

Abstract
It now appears likely that soluble oligomers of amyloid-β1-42 peptide, rather than insoluble fibrils, act as the primary neurotoxin in Alzheimer’s disease (AD). Consequently, compounds capable of altering the assembly state of these oligomers (referred to as ADDLs) may have potential for AD therapeutics. Phenolic compounds are of particular interest for their ability to disrupt Aβ oligomerization and reduce pathogenicity. This study has focused on oleocanthal (OC), a naturally-occurring phenolic compound found in extra-virgin olive oil. OC increased the immunoreactivity of soluble Aβ species, when assayed with both sequence- and conformation-specific Aβ antibodies, indicating changes in oligomer structure. Analysis of oligomers in the presence of OC showed an upward shift in MW and a ladder-like distribution of SDS-stable ADDL subspecies. In comparison with control ADDLs, oligomers formed in the presence of OC (Aβ-OC) showed equivalent colocalization at synapses but exhibited greater immunofluorescence as a result of increased antibody recognition. The enhanced signal at synapses was not due to increased synaptic binding, as direct detection of fluorescently-labeled ADDLs showed an overall reduction in ADDL signal in the presence of OC. Decreased binding to synapses was accompanied by significantly less synaptic deterioration assayed by drebrin loss. Additionally, treatment with OC improved antibody clearance of ADDLs. These results indicate oleocanthal is capable of altering the oligomerization state of ADDLs while protecting neurons from the synaptopathological effects of ADDLs and suggest OC as a lead compound for development in AD therapeutics.
Keywords: Alzheimer’s disease, amyloid beta, oligomer, olive oil, synapse
Introduction
Alzheimer’s disease (AD) is a dementia that generally manifests spontaneously in later adulthood as a result of synaptic and neuronal loss, putatively due to beta-amyloid (Aβ) oligomers (oAβs) binding to postsynaptic sites (Terry et al., 1991; Lacor et al., 2007). Aβ was suggested to be the initiating factor in AD by Hardy and Higgins in 1992 in their “amyloid cascade hypothesis” in which they proposed that Aβ plaques were the toxins responsible for the neurodegeneration and dementia observed in AD (Hardy & Higgins, 1992). Subsequent experiments have implicated soluble Aβ oligomers (also referred to as ADDLs) as the toxic species responsible for the development of AD-associated pathology: abnormal dendritic spine morphology, synaptic receptor composition, and spine loss (Lacor et al., 2007); formation of reactive oxygen species (De Felice et al., 2007); tau hyperphosphorylation (De Felice et al., 2008), prolonged long-term depression (Wang et al., 2002); inhibition of long-term potentiation and cell death (Lambert et al., 1998). Oligomers from different sources have been shown to produce AD-consistent alterations in cell functioning. These sources include various synthetic preparations, naturally-secreted oAβs, transgenic mouse AD models, and human AD brain-derived extracts (Deshpande et al., 2006; Walsh et al., 2002; Oddo et al., 2003; Jacobsen et al., 2006; Knobloch et al., 2007; Lacor et al., 2004). These soluble oligomers have replaced insoluble plaques as the putative cause of AD in the new amyloid cascade hypothesis (Small, 1998; Hardy & Selkoe, 2002).
As the interaction of Aβ oligomers with neurons appears to be a critical step in the initiation of AD pathology, considerable research has focused on preventing the formation of toxic oligomeric species. Many natural compounds have been found to suppress pathology associated with oAβs (Yang et al., 2005; Wu et al., 2006). Studies using several phenolic compounds have shown their ability to prevent prefibrillar oligomerization, fibril formation, cytotoxicity in PC12 cells and in Tg2576 mice as well as cognitive decline typically seen in Tg2576 mice (De Felice et al., 2004; Yu et al., 2002; Wang et al., 2004; Wang et al., 2008; Ono et al., 2008). Salvianolic acid B, a polyphenolic compound derived from the root of Salvia miltiorrhiza, disrupts the aggregation of Aβ into fibrils and protects against the cytotoxic effects of high Aβ doses (Durairajan et al., 2008). Oleuropein, a chemical compound extracted from olive leaves, has exhibited non-covalent interaction with the Aβ1-40 peptide, although the structural or functional consequences of this interaction were not reported (Bazoti et al., 2006; Bazoti et al., 2008).
Oleocanthal (OC) is an olive oil-derived compound with several potentially neuroprotective properties: it is an amphipathic chemical compound similar to oleuropein, suggesting its ability to interact with Aβ and possibly alter oligomer structure or function; it exhibits non-selective COX inhibition, and it is an antioxidant (Beauchamp et al., 2005; Smith et al., 2005). Ibuprofen, another non-selective COX inhibitor, has been shown to reduce Aβ1-42 production in vitro and intraneuronal Aβ, cognitive deficits, and hyperphosphorylated tau in a transgenic mouse model of AD (Blasko et al., 2001; McKee et al., 2008). Additionally, due to the putative role of oxidative insult in the onset and perpetuation of AD, studies have indicated that diets rich in antioxidants can reduce risk of AD generation, while the traditional Mediterranean diet, rich in olive oil and monounsaturated fats, protects against age-related cognitive decline (Engelhart et al., 2002; Solfrizzi et al., 2006; Abdul et al., 2008). All of these properties make OC a candidate as a structural modifier of oAβs, and thus a possible AD therapeutic.
