

Many classes of biomolecules derive their biological activity from the synergistic effects of carbohydrate and non-carbohydrate (aglycone) functionality.[1] From the standpoint of chemical synthesis, the assembly of the aglycone and attachment of a carbohydrate (glycosylation) are viewed as distinct operations. As a result, the independent synthesis of a suitably-protected aglycone and a suitably-protected carbohydrate is typically followed by a separate sequence involving Lewis acid activation of the sugar anomeric substituent and assembly of the O-glycoside bond via addition of a hydroxyl-bearing aglycone.[2] The powerful glycal oxidation method similarly involves addition of a hydroxyl nucleophile to the electrophilic anomeric position.[3] An important complement to these strategies involves intramolecular aglycone delivery.[4] Seminal work from Hindsgaul[5] and Ogawa[6] using acetal linkages and from Stork[7] and Bols[8] using silane linkages demonstrated that intramolecular aglycone delivery strategies provide a powerful entry to cis-1,2 glycoside linkages, namely the synthetically challenging β-mannose and α-glucose configurations. Whereas the intramolecular strategies involve glycoside bond assembly directly from an acetal- or silyl-protected hydroxyl, preparation of the tethered aglycone-carbohydrate assembly is ultimately derived from a free hydroxyl on the aglycone earlier in the synthesis. An alternate strategy involving O-alkylation of a C-1-O-hemiacetal nucleophile with an electrophilic aglycone provides a powerful entry to glycoconjugates and oligosaccharides, although this method also requires that potentially nucleophilic sites in the aglycone are protected.[9] As a complement to all of the above strategies, a glycosylation method that does not require a nucleophilic free hydroxyl on the aglycone at any point in the synthesis, and that tolerates spectator free hydroxyls on the aglycone, could have important implications as a strategy for native glycoside bond construction that is orthogonal to conventional glycosylation methods.[10]
With this challenge in mind, we were attracted to the notion of developing transition metal-catalyzed hydrosilylations of ketones by silyl hydride reagents that possess glycosyl donors as a silyl substituent. Such a strategy could allow site-selective installation of installing stereochemical features of the aglycone via reduction of the carbonyl. Herein we describe the efficient synthesis of “sugar silanes” to enable such a strategy, along with the first examples of the direct glycosylation of ketones as a fundamentally new and orthogonal strategy for preparing glycoside bonds.
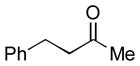
Glycosyl donor reagents that possess a C-2 free hydroxyl, protecting groups at the 3, 4, and 6-positions, and a thioalkyl anomeric substituent are readily prepared in the glucose or mannose configurations by known procedures.[11] The C-2 hydroxyl may be silylated in near quantitative yield using commercially available Me2SiHCl and Et3N in dichloromethane to afford reagents 1 or 2 (Scheme 1). The synthesis of 1 or 2 may be performed on multigram scale, requiring only a single chromatographic purification. While 1 and 2 are not stable to chromatography, they are obtained in pure form and may be stored for months with comparable results in subsequent reactions compared with reagents that are freshly prepared.
Scheme 1.

Preparation of Sugar Silane Reagents
In this intial study, we first examined ketone hydrosilylations with reagents 1a and 1b using either nickel[12] or copper[13] catalysts with IMes as the ligand, generated from 1,3-dimesitylimidazolium chloride, and KO-tBu or NaO-tBu. A variety of unhindered ketones underwent efficient couplings with the catalyst derived from Ni(COD)2 and IMes in THF, using Ti(O-iPr)4 as a Lewis acidic additive (Table 1) to afford hydrosilylation products 4 (starting from glucose) or 5 (starting from mannose). Control experiments demonstrated that no conversion was observed in the absence of the nickel catalyst. The procedure (Scheme 2) was generally effective with unhindered ketones, whereas the corresponding copper-IMes catalyst, generated in toluene following the procedure from Nolan,[13] was more effective with hindered ketones. Once substrates 4 or 5 were prepared via hydrosilylation, intramolecular glycosylation was carried out using N-iodosuccinimide and trimethylsilyltriflate with 2,6-di-t-butyl-4-methylpyridine (2,6-DTBMP) in dichloromethane at −40 to 0 °C to produce α-glucosides 6 (from 4) or β-mannosides 7 (from 5).[14] As described below, both ethylthio and phenylthio sugar silanes 1a/b and 2a/b were similarly effective in hydrosilylations, although the phenylthio donors were more effective in subsequent intramolecular glycosylations when hindered ketones were employed.
