

Abstract
In this study, we present a fully automated tool, called IDEAL-Q, for label-free quantitation analysis. It accepts raw data in the standard mzXML format as well as search results from major search engines, including Mascot, SEQUEST, and X!Tandem, as input data. To quantify as many identified peptides as possible, IDEAL-Q uses an efficient algorithm to predict the elution time of a peptide unidentified in a specific LC-MS/MS run but identified in other runs. Then, the predicted elution time is used to detect peak clusters of the assigned peptide. Detected peptide peaks are processed by statistical and computational methods and further validated by signal-to-noise ratio, charge state, and isotopic distribution criteria (SCI validation) to filter out noisy data. The performance of IDEAL-Q has been evaluated by several experiments. First, a serially diluted protein mixed with Escherichia coli lysate showed a high correlation with expected ratios and demonstrated good linearity (R2 = 0.996). Second, in a biological replicate experiment on the THP-1 cell lysate, IDEAL-Q quantified 87% (1,672 peptides) of all identified peptides, surpassing the 45.7% (909 peptides) achieved by the conventional identity-based approach, which only quantifies peptides identified in all LC-MS/MS runs. Manual validation on all 11,940 peptide ions in six replicate LC-MS/MS runs revealed that 97.8% of the peptide ions were correctly aligned, and 93.3% were correctly validated by SCI. Thus, the mean of the protein ratio, 1.00 ± 0.05, demonstrates the high accuracy of IDEAL-Q without human intervention. Finally, IDEAL-Q was applied again to the biological replicate experiment but with an additional SDS-PAGE step to show its compatibility for label-free experiments with fractionation. For flexible workflow design, IDEAL-Q supports different fractionation strategies and various normalization schemes, including multiple spiked internal standards. User-friendly interfaces are provided to facilitate convenient inspection, validation, and modification of quantitation results. In summary, IDEAL-Q is an efficient, user-friendly, and robust quantitation tool. It is available for download.
Quantitative analysis of protein expression promises to provide fundamental understanding of the biological changes or biomarker discoveries in clinical applications. In recent years, various stable isotope labeling techniques, e.g. ICAT (1), enzymatic labeling using 18O/16O (2, 3), stable isotope labeling by amino acids in cell culture (4), and isobaric tagging for relative and absolute quantitation (2, 5), coupled with LC-MS/MS have been widely used for large scale quantitative proteomics. However, several factors, such as the limited number of samples, the complexity of procedures in isotopic labeling experiments, and the high cost of reagents, limit the applicability of isotopic labeling techniques to high throughput analysis. Unlike the labeling approaches, the label-free quantitation approach quantifies protein expression across multiple LC-MS/MS analyses directly without using any labeling technique (7–9). Thus, it is particularly useful for analyzing clinical specimens in highly multiplexed quantitation (10, 11); theoretically, it can be used to compare any number of samples. Despite these significant advantages, data analysis in label-free experiments is an intractable problem because of the experimental procedures. First, although high reproducibility in LC is considered a critical prerequisite, variations, including the aging of separation columns, changes in sample buffers, and fluctuations in temperature, will cause a chromatographic shift in retention time for analytes in different LC-MS/MS runs and thus complicate the analysis. In addition, under the label-free approach, many technical replicate analyses across a large number of samples are often acquired; however, comparing a large number of data files further complicates data analysis and renders lower quantitation accuracy than that derived by labeling methods. Hence, an accurate, automated computation tool is required to effectively solve the problem of chromatographic shift, analyze a large amount of experimental data, and provide convenient user interfaces for manual validation of quantitation results.
The rapid emergence of new label-free techniques for biomarker discovery has inspired the development of a number of bioinformatics tools in recent years. For example, Scaffold (Proteome Software) and Census (12) process PepXML search results to quantify relative protein expression based on spectral counting (13–15), which uses the number of MS/MS spectra assigned to a protein to determine the relative protein amount. Spectral counting has demonstrated a high correlation with protein abundance; however, to achieve good quantitation accuracy with the technique, high speed MS/MS data acquisition is required. Moreover, manipulations of the exclusion/inclusion strategy also affect the accuracy of spectral counting significantly. Because peptide level quantitation is also important for post-translational modification studies, the accuracy of spectral counting on peptide level quantitation deserves further study.
Another type of quantitation analysis determines peptide abundance by MS1 peak signals. According to some studies, MS1 peak signals across different LC-MS/MS runs can be highly reproducible and correlate well with protein abundance in complex biological samples (7–9). Quantitation analysis methods based on MS1 peak signals can be classified into three categories: identity-based, pattern-based, and hybrid-based methods (16). Identity-based methods (7–9) depend on the results of MS/MS sequencing to identify and detect peptide signals in MS1 data. However, because the data acquisition speed of MS scanning is insufficient, a considerable number of low abundance peptides may not be selected for limited MS/MS sequencing. Only a few peptides can be repetitively identified in all LC-MS/MS runs and subsequently quantified; thus, only a small fraction of identified peptides are quantified, resulting in a small number of quantifiable peptides/proteins.