The current study explores the ability of OC to alter the structure of forming or pre-formed ADDLs and assesses the functional changes in ADDLs produced by these modifications using primary hippocampal neuron cultures. The results show that with OC, oAβs show altered structure, increased immunoreactivity, and lowered binding and synaptic toxicity. These changes have been found to facilitate the clearance of oAβs from synapses using oligomer-specific antibodies. OC appears to be a candidate for a lead compound in developing AD therapeutics.
Methods
Preparation of Aβ-Derived Diffusible Ligands (ADDLs)
Aβ1-42 peptides (American Peptide) were solubilized at room temperature in DMSO at a concentration of 5 mM. Cold Ham’s F12 media (Caisson, HF12-02), containing various concentrations of OC (100 μM to make Aβ-OC) or an equivalent volume of DMSO (to make ADDLs), was added to make a 100 μM solution of Aβ. After 24 h incubation at 4°C, the Aβ prep was centrifuged at 14,000 × g for 10 minutes to remove any large insoluble Aβ aggregates. The supernatant was kept and used as the ADDL preparation.
Preparation of Fluorescently-Labeled ADDLs
Lyopholized Aβ1-42 peptides with and without carboxyfluorescein (FAM) conjugated were solubilized at room temperature in DMSO and mixed at a 1:4 ratio, respectively, to a final concentration of 5 mM. Cold Ham’s F12 was added to make a 100 μM solution of Aβ. After 24 h incubation at 4°C, the Aβ prep was centrifuged at 14,000 × g for 10 minutes to remove any large insoluble Aβ aggregates. The supernatant was kept and used as the ADDL preparation. 50 μl aliquots were then dried using the low setting of a DNA Speed Vac and stored in dark at room temperature for future use. Aliquots were reconstituted in sterile water prior to use.
Dot Immunoblot Assay
0.5 mg lyophilized aliquots of OC were solubilized in DMSO to a concentration of 11 mM. The OC stock was then diluted in F12 10-fold, and dilutions in F12 containing 10% DMSO were performed to create the appropriate stocks for each molar concentration of OC used. Once all OC solutions were made with the appropriate concentrations, different Aβ solutions - either monomeric (lyophilized Aβ1-42 solubilized in DMSO and used immediately), oligomeric (ADDLs formed as described above), or fibrillar (lyophilized Aβ1-42 solubilized in water and incubated at 37°C for 5 days) - were added to a concentration at which the Aβ1-42 monomer would be present at 100 nM. After 15 minutes at 37°C, solutions were serially diluted and 1 μl samples were spotted in triplicate onto a nitrocellulose membrane. Immunoblotting was carried out as described below.
SDS-PAGE
Protein samples were run on 4-20% Tris-Glycine gels (Invitrogen, EC60285) using a running buffer containing 25 mM Tris, pH 8.3, 192 mM glycine, and 0.1% SDS. Silver staining was performed using the SilverXpress Silver Staining Kit (Invitrogen). For western blots, transfers were carried out using a transfer buffer containing 25 mM Tris, pH 8.3, and 192 mM glycine. 5% nonfat dry milk in 20 mM Tris-HCl, pH 7.4, 0.8% NaCl, 0.1% Tween-20 was used as blocking solution for dot and western blot analyses. Anti-oAβ conformation-specific antibodies NU1 (1.5 μg/ml; Lambert et al., 2007) and M89 (1:500; generated similar to Lambert et al., 2001) and anti-Aβ sequence-specific antibodies 6E10 (1:500; Signet, SIG-39320) and 4G8 (0.5 μg/ml; Signet, SIG-39220) were used in western and dot blots. Secondary labeling was carried out with HRP-conjugated antibodies (CGE), which were then developed with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific). Blots were visualized on a Kodak Image Station 440CF and analyzed with ImageJ (NIH).
Cell Treatments
Primary hippocampal cultures aged 18 days in vitro (DIV) were pretreated with 0, 0.1, 1, 10, or 100 μM OC or an equivalent DMSO control for 5 minutes before addition of ADDLs, Aβ-OC, or an equivalent DMSO control under the following conditions: hotspot assay - 500 nM for 3 h; drebrin loss - 500 nM for 24 h; tau phosphorylation - 500 nM for 4 h. Treated cells were washed twice with 1 ml warm neurobasal media. The third wash was left on and 1 ml of 3.7% formaldehyde was added for 10 minutes. This first fixing solution was removed and an additional ml of 3.7% formaldehyde was added for 10 minutes. Cells were then washed 5x with 1 ml PBS and stored at 4°C for no more than 2 weeks before immunocytochemical assays were performed.
Immunocytochemistry
For all immunocytochemical assays, the blocking solution contained 10% normal goat serum (NGS) diluted in PBS. To visualize intracellular epitopes, 0.1% triton X-100 was added to the blocking solution. Antibodies against the following were used: oAβs (conformation-specific) [NU1 (1.5 μg/ml) & M89 (1:500)], drebrin (1:100; MBL, D029-3), and PSD-95 (1:500; Affinity BioReagents, MA1-046). All Alexa-conjugated secondary antibodies were used at 1:500 in 10x diluted block for 1.5 h at RT. Coverslips were mounted in ProLong Gold antifade reagent with DAPI (Invitrogen), allowed to dry, and imaged with one of the following: Nikon Eclipse TE2000-U epifluorescent microscope, Leica DMRXE confocal microscope, or Leica IRE2 confocal microscopes. Images were then analyzed using either ImageJ, MetaMorph (Molecular Devices), Volocity (Improvision), or ImageSurfer (www.imagesurfer.org). Colocalization analysis was performed using the JaCoP plug-in in ImageJ (Bolte & Cordelieres, 2006).