Table 1.
Ketone Glycosylations.
| entry | Ketone 3 | Silyl ether 4 or 5c (% yield) | Glycoside 6 or 7c,d (% yield) |
|---|---|---|---|
 |
 |
 |
|
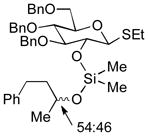
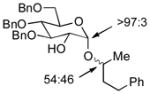
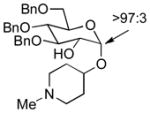
| 1 | 3a | 4a (97 %)a | 6a (97 %) |
 |
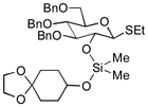 |
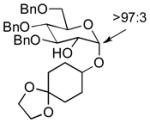 |
|
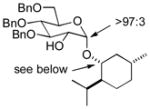
| 2 | 3b | 4b (96 %)a | 6b (82 %) |
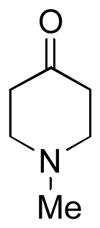 |
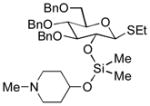 |
 |
|
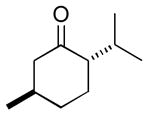
| 3 | 3c | 4c (99 %)a | 6c (70 %) |
 |
 |
 |
|
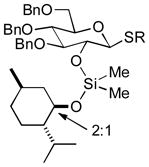
| 4 | 3d | 4d R = Et, 68 %)b | 6d (20 %, 4:1) |
| 5 | 3d | 4e (R = Ph, 64 %)b | 6d (72 %, 5:1) |
 |
 |
||
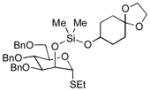
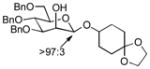
| 6 | 3b | 5a (86 %)a | 7a (58 %) |
 |
 |
||
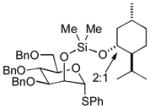
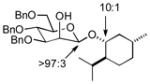
| 7 | 3d | 5b (75 %)b | 7b (74 %) |
Method A was employed: Ni(COD)2 (10 mol %), IMes·HCl (10 mol %), KO-tBu (10 mol %), Ti(O-iPr)4 (1.1 equiv), silane 1 or 2 (1.1 equiv), ketone (1.0 equiv), THF (0.1 M), rt, 3–13 h.
Method B was employed: CuCl (5 mol %), IMes·HCl (5 mol %), NaO-tBu (10 mol %), silane 1 or 2 (1.1 equiv), ketone (1.0 equiv), toluene (0.12 M), rt, 4–8 h.
In cases where diastereomeric mixtures are present, the major isomer is depicted.
Glycosylation procedure: Compound 4 or 5 (1.0 equiv), N-iodosuccinimide (1.3 equiv), 2,6-di-t-butyl-4-methylpyridine (2,6-DTBMP, 2.0 equiv), trimethylsilyl triflate (1.2 equiv), CH2Cl2, −40 to 0 °C, then nBu4NF.
Scheme 2.

Strategy for Conversion of Ketones to Glycosides.