In contrast to identity-based methods, pattern-based methods (17–23), including the publicly available MSight (20), MZmine (21, 22), and msInspect (23), tend to quantify all peptide peaks in MS1 data to increase the number of quantifiable peptides. These methods first detect all peaks in each MS1 data and then align the detected peaks across different LC-MS/MS runs. However, in pattern-based methods, efficient detection and alignment of the peaks between each pair of LC-MS/MS runs are a major challenge. To align the peaks, several methods based on dynamic programming or image pattern recognition have been proposed (24–26). The algorithms applied in these methods require intensive computation, and their computation time increases dramatically as the number of compared samples increases because all the LC-MS/MS runs must be processed. Therefore, pattern-based approaches are infeasible for processing a large number of samples. Furthermore, pattern recognition algorithms may fail on data containing noise or overlapping peptide signal (i.e. co-eluting peptides). The hybrid-based quantitation approach (16, 27–30) combines a pattern recognition algorithm with peptide identification results to align shifted peptides for quantitation. The pioneering accurate mass and time tag strategy (27) takes advantage of very sensitive, highly accurate mass measurement instruments with a wide dynamic range, e.g. FTICR-MS and TOF-MS, for quantitation analysis. PEPPeR (16) and SuperHirn (28) apply pattern recognition algorithms to align peaks and use the peptide identification results as landmarks to improve the alignment. However, because these methods still align all peaks in MS1 data, they suffer the same computation time problem as pattern-based methods.
To resolve the computation-intensive problem in the hybrid approach, we present a fully automated software system, called IDEAL-Q, for label-free quantitation including differential protein expression and protein modification analysis. Instead of using computation-intensive pattern recognition methods, IDEAL-Q uses a computation-efficient fragmental regression method for identity-based alignment of all confidently identified peptides in a local elution time domain. It then performs peptide cross-assignment by mapping predicted elution time profiles across multiple LC-MS experiments. To improve the quantitation accuracy, IDEAL-Q applies three validation criteria to the detected peptide peak clusters to filter out noisy signals, false peptide peak clusters, and co-eluting peaks. Because of the above key features, i.e. fragmental regression and stringent validation, IDEAL-Q can substantially increase the number of quantifiable proteins as well as the quantitation accuracy compared with other extracted ion chromatogram (XIC)1 -based tools. Notably, to accommodate different designs, IDEAL-Q supports various built-in normalization procedures, including normalization based on multiple internal standards, to eliminate systematic biases. It also adapts to different fractionation strategies for in-depth proteomics profiling.
We evaluated the performance of IDEAL-Q on three levels: 1) quantitation of a standard protein mixture, 2) large scale proteome quantitation using replicate cell lysate, and 3) proteome scale quantitative analysis of protein expression that incorporates an additional fractionation step. We demonstrated that IDEAL-Q can quantify up to 89% of identified proteins (703 proteins) in the replicate THP-1 cell lysate. Moreover, by manual validation of the entire 11,940 peptide ions corresponding to 1,990 identified peptides, 93% of peptide ions were accurately quantified. In another experiment on replicate data containing huge chromatographic shifts obtained from two independent LC-MS/MS instruments, IDEAL-Q demonstrated its robust quantitation and its ability to rectify such shifts. Finally, we applied IDEAL-Q to the THP-1 replicate experiment with an additional SDS-PAGE fractionation step. Equipped with user-friendly visualization interfaces and convenient data output for publication, IDEAL-Q represents a generic, robust, and comprehensive tool for label-free quantitative proteomics.
EXPERIMENTAL PROCEDURES
Sample Preparation
Materials
Triethylammonium bicarbonate (TEABC) was purchased from Sigma-Aldrich. The BCA™ protein assay reagent kit was obtained from Pierce. Ammonium persulfate and N,N,N′,N′-tetramethylenediamine were purchased from Amersham Biosciences. TFA, formic acid (FA), and HPLC grade ACN were purchased from Sigma-Aldrich. Modified, sequencing grade trypsin was purchased from Promega (Madison, WI). Standard protein mixture 1 (25 fmol/μl glycogen phosphorylase, serum albumin precursor, enolase 1, and alcohol dehydrogenase 1) and mixture 2 (12.5 fmol/μl glycogen phosphorylase, 200 fmol/μl serum albumin precursor, 50 fmol/μl enolase 1, and 25 fmol/μl alcohol dehydrogenase 1) were purchased from Waters Corp.; 0.4 fmol/μl Escherichia coli was also purchased from Waters Corp.