Results
OC alters the immunoreactivity and structure of Aβ1-42 oligomers
Our first goal was to determine if OC could block the oligomerization of monomeric Aβ1-42 into ADDLs, as the soluble oligomers of Aβ appear to be the toxic agents responsible for the initiation of AD. We assessed oligomerization by dot blot immunoassay after adding Aβ1-42 to solutions containing increasing amounts of OC. Oligomers were detected by the previously characterized conformation-specific antibody NU1 (Lambert et al., 2007). Contrary to our expectations, Aβ1-42 immunoreactivity increased in the presence of OC at doses as low as 10 nM (Figure 1). Similar results on Aβ1-42 oligomerization were observed in dot blots using the antibodies M89, 4G8, and 6E10 (Figure 2). Antibody reactivity increased for both conformation- and sequence-specific antibodies, indicating an alteration in Aβ oligomerization.
Figure 1. Oleocanthal increases immunoreactivity of soluble, but not fibrillar Aβ.
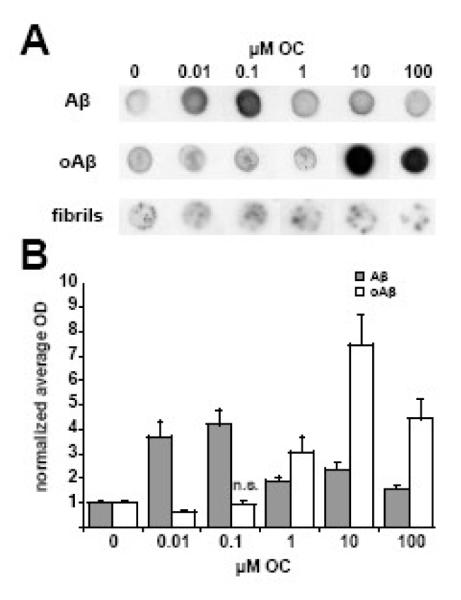
Monomeric, oligomeric, or fibrillar Aβ were added at 100 nM to increasing concentrations of OC solutions. Immunoreactivity was then assayed in a dot immunoblot assay. A significant increase in NU1 reactivity for both monomeric and oligomeric Aβ was observed after a 15 minute incubation. All OC incubations were significantly different than control except oligomeric Aβ with 0.1 μM OC. (A) Representative dot blot. (B) Mean ± SEM for five separate experiments.
Figure 2. Increased immunoreactivity of soluble Aβ brought on by OC is evident with multiple Aβ antibodies.

Monomeric and oligomeric Aβ were added at a concentration of 100 nM to solutions containing different OC concentrations. Immunoreactivity was assayed by dot blot using three Aβ antibodies with different epitope specificity. (A) Representative dot blot for 6E10 staining. (B) Box plots show the average integrated optical density ± SEM (arbitrary units) for ADDLs or monomeric Aβ incubated with oleocanthal for 15′ with 6E10. As observed with NU1, 6E10 immunoreactivity was increased with the addition of oleocanthal. Other antibodies (4G8 and NU2) were also tested and found to produce the same trend toward higher immunoreactivity.
The next experiment tested the interaction of OC with ADDLs rather than monomeric Aβ1-42. Results from dot blot analysis on preformed ADDLs were similar to that of Aβ1-42. However, to produce the increased immunoreactivity, higher OC concentrations were required (Figure 1). OC thus seemed less able to interact productively with Aβ1-42 as the aggregation state increased. To further investigate this idea, we carried out dot blot analysis using fibrillar Aβ1-42, the most aggregated form of Aβ1-42, in place of ADDLs. Fibrillar Aβ1-42 showed minimal to no change in immunoreactivity (Figure 1). The marginal changes observed in some experiments are most likely attributed to OC interactions with the limited pool of soluble oligomers present in the fibrillar preparation. These results indicate that OC is capable of changing the oligomer structure of soluble Aβ1-42 species during and after monomeric Aβ1-42 oligomerization.
This OC-induced change in immunoreactivity potentially could be associated with modifications in the size of Aβ oligomers (oAβs). To address this possibility, SDS-PAGE analysis was carried out on oAβ preparations with OC added during or after oligomerization. When added during oligomerization, OC was present at equimolar ratios. However, as OC appeared to be less able to interact with oAβ compared to monomer, preformed ADDLs were incubated with a 10-fold molar excess of OC (100 nM Aβ, 1 μM OC). In samples containing OC, silver stain analysis revealed a striking ladder-like distribution of oligomers (Figure 3A). Laddering occurred when OC was present during the 24 h oligomerization period (Aβ-OC) or following a 1 h incubation with preformed ADDLs (Figure 3A). At 2 h, ADDLs incubated with OC showed a shift toward high MW species, with a reduction of smaller species. OC could have produced this change in SDS-PAGE profile by generating larger oligomers from small species (<18 kDa) or by enhancing SDS-stability of detergent-generated fragments of larger oligomers. Previous work has shown it is possible to separate large from small oligomers using ultrafiltration (Lacor et al., 2007). We therefore used a 50 kDa filter to separate ADDLs incubated with OC into large and small oligomers and compared the SDS-PAGE profile of the filtrate and retentate. As shown in Figure 3B, the SDS-stable bands seen in the presence of OC originate from oligomers >50 kDa. Additionally, the filtrate fractions had no Coomassie-detectable proteins and were nearly absent of oAβ bands. This, along with the an enlarged pellet in the Aβ-OC preparation indicated that OC generated larger oAβs, possibly to the point of insolubility (data not shown). The reduced solubility may lead to a decrease in toxicity. When assayed after a 1 h incubation, we see a greater shift toward high MW species and a reduction of smaller species within the oAβ ladder (Figure 3B), raising the possibility that SDS-stability of the native oligomers may increase over time. Our results indicate that OC changes the oligomeric state of ADDLs, with a time- and concentration-dependent trend toward higher MW species and increased SDS stability of fragments generated from larger oAβs.