As a first example, coupling with benzyl acetone proceeded in high yield with the Ni(0)-IMes catalyst to afford a 54:46 mixture of diastereomers of 4a epimeric at the newly formed stereogenic center, thus illustrating that sugar chirality has little impact on the diastereoselectivity of the hydrosilylation (Table 1, entry 1). Glycosylation of 4a provided α-glucoside 6a in 97 % isolated yield with excellent diastereoselectivity at the anomeric position. Cyclic acetals and basic tertiary amines were tolerated in high yielding transformations with the Ni(0)-IMes catalyst to produce 4b and 4c (entries 2–3), and glycosylation of these substrates afforded the α-glucosides 6b and 6c in high yield and excellent diastereoselectivity. Hydrosilylations of (−)-menthone (3d) were low yielding with the nickel catalyst system (ca. 20–25 % yield); however, the more reactive Cu-IMes catalyst led to faster and higher yielding reactions (entry 4). Using either ethylthio or phenylthio sugar silanes 1a or 1b, hydrosilylations using 3d were effective to produce 4d or 4e in good yield with 2:1 diastereoselectivity. Subsequent glycosylations, however, were much higher yielding with phenylthioglycosyl donor 4e, which generated product 6d in 72 % isolated yield, compared with 20 % isolated yield from 4d. The enhancement of diastereoselectivity observed in the glycosylation is derived from significantly different rates of glycosylation of the two diastereomers of 4d or 4e. Efficiency of the process in β-mannosylations was next examined. Hydrosilylation of ketone 3b with mannose silane 2a was efficient with the Ni-IMes catalyst, affording product 5a in 86% isolated yield (entry 5). Glycosylation of 5a was moderately effective to generate exclusively the β-mannoside 7a in 58 % isolated yield with excellent control of the anomeric stereochemistry. Hydrosilylation of (−)-menthone (3d) with mannose silane 2b using the Cu-IMes catalyst afforded 5b in 75 % isolated yield with 2:1 diastereoselectivity (entry 6), and subsequent glycosylation afforded β-mannoside 7b in 74 % isolated yield and excellent control of the anomeric configuration. Enhancement of the diastereomeric ratio derived from carbonyl reduction was again noted as described above (entry 4). These examples suggest that a broad range of ketones may be efficiently converted to either α-glucosides or β-mannosides.[15,16]
As noted above, an important implication of a glycosylation procedure that does not require addition of a free hydroxyl on the aglycone to a glycosyl donor is its potential to allow site selective glycosylation of aglycones that possess unprotected hydroxyls.[17,18] Silanes are well known to undergo ketone hydrosilylations[13] and alcohol dehydrogenative silylations[19] with a broad range of transition metal catalysts, although remarkably little quantitative data is available regarding the relative rates of the two processes. Our initial examinations of the Cu-IMes catalyst employed herein illustrated that addition of sugar silanes 1 and 2 are efficient with both ketones (hydrosilylation) and alcohols (dehydrogenative silylation), but generally fastest with unhindered hydroxyls. Alternatively, with the Ni-IMes catalyst, hydrosilylations of unhindered ketones proceed much more rapidly than dehydrogenative silylations of alcohols.[20]
In order to illustrate the opportunity for site-selective glycosylation of a hydroxy ketone, dihydrotestosterone (7) was subjected to the nickel-catalyzed hydrosilylation procedure, and only the ketone functionality was affected (Scheme 3). Starting with glucosilane 1a, silyl ether 8a was prepared in 89% isolated yield with 5:1 diastereoselectivity. Treatment of 8a to the conditions for intramolecular glycosylation afforded α-glucoside 9a in 95% isolated yield with complete control of anomeric configuration. Purification of the products of intramolecular glycosylation involves treatment with nBu4NF, so any competitive silylation of the free hydroxyl by TMSOTf during the glycosylation event is inconsequential since the site selectivity is derived from the previous hydrosilylation event. Using the same hydroxyketone 7 combined with mannosilane 2a, efficient nickel-catalyzed site-selective hydrosilylation proceeds to generate product 8b in 80 % isolated yield with 6:1 diastereoselectivity. As anticipated based on the lack of impact of sugar structure in controlling the hydrosilylation diastereoselectivity (Table 1, entry 1), diastereoselectivities involving hydrosilylation of chiral substrate 7 were comparable with both gluco- and mannosilanes 1a and 2a. Treatment of compound 8b to the glycosylation conditions afforded β-mannoside 9b in 92 % isolated yield with complete control of anomeric stereochemistry.
Scheme 3.

Hydroxyketone Site-selective Glycosylation.
