell J, Argentieri D, Hageman W. J. Med. Chem. 1994;37:3663. doi: 10.1021/jm00047a023. [DOI] [PubMed] [Google Scholar]; (b) Stewart AO, Bhatia PA, Martin JG, Summers JB, Rodriquez KE, Martin MB, Holms JH, Moore JL, Craig RA, James DT, Ratajczyk K, Mazdiyasni H, Kerdesky FAJ, DeNinno SL, Maki RG, Bouska JB, Young PR, Lanni C, Bell RL, Carter GW, Brooks CDW. J. Med. Chem. 1997;40:1955. doi: 10.1021/jm9700474. [DOI] [PubMed] [Google Scholar]; (c) Bosiak MJ, Krzemiński MP, Jaisankarand P, Zaidlewicz M. Tetrahedron: Asymmetry. 2008;19:956. [Google Scholar]
- 6.Marchand B, Gargouil YM. FR Pat 2588260. 1987 CAN 108:37647. [Google Scholar]
- 7.(a) Paizs C, Tosa M, Bódai V, Szakács G, Kmecz I, Simándi B, Majdik C, Novák L, Irimie F-D, Poppe L. Tetrahedron: Asymmetry. 2003;14:1943. [Google Scholar]; (b) Toşa M, Pilbák S, Moldovan P, Paizs C, Szatzker G, Szakács G, Novák L, Irimie F-D, Poppe László. Tetrahedron: Asymmetry. 2008;19:1844. [Google Scholar]; (c) Drueckhammer DG, Barbas CF, III, Nozaki K, Wong CH, Wood C, Ciufolini Y, Ciufolini MA. J. Org. Chem. 1988;53:1607. [Google Scholar]; (d) Fantin G, Fogagnolo M, Medici A, Pedrini P, Poli S, Gardini F. Tetrahedron: Asymmetry. 1993;4:1607. [Google Scholar]; (e) Boyd DR, Sharma ND, Boyle R, Malone JF, Chima J, Dalton H. Tetrahedron: Asymmetry. 1993;4:1307. [Google Scholar]; (f) Szigeti M, Toke ER, Turoczi MC, Nagy V, Szakacs G, Poppe L. ARKIVOC. 2008;3:54. [Google Scholar]; (g) Kusakabe M, Kitano Y, Kobayashi Y, Sato F. J. Org. Chem. 1989;54:2085. [Google Scholar]
- 8.(a) For an extensive review see: Nakamura K, Yamanaka R, Matsuda T, Harada T. Tetrahedron: Asymmetry. 2003;14:2659. Martín-Matute B, Edin M, Bogár K, Kaynak FB, Bäckvall J-E. J. Am. Chem. Soc. 2005;127:8817. doi: 10.1021/ja051576x. Toşa MI, Podea PV, Paizs C, Irimie FD. Tetrahedron: Asymmetry. 2008;19:2068.
- 9.(a) Evans DA, Michael FE, Tedrow JS, Campos KR. J. Am. Chem. Soc. 2003;125:3534. doi: 10.1021/ja012639o. [DOI] [PubMed] [Google Scholar]; (b) Xu Y, Clarkson GC, Docherty G, North CL, Woodward G, Wills M. J. Org. Chem. 2005;70:8079. doi: 10.1021/jo051176s. [DOI] [PubMed] [Google Scholar]; (c) Koizumi M, Yoshida M, Noyori R. Org. Lett. 2000;2:1749. doi: 10.1021/ol0000814. [DOI] [PubMed] [Google Scholar]
- 10.(a) Daverio P, Zanda M. Tetrahedron: Asymmetry. 2001;12:2225. [Google Scholar]; (b) Kanth JVB, Brown HC. Tetrahedron. 2002;58:1069. [Google Scholar]; (c) Matteson DS. Stereodirected Synthesis with Organoboranes. Berlin: Springer-Verlag; 1995. [Google Scholar]; (d) Singh VK. Synthesis. 1992;7:605. [Google Scholar]; (e) Cherng Y-J, Fang J-M, Lu T-J. J.Org. Chem. 1999;64:3207. doi: 10.1021/jo982403b. [DOI] [PubMed] [Google Scholar]; (f) Brown HC, Chandrasekharan J, Ramachandran PV. J. Am. Chem. Soc. 1988;110:1539. [Google Scholar]; (g) Mastranzo VM, Quintero L, Anaya de Parrodi C, Juarist E, Walsh PJ. Tetrahedron. 2004;60:1781. [Google Scholar]
- 11.Cho BT. Chem. Soc. Rev. 2009;443 doi: 10.1039/b811341f. [DOI] [PubMed] [Google Scholar]; (b) Cho BT. Tetrahedron. 2006;62:7621. [Google Scholar]; (c) Glushkov VA, Tolstikov AG. Russ. Chem. Rev. 2004;73:581. [Google Scholar]; (d) Corey EJ, Helal CJ. Angew. Chem. Int. Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 12.(a) Quallich JG, Woodall TM. Tetrahedron Lett. 1993;34:785. [Google Scholar]; (b) Mathre DJ, Thompson AS, Douglas AW, Hoogsteen K, Carroll JD, Corley EG, Grabowski EJJ. J. Org. Chem. 1993;58:2880. [Google Scholar]; (c) Bichlmaier I, Siiskonen A, Finel M, Yli-Kauhaluoma J. J. Med. Chem. 2006;49:1825. doi: 10.1021/jm051142c. [DOI] [PubMed] [Google Scholar]
- 13.(a) Stepanenko V, Ortiz-Marciales M, Correa W, De Jesús M, Espinosa S, Ortiz L. Tetrahedron: Asymmetry. 2006;17:112. [Google Scholar]; (b) Ortiz-Marciales M, Stepanenko V, Correa W, De Jesús M, Espinosa S. U.S. Patent Application 11/512,599. 2006 Aug 30; [Google Scholar]; (c) Stepanenko V, De Jesús M, Correa W, Vázquez C, Guzman I, Vazquez C, De la Cruz W, Ortiz-Marciales M, Barnes CL. Tetrahedron Lett. 2007;48:5799. doi: 10.1016/j.tetlet.2007.06.086. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Stepanenko V, Ortiz-Marciales M, De Jesús M, Correa W, Vázquez C, Ortiz L, Guzmán I, De la Cruz W. Tetrahedron: Asymmetry. 2007;18:2738. doi: 10.1016/j.tetasy.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Stepanenko V, Ortiz-Marciales M, Barnes CL, Garcia C. Tetrahedron Lett. 2009;50:995. doi: 10.1016/j.tetlet.2008.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Huang K, Ortiz-Marciales M, Correa W, Pomales E, López XY. J. Org. Chem. 2009;74:4195. doi: 10.1021/jo900666r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demir AS, Sesenoglu Ö, Gerçek-Arkin Z. Tetrahedron: Asymmetry. 2001;12:2309. [Google Scholar]
- 15.(a) Alexakis A, Frutos JC, Mutti S, Mangeney P. J. Org. Chem. 1992;57:1224. [Google Scholar]; (b) Alexakis A, Frutos JC, Mutti S, Mangeney P. J. Org. Chem. 1994;59:3326. [Google Scholar]
- 16.Corey EJ, Bakshi RK. Tetrahedron Lett. 1990;31:611. [Google Scholar]