Figure 3. OC shifts the oligomeric distribution of ADDLs to higher MW species in SDS-PAGE.
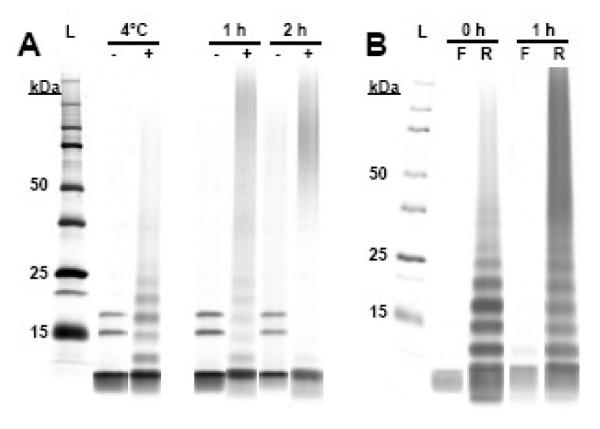
(A) ADDLs (4°C -) were heated at 37°C for 1 or 2 hours with a DMSO control (-) or OC (+). At 1 h, OC produces a ladder-like distribution of oAβs (1 h +) similar to Aβ-OC (4°C +) that moves toward higher MW species at 2 h (2 h +), while ADDLs heated in the absence of OC show only a marginal shift toward high MW species and no laddering (1 h - & 2 h -). (B) The Aβ ladder is the breakdown product of SDS-unstable higher MW species. The 50 kDa filtrate (F) shows little to no oAβs after 0, 1, and 2 h incubation with a 10-fold molar excess with OC. The >50 kDa retentate (R) contains the ladder-like oAβ SDS-PAGE distribution initially and after 1 h that disappears after a 2 h incubation. These results indicate that OC initially enhances the SDS stability of ADDL subspecies and later ADDLs as a whole.
OC reduces initial ADDL binding and subsequent pathology
After observing altered oligomer structures in the presence of OC, we tested the ability of Aβ-OC to act as pathogenic ligands. It has previously been established that oligomers of particular sizes are able to bind at synapses, an action thought to be a crucial first step in the initiation of AD (Lacor et al., 2007; for review, see Viola et al., 2008). Our first goal was to determine whether the altered structure of Aβ-OC resulted in altered binding. Primary hippocampal cultures were treated with ADDLs or Aβ-OC and the degree to which oAβs colocalized with the postsynaptic marker PSD-95 was assessed. Consistent with previously published data, ADDLs showed strong colocalization with PSD-95 (87 ± 3%; Lacor et al., 2004). Aβ-OC showed a statistically equivalent degree of colocalization (89 ± 2%; Figure 4), indicating a binding pattern indistinguishable from that of ADDLs.
Figure 4. Specific binding of Aβ-OC to synapses shows greatly enhanced immunoreactivity compared to ADDLs.

PSD-95 (magenta) was used as a synaptic marker to determine if the pattern of binding was similar between ADDLs or Aβ-OC (both in green) added at 500 nM for 3 h before fixation. When comparing the levels of ADDLs and Aβ-OC detected by M89, we observe much brighter puncta with Aβ-OC that, as a result, appear larger and more abundant than ADDL puncta. Lower panels show a 2x zoomed image of a process bound with ADDLs or Aβ-OC. In 50 fields taken for each condition in 3 separate experiments, no statistical difference was found in the degree of colocalization with PSD-95 between ADDLs (87 ± 3%) and Aβ-OC (89 ± 2%).
Although the binding patterns were indistinguishable, levels of oAβ immunofluorescence showed striking differences between ADDLs and Aβ-OC. We observed a marked increase in immunofluorescence in Aβ-OC (Figure 4). The enhanced antibody reactivity of oAβs formed in the presence OC observed in dot blot assays thus remained after synaptic binding and was observed at all time points assayed (data not shown).
Enhanced immunofluorescence could have resulted from either increased synaptic binding of oAβs or alterations in oligomer structure that facilitate antibody recognition. To distinguish these possibilities, we assayed binding using direct detection of fluorescently-conjugated oAβs with OC present either during or after oligomer formation. Interestingly, the increased binding of Aβ-OC observed using immunofluorescence was not found with direct detection, indicating bound Aβ-OC molecules were more accessible to antibody than control ADDLs. Synaptic binding of Aβ-OCs in fact appeared slightly reduced compared to ADDLs, although the difference was not statistically significant (Figure 5).
Figure 5. All oligomers show reduced binding to hippocampal neurons in the presence OC when detected directly with FAM-conjugated Aβ.