Since the above example (Scheme 3) involves functionalization of an inherently biased substrate with a highly hindered free hydroxyl, we examined the site selectivity of a simpler substrate 10, which possess both an unhindered ketone and a primary hydroxyl. In this instance, we found highly complementary behavior of the nickel and copper catalytic systems. Treatment of 10 with glucosilane 1a using the Ni-IMes catalyst led to clean ketone hydrosilylation, affording product 11 in 86 % isolated yield, whereas the corresponding reaction of 10 and 1a with the Cu-IMes catalyst afforded product 12 from dehydrogenative silylation of the alcohol in 57 % isolated yield, along with 7 % yield of the bis-silylated product derived from reaction of both the ketone and alcohol. Products 11 and 12 were then converted to glycosides 13 and 14 by the standard procedure described above. This catalyst-controlled reversal of chemoselectivity in hydroxyketone functionalization with silanes is unprecedented to our knowledge.[21]
In summary, a new method has been developed that allows the conversion of ketones to native glycoside bonds without the intermediacy of free alcohols. Starting from a hydroxy ketone, the site-selective installation of a glycoside bond at only the ketone or at only the alcohol is possible based on catalyst structure without separate steps involving the protection and deprotection of the alcohol functionality. Additionally, generation of a new stereogenic center, subject to substrate-controlled diastereoselection, is possible during the hydrosilylation-glycosylation sequence, thus allowing aglycone tailoring and glycoside bond installation to be accomplished in a single strategy. We anticipate that these advances will facilitate the rapid synthesis of various classes of synthetic and natural product-derived glycoconjugates. Application of this concept to other catalytic processes involving sugar silanes, including C-C bond-forming processes, is in progress.
Experimental Section
General Procedure for the Ni(COD)2/IMes Promoted Hydrosilylation of Ketones: A solid mixture of Ni(COD)2 (10%), IMes·HCl (10%), and KO-tBu (10%) was dissolved in dry THF (0.02M) at rt under an inert atmoshphere (N2), and stirred for 10–15 minutes until the catalyst mixture was a dark blue color. Ti(O-iPr)4 (1.1–2.2 equiv) was then added to the catalyst mixture followed by the addition of the sugar silane (1.1 equiv), and ketone (1.0 equiv) as a solution in dry THF (0.2M). Upon completion of the reaction, as monitored by TLC, the reaction mixture was filtered through a short plug of silica gel with a mixture of EtOAc/hexanes and concentrated by rotary evaporation. The resulting residue was purified via flash chromatography (SiO2) to afford the desired product. Note – When doing the site-selective hydrosilylation of a ketone in the presense of a free hydroxyl group, the use of 2.2 equiv of Ti(O-iPr)4, and a 0.05 M solution in THF results in higher yields of the desired product.
General Procedure for the CuCl/IMes Promoted Hydrosilylation of Ketones: A solid mixture of CuCl (5%), IMes·HCl (5%) and KO-tBu (10%) was dissolved in dry toluene (0.015M) at rt under an inert atmosphere (N2), and stirred for 20 minutes. A mixture of ketone (1.0 equiv) and silane (1.1 equiv) was dissolved in dry toluene (0.2M), the catalyst was then added to this mixture as a solution in a minimum of dry toluene. Upon completion of the reaction, as monitored by TLC, the reaction mixture was filtered through a short plug of silical gel with a mixture of EtOAc/hexanes and concentrated by rotary evaporation. The resulting residue was purified via flash chromatography (SiO2) to afford the desired product.
Supplementary Material
Scheme 4.

Catalyst-Controlled Site Reversal.