Cell Culture
THP-1 (human acute monocytic leukemia, The American Type Culture Collection) cell lines were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin G at 37 °C in a 5% CO2 atmosphere. Cell lines were harvested, washed three times with PBS, and lysed in lysis buffer (0.25 m Tris-HCl, pH 6.8, 1% SDS).
Preparative SDS-PAGE Separation
The protein concentrations in cell lysate were determined by BCA assay (Pierce) before tryptic digestion. For large scale identification of the THP-1 cell line, 70 μg of cell lysate was separated by 10% SDS-PAGE (31) (0.5 cm × 4.0 cm × 0.75 mm). The remaining gel was then excised into five gel slices based on molecular weight. Each slice was cut into pieces, washed with Milli-Q water, and destained twice with 25 mm TEABC, pH 8 in 50% (v/v) ACN for 15 min after which the slices were dehydrated with 100% ACN and dried for 10 min under vacuum. Next, the dry gel pieces were rehydrated in 25 mm TEABC, pH 8 containing an additional β-casein as the internal standard prior to in-gel digestion (in this study, we used β-casein as an internal standard to normalize XIC area readings of endogenous peptides). Following the addition of trypsin (10 ng/μl), the gel pieces were incubated at 37 °C overnight. The tryptic peptides were then extracted twice with 5% (v/v) FA in 50% (v/v) ACN for 30 min, dried completely under vacuum, and stored at −30 °C.
LC-MS/MS and Protein Identification
Samples were reconstituted in 4 μl of buffer A (0.1% FA in H2O) and analyzed by LC-MS/MS (Waters Q-TOF™ Premier from Waters Corp.). Samples were injected into a 2-cm × 180-μm capillary trap column and separated by a 25-cm × 75-μm nanoACQUITY™ 1.7-μm Bridged Ethyl Hybrid C18 column using the nanoACQUITY Ultra Performance LC™ system (Waters Corp.). The column was maintained at 35 °C, and bound peptides were eluted with a linear gradient of 0–80% buffer B (buffer A, 0.1% FA in H2O; buffer B, 0.1% FA in ACN) for 120 min. The MS system was operated in ESI positive V mode with a resolving power of 10,000. The NanoLockSpray source was used for accurate mass measurement, and the lock mass channel was sampled every 30 s. The mass spectrometer was calibrated with a synthetic human [Glu1]-fibrinopeptide B solution (1 pmol/μl; from Sigma-Aldrich) delivered through the NanoLockSpray source. Data were acquired via data-directed analysis. The method included a full sequential MS scan (m/z 400–1600, 0.6 s) and three MS/MS scans (m/z 100–1990, 1.2 s per scan) on the three most intense ions present in the full-scan mass spectrum.
Raw MS/MS data were converted into peak lists using Distiller (Matrix Science, London, UK; version 2.0) with the default parameters. All MS/MS samples were analyzed using Mascot (Matrix Science; version 2.2.1). Mascot was set up to search the ipi_HUMAN_3.29 database (version 3.29; 68,161 entries) for the THP-1 cell line and the Swisssprot_Metazoa_Animals database (version 54.2; 17,170 entries) for standard proteins, assuming trypsin as the digestion enzyme. Mascot was set up to search with a fragment ion mass tolerance of 0.1 Da and a parent ion tolerance of 0.1 Da. Two missed cleavages were allowed during trypsin digestion. Oxidation (Met) was selected as a variable modification. To determine the false discovery rate of protein identification, we repeated the search using identical search parameters and validation criteria on a randomized decoy database created by Mascot. We accepted identified peptides with Mascot scores above the statistically significant threshold (p < 0.05). The unique MS/MS spectra and assignment of identified peptides are shown in the supplemental figures.
Protein Quantitation by IDEAL-Q
IDEAL-Q quantifies label-free experiments with multiple LC-MS/MS runs and different fractionation strategies, such as strong cation exchange and SDS-PAGE. The numbers of samples, fractions, and runs are unrestricted. As shown in Fig. 1, the workflow involves four steps: 1) data preparation and construction of the ID database; 2) processing of data from each LC-MS/MS run, 3) peptide level processing, and 4) protein level processing.
Fig. 1.

Workflow of IDEAL-Q.
Data Preparation and Construction of ID Database
IDEAL-Q accepts spectral data in the mzXML format and peptide and protein identification results from different pipelines, namely Mascot, SEQUEST, and X!Tandem followed by PeptideProphet and ProteinProphet. It also provides two filtering criteria as user options: 1) a confidence score threshold to filter out low confidence identification results and 2) an elution time range to determine the range of peptides that should be included.