oAβs (either ADDLs or Aβ-OC) were added at 500 nM, and OC was present at 700 nM (ADDL+OClow and Aβ-OC) or 10 μM (ADDL+OChigh). oAβs were detected directly using FAM-conjugated Aβ. (A) oAβ positive puncta are pictured in pseudocolor (purple to yellow from dim to bright) for one branch in each condition. (B) Intensity of an entire field is plotted along the z-axis using a height and color scale. ADDLs show the most intense puncta, while increased concentrations of OC show a decrease in ADDL puncta number and brightness. (C) The average number of oAβ-positive pixels per length (25 μm per region analyzed) plus 1 SEM were calculated from max projections. While Aβ-OC shows a trend in reduced ADDL-positive pixels, ADDL+OClow and ADDL+OChigh both show significantly less Aβ load compared to the OC-free ADDL treatment. [*p < 0.05]
Effects of OC on oAβ binding were particularly obvious when we treated neurons with OC before adding ADDLs (Figure 5B). In cells given 10 μM OC followed by fluorescently-labeled ADDLs (500 nM), ADDL puncta were significantly reduced in number and brightness (Figure 5A). When assessed by immunofluorescence, ADDL binding following OC pretreatment also showed striking reduction (Figure 6). Decreased ADDL binding was evident with OC treatments as low as 1 μM. Thus maintaining neurons in OC for even a short period before exposure to ADDLs can result in effective lowering of ADDL binding.
Figure 6. OC preincubation with primary hippocampal cultures precludes accumulation of ADDLs at synapses.

Cells received a 5 minute OC preincubation at 100 (A), 10 (B), 1 (C), and 0 (D) μM. We observed a dose-dependent decrease in 500 nM ADDL binding after 3 h. ADDLs were detected with NU1 and show a 2-3-fold reduction in Aβ load with 10 & 100 μM OC preincubation, despite the OC-induced increased immunoreactivity expected from previous experiments.
Our next experiment was designed to determine if the reduced binding protected hippocampal neurons from downstream effects of preformed ADDLs. The effect of OC on ADDL-induced dendritic spine deterioration was assayed by drebrin loss. Drebrin, a spine-associated actin binding protein involved in spinogenesis and the regulation of dendritic spine morphology (Takahashi et al., 2003; Aoki et al., 2005), is depleted in hippocampal neurons following ADDL binding (Lacor et al., 2007). Using confocal microscopy, we measured the 3-dimensional intensity and volume of drebrin immunofluorescence. At 24 hours, 10 μM OC pretreatment protected against the oAβ-induced reduction in drebrin signal (Figure 7A&B). At this concentration, OC reduced the drebrin volume loss observed in control ADDLs by 80 ± 8%. Drebrin intensity in the presence of 10 μM OC was indistinguishable from control. To determine if the volume differences were due to a reduction in all spines or spines of specific sizes, we further examined the distribution of volumes for each oAβ treatment. ADDLs caused an increase in smaller spines as well as a reduction in larger spines, with both showing a 38-42% difference compared to vehicle (Figure 7C). Significantly, this altered distribution was blocked by OC treatment. Statistically, there were no differences in the proportion of either larger or smaller spines in the presence of OC (Figure 7C). These results indicate that OC is capable of protecting spines from ADDL-induced synaptic deterioration.
Figure 7. OC protects against synaptic deterioration as measured by drebrin loss.

The average drebrin object volume (A) and intensity (B) for each condition is shown normalized to the vehicle. OC at high concentrations (10 μM) showed a protective effect against the ADDL-induced reductions in drebrin following 24 h treatments with 500 nM oAβs. Low levels of OC (700 nM) showed an intermediate protective effect. (C) The distribution of spine sizes is similar to vehicle for OC-treated conditions. The proportion of large (>1.30 μm3) and small (<0.2 μm3) drebrin objects show that only in the absence of OC does the proportion of drebrin volumes change significantly from vehicle. (D) Representative images of the drebrin staining for each condition are shown. At least 5000 drebrin objects were analyzed for each condition. [*p < 0.05; **p < 0.001]
Enhanced antibody sensitivity allows greater clearance of ADDLs from synapses
Immunotherapies are currently being investigated for the treatment of AD, and it would be of value to increase the efficacy of antibody action (for review, see Jicha, 2009). The increased immunoreactivity of oAβs in the presence of OC raises the possibility that OC could enhance immunotherapy by making synapse-bound ADDLs more accessible to antibody. We treated primary hippocampal cultures for 30′ with 500 nM OC or vehicle, followed by a 30′ ADDL incubation (250 nM as calculated by Aβ monomer), followed by a 24 h incubation with the oligomer-specific antibody NU1 or vehicle. As seen in Figure 8, the reduction in ADDLs observed bound to neurons following NU1 treatment was greater in the presence of OC. The data thus indicate the potential for the use of OC to facilitate AD immunotherapeutics.
Figure 8. OC promotes the clearance of ADDLs by oligomer-specific antibodies.

Hippocampal cultures were treated with fluorescently-conjugated ADDLs following a pretreatment with OC or vehicle. After 30 minutes, the oligomer-specific antibody NU1 was added to reverse and prevent further binding. (A) NU1 reduction in detectable ADDLs is much greater in the presence of OC. (B) Quantification of the ADDL puncta per 50 microns plotted as the mean of 60 branches ± 1 SEM. The reduction in synaptic binding by OC is transient, as at 24 h the presence of OC has no effect on the binding of ADDLs. However, the removal of ADDLs by NU1 was enhanced by OC compared to the OC-free condition [p < 5×10-7].