Acknowledgments
The authors wish to acknowledge support from the National Institutes of Health (GM 57014) and from Thermo Fisher for a pilot project grant administered by the University of Michigan Life Sciences Institute.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.(a) Wong C-H, editor. Carbohydrate-Based Drug Discovery. Vol. 1 Wiley; Weinheim: 2003. [Google Scholar]; (b) Klyosov AA, Witczak ZJ, Platt D, editors. Carbohydrate Drug Design; ACS Symposium Series 932; Washington, DC: American Chemical Society; 2006. [Google Scholar]; (c) Galonic DP, Gin DY. Nature. 2007;446:1000–1007. doi: 10.1038/nature05813. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pratt MR, Bertozzi CR. Chem Soc Rev. 2005;34:58–68. doi: 10.1039/b400593g. [DOI] [PubMed] [Google Scholar]; (e) Herzner H, Reipen T, Schultz M, Kunz H. Chem Rev. 2000;100:4495–4538. doi: 10.1021/cr990308c. [DOI] [PubMed] [Google Scholar]; (f) Buskas T, Ingale S, Boons GJ. Glycobiology. 2006;16:113R–136R. doi: 10.1093/glycob/cwj125. [DOI] [PubMed] [Google Scholar]; (g) Borman S. Chem Eng News. 2006 September 4;84:17–26. [Google Scholar]
- 2.For reviews: Toshima K, Tatsuta K. Chem Rev. 1993;93:1503–1531.Cumpstey I. Carbohydrate Res. 2008;343:1553–1573. doi: 10.1016/j.carres.2008.04.031.For representative state-of-the-art glycosylation methods: Kahne D, Walker S, Cheng Y, Van Engen D. J Am Chem Soc. 1989;111:6881–6882.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223.Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5584.Schmidt RR, Michel J. Angew Chem Int Ed Engl. 1980;19:731–732.Mukaiyama T, Murai Y, Shoda SI. Chem Lett. 1981:431–432.Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523–1527. doi: 10.1126/science.1057324.
- 3.(a) Friesen RW, Danishefsky SJ. J Am Chem Soc. 1989;111:6656–6660. [Google Scholar]; (b) Halcomb RL, Danishefsky SJ. J Am Chem Soc. 1989;111:6661–6666. [Google Scholar]
- 4.(a) Jung KH, Müller M, Schmidt RR. Chem Rev. 2000;100:4423–4442. doi: 10.1021/cr990307k. [DOI] [PubMed] [Google Scholar]; (b) Zhu X, Schmidt RR. Angew Chem Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]; (c) Fairbanks AJ. Synlett. 2003:1945. [Google Scholar]; (d) Cumpstey I. Carb Res. 2008;343:1553. doi: 10.1016/j.carres.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Barresi F, Hindsgaul O. J Am Chem Soc. 1991;113:9376–9377. [Google Scholar]
- 6.Ito Y, Ogawa T. Angew Chem. 1994;33:1765–1767. [Google Scholar]
- 7.(a) Stork G, Kim G. J Am Chem Soc. 1992;114:1087–1088. [Google Scholar]; (b) Stork G, La Clair JJ. J Am Chem Soc. 1996;118:247–248. [Google Scholar]
- 8.(a) Bols M. J Chem Soc, Chem Commun. 1992:913–914. [Google Scholar]; (b) Bols M. Tetrahedron. 1993;49:10049–10060. [Google Scholar]; (c) Bols M. J Chem Soc, Chem Commun. 1993:791–792. [Google Scholar]; (d) Bols M, Hansen HC. Chem Lett. 1994:1049–1052. [Google Scholar]
- 9.(a) Schmidt RR. Angew Chem Int Ed. 1986;25:212–235. [Google Scholar]; (b) Ryan DA, Gin DY. J Am Chem Soc. 2008;130:15228–15229. doi: 10.1021/ja804589j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.We use the term “orthogonal” to refer to the glyosyl acceptor. Orthogonal glycosyl donors have previously been described: Kanie O, Ito Y, Ogawa T. J Am Chem Soc. 1994;116:12073.