Discussion
Using a combination of dot immunoblot, SDS-PAGE, and confocal immunofluorescence microscopy, we have shown OC to alter oAβ structure in such a way that increases immunoreactivity and SDS stability. Additionally, we have found that OC treatments reduce the binding of ADDLs and the subsequent deterioration of dendritic spines while enhancing the clearance of ADDLs from synapses using oligomer-specific antibodies.
OC was found to interact with soluble Aβ species and alter oAβ structure at doses as low as 10 nM. Such an interaction was likely based on the phenolic properties of OC (Yu et al., 2002; Smith et al., 2005), although no prior studies have investigated this possibility. The structural changes that occurred permitted better antibody detection by both conformation- and sequence-specific Aβ antibodies (Figures 1 & 2). Antibodies with different epitopes all showed enhanced immunoreactivity. As yet, the underlying structural basis responsible for changes in immunoreactivity have not been determined.
Structural changes following OC incubation were also evident in the ladder-like distribution of oAβs visible in SDS-PAGE. Fractionation experiments showed the oAβ ladder comprised SDS-stable fragments derived from native oAβs > 50 kDa (Figure 3B). OC alters oAβ structure quickly, as laddering was apparent even when OC was added immediately prior to fractionation and gel loading (Figure 3B). Within the laddered region, distinct bands are observed for each oAβ n-mer up to 10 (Figure 3). One possible explanation is that the oAβ ladder is the result of substoichiometic associations of OC with oAβs, which stochastically stabilize peptide interactions within native oligomers. These interactions could generate the integer-multiple of SDS-stable fragments that appear in SDS-PAGE. OC stabilization of adjacent peptide interactions would then stabilize larger oligomers, explaining the concentration-dependent shift toward higher MW species. The more stable Aβ-Aβ interactions would most likely come about through better alignment of the β-sheets, the proposed interfaces through which oligomer subunits interact with one another and confer their toxicity (Yu et al., 2009; Cerf et al., 2009). Observations of a more pronounced pellet during Aβ-OC preparation compared to ADDLs combined with preliminary size exclusion chromatography data indicate that OC produces a shift to high MW oAβs (data not shown). Consistent with this notion, higher concentrations of OC replace the oAβ ladder, and even the monomer band, with strictly high MW oAβs (data not shown). Further analysis of changes in oligomeric structure occurring in the presence of OC can be approached with a newly developed tool combining high-resolution localized surface plasmon resonance (HR-LSPR) combined with matrix assisted laser desorption ionization mass spectroscopy (MALDI-MS; Anker et al., 2009).
The disruptive effect of oleocanthal on oligomer structure, which results in an increased size, is somewhat surprising given that studies with other polyphenolic compounds have shown prevention of oligomer formation. Most groups show only a reduction in MW in the presence of polyphenols (Wang et al., 2008; Ho et al., 2009). Ono et al. (2008) used SDS-PAGE following PICUP (photo-induced cross-linking of unmodified proteins) to analyze the effects of the grape seed polyphenolic extract MegaNatural-AZ (MN) on Aβ oligomerization. While they came to the same conclusion that MW was reduced, a noticeable band at ~60 kDa was present in oligomer samples formed with 250 μM MN, but not 25 μM MN or in its absence. This is consistent with our observation of higher MW bands in the presence of OC. The use of different oligomerization protocols may account for the differing alterations in oligomerization, although it is likely that additional chemical groups on the phenolic compounds can produce different outcomes.
A striking observation was the increased antibody detection of Aβ-OC compared to ADDLs. As seen in Figure 4, antibody-detection showed a marked increase in oAβ signal for Aβ-OC compared to ADDLs, although we found no difference when using direct-detection of fluorescently-conjugated oAβs (Figure 5). Oligomers formed in the presence of OC thus do not exhibit increased synaptic binding or downstream pathology, only immunoreactivity. This may provide a way to enhance immunotherapy, as ADDLs will be more easily detected and removed by the antibodies.
When OC interacted with preformed ADDLs, the results were similar to that of Aβ-OC, although with a much greater reduction in ADDL initial binding and subsequent pathology (Figures 5-7). These changes are likely due to prevention of binding by altered oligomer structure or changes in binding sites. It is known that oAβs of different structure have different binding patterns and pathogenic capabilities: ADDLs >50 kDa bind to hippocampal neurons in much greater abundance than ADDLs <50 kDa (Lacor et al., 2007); Aβ*56 has been identified as the major toxic species in Tg2576 mice (Lesne et al., 2006), although there is evidence that smaller oAβs are capable of producing functional alterations in neuronal functioning (Walsh et al., 2002). Changes in ADDL binding sites have also been recently reported (De Felice et al., 2009).
OC concentrations that reduced binding to synapses also protected hippocampal neurons from ADDL-induced synaptic deterioration assessed by drebrin loss at 24 h (Figure 7). The level of drebrin is reported to be correlated with both spine maturity and the amount of spine transmission, particularly at glutamatergic synapses, making it a good indicator of spine functionality (Ivanov et al., 2009). It is unknown whether the protective effect observed in the drebrin assay is the result of lowered synaptic binding or an alteration of ADDLs that reduces their pathological potency. Observations from long-term binding studies show that effects of OC on ADDL binding are relatively short-lived and no longer observed at 24 h (Figure 8), suggesting that reduced ADDL-binding alone may not explain the reduction in drebrin loss. It is likely that in addition to the modifications in oligomer structure observed in the presence of OC, ADDL binding sites and/or their downstream effectors are altered by OC to prevent ADDL pathology.
Immunotherapy is currently being developed for the treatment of AD. Active immunization trials of aggregated Aβ have been shown to boost the titer of Aβ antibodies leading to decreased Aβ burden and reduced cognitive deterioration (Hock et al., 2003; Nicoll et al., 2003). However, aseptic meningoencephalitis in a subset of patients forced early termination of the trials (Orgogozo et al., 2003). Passive immunization, in which Aβ-recognizing antibodies are given to augment patient titers, shows promise in mouse models and is currently in Phase III human trials (Bard et al., 2000; Grundman & Black, 2008). Methods to boost antibody recognition of oAβs would be advantageous in passive immunization. The enhanced immunoreactivity and removal of ADDLs by the oligomer-specific antibody NU1 imply that OC could be a potential enhancer of AD immunotherapy. Additionally, OC-increased antibody sensitivity could be of value for diagnostics using antibody detection of oAβ levels to assess AD risk (Georganopoulou et al., 2005).
Results presented here indicate oleocanthal is capable of altering the oligomerization state of ADDLs while protecting neurons from the synaptopathological effects of ADDLs. Beneficial effects were indicated directly by the blocking of synaptic deterioration, as well as indirectly by enhanced antibody clearance of ADDLs. In addition to reducing the effects of oAβs, OC has been shown to have antioxidant and anti-inflammatory properties that further implicate a benefit in AD prevention (Beauchamp et al., 2005; Smith et al., 2005). Overall, these results are consistent with findings suggesting potential benefits of phenolic compounds capable of altering Aβ aggregation states and providing neuroprotective benefits, and suggest that optimized derivatives of OC may show potential as therapeutic drugs.
Acknowledgements
This study was supported by grants 5 T32 AG020418, RO1 AG022547, RO1 AG029460, IIRG-06-26989, and P50 DC06760. We would like to thank David Fang and Kevin Luo for preliminary HPLC analysis and Amos B. Smith, III for generating oleocanthal. We would also like to thank Garry Cooper and Jean Miller for valuable scientific and grammatical input.
Abbreviations
- AD
Alzheimer’s disease
- ADDLs
Aβ-derived diffusible ligands
- oAβs
oligomeric Aβ
- OC
oleocanthal
- Aβ-OC
ADDLs formed in the presence of OC
- ADDL+OC
preformed ADDLs treated with OC
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul HM, Sultana R, Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008 Nov 15;45(10):1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anker JN, Hall WP, Lambert MP, Velasco PT, Mrksich M, Klein WL, Van Duyne RP. Detection and Identification of Bioanalytes with High Resolution LSPR Spectroscopy and MALDI Mass Spectrometry. The Journal of Physical Chemistry. 2009;113(15):5891–5894. doi: 10.1021/jp900266k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki C, Sekino Y, Hanamura K, Fujisawa S, Mahadomrongkul V, Ren Y, Shirao T. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. J Comp Neurol. 2005;483(4):383–402. doi: 10.1002/cne.20449. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bazoti FN, Bergquist J, Markides KE, Tsarbopoulos A. Noncovalent interaction between amyloid-beta-peptide (1-40) and oleuropein studied by electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2006 Apr;17(4):568–575. doi: 10.1016/j.jasms.2005.11.016. [DOI] [PubMed] [Google Scholar]
- Bazoti FN, Bergquist J, Markides K, Tsarbopoulos A. Localization of the noncovalent binding site between amyloid-beta-peptide and oleuropein using electrospray ionization FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 2008;19(8):1078–1085. doi: 10.1016/j.jasms.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Beauchamp GK, Keast RSJ, Morel D, Lin J, Pika J, Han Q, Lee C, Smith AB, Breslin PAS. Phytochemistry: ibuprofen-like activity in extra-virgin olive oil. Nature. 2005 Sep 1;437(7055):45–46. doi: 10.1038/437045a. [DOI] [PubMed] [Google Scholar]
- Blasko I, Apochal A, Boeck G, Hartmann T, Grubeck-Loebenstein B, Ransmayr G. Ibuprofen decreases cytokine-induced amyloid beta production in neuronal cells. Neurobiol Dis. 2001;8(6):1094–1101. doi: 10.1006/nbdi.2001.0451. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(Pt 3):213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Cerf E, Sarroukh R, Tamamizu-Kato S, Breydo L, Derclaye S, Dufrene Y, Narayanaswami V, Goormaghtigh E, Ruysschaert J, Raussens V. Anti-parallel beta-sheet - a signature structure of the oligomeric amyloid-beta peptide. Biochem J. 2009 doi: 10.1042/BJ20090379. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007 Apr 13;282(15):11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Vieira MNN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao W, Ferreira ST, Klein WL. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci U S A. 2009;106(6):1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Vieira MNN, Saraiva LM, Figueroa-Villar JD, Garcia-Abreu J, Liu R, Chang L, Klein WL, Ferreira ST. Targeting the neurotoxic species in Alzheimer’s disease: inhibitors of Abeta oligomerization. FASEB J. 2004;18(12):1366–1372. doi: 10.1096/fj.04-1764com. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008 Sep;29(9):1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26(22):6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durairajan SSK, Yuan Q, Xie L, Chan W, Kum W, Koo I, Liu C, Song Y, Huang J, Klein WL, Li M. Salvianolic acid B inhibits Abeta fibril formation and disaggregates pre-formed fibrils and protects against Abeta-induced cytotoxicty. Neurochem Int. 2008 Mar-Apr;52(45):741–750. doi: 10.1016/j.neuint.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JCM, Breteler MMB. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002 Jun 26;287(24):3223–3229. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- Georganopoulou DG, Chang L, Nam J, Thaxton CS, Mufson EJ, Klein WL, Mirkin CA. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2005;102(7):2273–2276. doi: 10.1073/pnas.0409336102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundman M, Black R. O3-04-05: Clinical trials of bapineuzumab, a beta-amyloid-targeted immunotherapy in patients with mild to moderate Alzheimer’s disease Alzheimer’s and Dementia. 2008. p. T166.