- 11.(a) Callam CS, Lowary TL. J Org Chem. 2001;66:8961–8972. doi: 10.1021/jo010827r. [DOI] [PubMed] [Google Scholar]; (b) Bamhaoud T, Sanchez S, Prandi J. Chem Commun. 2000:659–670. [Google Scholar]; (c) Düffel A, Green LG, Ley SV, Miller AD. Chem Eur J. 2000;6:1416–1430. doi: 10.1002/(sici)1521-3765(20000417)6:8<1416::aid-chem1416>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 12.(a) Irrgang T, Schareina T, Kempe R. J Mol Cat A: Chem. 2006;257:48–52. [Google Scholar]; (b) Kong YK, Kim J, Choi S, Choi SB. Tetrahedron Lett. 2007;48:2033–2036. [Google Scholar]; (c) Chaulagain MR, Mahandru GM, Montgomery J. Tetrahedron. 2006;62:7560–7566. [Google Scholar]
- 13.Díez-González S, Nolan SP. Acc Chem Res. 2008;41:349–358. doi: 10.1021/ar7001655.Kaur H, Zinn FK, Stevens ED, Nolan SP. Organomet. 2004;23:1157–1160.Díez-González S, Kaur H, Zinn FK, Stevens ED, Nolan SP. J Org Chem. 2005;70:4784–4796. doi: 10.1021/jo050397v.See also: Lipshutz BH, Chrisman W, Noson K. J Organomet Chem. 2001;624:367–371.
- 14.(a) Ennis SC, Fairbanks AJ, Tennant-Eyles RJ, Yeates HS. Synlett. 1999:1387. [Google Scholar]; (b) Fügedi P, Garegg PJ, Lönn H, Norberg T. Glycoconjugate J. 1987;4:97. [Google Scholar]; (c) Veeneman GH, van Leeuwen SH, van Boom JH. Tetrahedron Lett. 1990;31:1331–1334. [Google Scholar]; (d) Konradsson P, Ododong UE, Fraser-Reid B. Tetrahedron Lett. 1990;31:4313–4316. [Google Scholar]; (e) Zhu T, Boons GJ. Org Lett. 2001;3:4201–4203. doi: 10.1021/ol016869j. [DOI] [PubMed] [Google Scholar]; (f) Geurtsen R, Coté F, Hahn MG, Boons GJ. J Org Chem. 1999;64:7828–7835. [Google Scholar]
- 15.Synthesis of C-glycosides via coupling processes starting with ketones is well precedented: Miquel N, Doisneau G, Beau JM. Angew Chem Int Ed. 2000;39:4111–4114. doi: 10.1002/1521-3773(20001117)39:22<4111::aid-anie4111>3.0.co;2-c.Miquel N, Doisneau G, Beau JM. Chem Commun. 2000:2347–2348. doi: 10.1002/1521-3773(20001117)39:22<4111::aid-anie4111>3.0.co;2-c.Brunckova J, Crich D. Tetrahedron. 1995;51:11945–11952.Herrera AJ, Rondón M, Suárez E. Synlett. 2007:1851–1856.Herrera AJ, Rondón M, Suárez E. J Org Chem. 2008;73:3384–3391. doi: 10.1021/jo702663w.
- 16.Using acetone or cyclohexanone as solvent, acetal-linked glycosides have been prepared: Aloui M, Fairbanks AJ. Chem Commun. 2001:1406–1407.
- 17.For a discussion of protecting group-free synthesis: Baran PS, Maimone TJ, Richter JM. Nature. 2007;446:404–408. doi: 10.1038/nature05569.
- 18.For an example of catalyst-controlled site-selective polyol derivatization: Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490.
- 19.(a) Lorenz C, Schubert U. Chem Ber. 1995;128:1267–1269. [Google Scholar]; (b) Schmidt DR, O’Malle SJ, Leighton JL. J Am Chem Soc. 2003;125:1190–1191. doi: 10.1021/ja0283201. [DOI] [PubMed] [Google Scholar]
- 20.For other nickel-catalyzed processes involving silanes that tolerate unprotected hydroxyls: Mahandru GM, Liu G, Montgomery J. J Am Chem Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n.Herath A, Montgomery J. J Am Chem Soc. 2008;130:8132–8133. doi: 10.1021/ja802844v.
- 21.The reversal of selectivity is not due solely to the presence of Ti(O-iPr)4 in the nickel-catalyzed procedure. Adding this reagent to the copper protocol led only to the corresponding isopropoxy sugar silane.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.