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992 Apr 10;256(5054):184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hock C, Maddalena A, Raschig A, Muller-Spahn F, Eschweiler G, Hager K, Heuser I, Hampel H, Muller-Thomsen T, Oertel W, Wienrich M, Signorell A, Gonzalez-Agosti C, Nitsch RM. Treatment with the selective muscarinic m1 agonist talsaclidine decreases cerebrospinal fluid levels of A beta 42 in patients with Alzheimer’s disease. Amyloid. 2003;10(1):1–6. doi: 10.3109/13506120308995249. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Esclapez M, Pellegrino C, Shirao T, Ferhat L. Drebrin A regulates dendritic spine plasticity and synaptic function in mature cultured hippocampal neurons. J Cell Sci. 2009;122(Pt 4):524–534. doi: 10.1242/jcs.033464. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu C, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103(13):5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA. Is passive immunization for Alzheimer’s disease ‘alive and well’ or ‘dead and buried’? Expert Opin Biol Ther. 2009;9(4):481–491. doi: 10.1517/14712590902828285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Farinelli M, Konietzko U, Nitsch RM, Mansuy IM. Abeta oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. J Neurosci. 2007;27(29):7648–7653. doi: 10.1523/JNEUROSCI.0395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24(45):10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007 Jan 24;27(4):796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998 May 26;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79(3):595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007 Jan;100(1):23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440(7082):352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008 May 1;1207:225–236. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JAR, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9(4):448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Orgogozo J, Gilman S, Dartigues J, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Ono K, Condron MM, Ho L, Wang J, Zhao W, Pasinetti GM, Teplow DB. Effects of grape seed-derived polyphenols on amyloid beta-protein self-assembly and cytotoxicity. J Biol Chem. 2008 Nov 21;283(47):32176–32187. doi: 10.1074/jbc.M806154200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A, Kabat J, Novak M, Wu Q, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys. 1998;357(2):299–309. doi: 10.1006/abbi.1998.0813. [DOI] [PubMed] [Google Scholar]
- Small DH. The Sixth International Conference on Alzheimer’s disease, Amsterdam, The Netherlands, July 1998. The amyloid cascade hypothesis debate: emerging consensus on the role of A beta and amyloid in Alzheimer’s disease. Amyloid. 1998 Dec;5(4):301–304. doi: 10.3109/13506129809007304. [DOI] [PubMed] [Google Scholar]
- Smith ABr, Han Q, Breslin PAS, Beauchamp GK. Synthesis and assignment of absolute configuration of (-)-oleocanthal: a potent, naturally occurring non-steroidal anti-inflammatory and anti-oxidant agent derived from extra virgin olive oils. Org Lett. 2005 Oct 27;7(22):5075–5078. doi: 10.1021/ol052106a. [DOI] [PubMed] [Google Scholar]
- Solfrizzi V, Colacicco AM, D’Introno A, Capurso C, Torres F, Rizzo C, Capurso A, Panza F. Dietary intake of unsaturated fatty acids and age-related cognitive decline: a 8.5-year follow-up of the Italian Longitudinal Study on Aging. Neurobiol Aging. 2006 Nov;27(11):1694–1704. doi: 10.1016/j.neurobiolaging.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23(16):6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991 Oct;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Viola KL, Velasco PT, Klein WL. Why Alzheimer’s is a disease of memory: the attack on synapses by A beta oligomers (ADDLs) J Nutr Health Aging. 2008;12(1):51S–7S. doi: 10.1007/BF02982587. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang H, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002 Jan 11;924(2):133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Zhao W, Ono K, Rosensweig C, Chen L, Humala N, Teplow DB, Pasinetti GM. Grape-derived polyphenolics prevent Abeta oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J Neurosci. 2008 Jun 18;28(25):6388–6392. doi: 10.1523/JNEUROSCI.0364-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chang L, Klein WL, Thatcher GRJ, Venton DL. Per-6-substituted-per-6-deoxy beta-cyclodextrins inhibit the formation of beta-amyloid peptide derived soluble oligomers. J Med Chem. 2004 Jun 17;47(13):3329–3333. doi: 10.1021/jm034224e. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wu Z, Butko P, Christen Y, Lambert MP, Klein WL, Link CD, Luo Y. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci. 2006;26(50):13102–13113. doi: 10.1523/JNEUROSCI.3448-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280(7):5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Yu J, Bakhos L, Chang L, Holterman MJ, Klein WL, Venton DL. Per-6-substituted beta-cyclodextrin libraries inhibit formation of beta-amyloid-peptide (A beta)-derived, soluble oligomers. J Mol Neurosci. 2002 Aug-Oct;19(12):51–55. doi: 10.1007/s12031-002-0010-x. [DOI] [PubMed] [Google Scholar]
- Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET. Structural characterization of a soluble amyloid beta-peptide oligomer. Biochemistry. 2009;48(9):1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]