Confidently identified peptides in the search results are then used to construct an ID database of the identification results from all LC-MS/MS runs. The identified peptide ions are deemed to be the same entry in the database if their following attributes are the same: 1) sequence, 2) precursor m/z value (tolerance range, ±0.2 Da), 3) charge state, 4) modification, and 5) modification site. Note that, in this study, we treat a peptide with different charge states as different peptides. The protein list and associated peptide lists are constructed in the database. If a peptide is only identified in some of the LC-MS/MS runs, information about the peptide will be assigned to the LC-MS/MS runs in which the peptide is not identified. The failure to identify a peptide in some runs is probably because of a low identification score or because the precursor m/z was not selected for MS/MS sequencing. In such runs, the peptide is denoted as an unidentified peptide. Therefore, each LC-MS/MS run contains identified and unidentified peptides. Note that the peptides cannot be quantified by IDEAL-Q if they were not identified by MS/MS in any of the MS runs.
Processing of Each LC-MS/MS Run Data
In each LC-MS/MS run, IDEAL-Q sequentially processes all peptides, both identified and unidentified, to quantify as many peptides as possible. For an identified peptide, IDEAL-Q uses the identified retention time to detect the peak cluster. However, for an unidentified peptide, IDEAL-Q uses the proposed ID-based elution time prediction by fragmental regression (IDEAL) algorithm (described under “IDEAL Algorithm”) to predict the retention time of the peptide in the current run after which it detects the peak cluster based on the predicted time. A detected peak cluster of an unidentified peptide is called an assigned peptide. The detected peak clusters of both identified and assigned peptides are validated by the SCI criteria (described under “SCI: Three-dimensional Peptide Cluster Validation”). A peak cluster that passes SCI validation is used to construct the XIC for quantifying peptide abundance in the current run. IDEAL-Q automatically detects the retention time and m/z ranges to construct the XIC. Various normalization procedures are provided in IDEAL-Q (described under “Peptide Abundance Determination and Normalization at LC-MS/MS Run Level”). Users can decide whether or not to perform normalization.
IDEAL Algorithm for Peptide Elution Time Prediction
Reproducible LC separation is a crucial prerequisite in most XIC-based label-free strategies. However, because of variations in LC, a peptide rarely elutes at the same time in replicate experiments, which could lead to construction of an incorrect XIC and thereby render an incorrect quantitation result. To quantify an unidentified peptide, its elution time needs to be accurately predicted from experimental data containing chromatographic shifts caused by possible variations in LC. The IDEAL algorithm tries to accurately predict the elution time of each unidentified peptide.
The algorithm consists of two parts: a linear regressionNo validation function and a fragmental refining function. For any two LC-MS/MS runs, a linear regression model is constructed by only using peptides identified in both runs. Let xi and yi represent the identified elution time of peptide i in the two LC-MS/MS runs, respectively. The linear regression model is defined as
where b = ȳ − ax̄ and a = Σ((xi − x̄)(yi − ȳ))/Σ(xi − x̄)2. The constructed regression model, which represents the correlation of the peptide elution time in both LC-MS/MS runs, is used to estimate the elution time of an unidentified peptide in one run given the elution time of the peptide identified in the other run.
However, in some cases, the elution time predicted by the regression model may deviate from the actual elution time beyond a certain tolerance range. Therefore, to rectify the prediction error of f(x), we introduce a fragmental refining function F(x) given that x represents the elution time of the peptide identified in one run. F(x) is determined by the deviations between yi (the actual elution time) and f(xi) (the predicted elution time) with all xi in the range of [x − 2, x + 2] as defined in Equation 2,
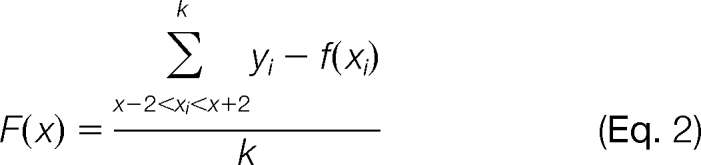 |
where k is the number of (xi, yi) pairs in the range. Given the identified elution time, x, of a peptide in a reference LC-MS/MS run, the IDEAL algorithm takes f(x) + F(x) as the predicted elution time of the unidentified peptide in a specific run.
Because an unidentified peptide generally has multiple reference LC-MS/MS runs in label-free experiments, i.e. it is unidentified in the current run but identified in several other runs, IDEAL-Q uses the weighted average of all predictions derived by different reference runs to determine the final prediction of the elution time as
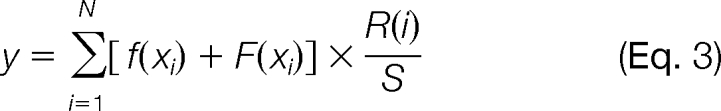 |
where S = Σi = 1N R(i). We use the R2 value of the prediction model as the weight factor R(i), i.e. the more accurate the prediction model, the higher the assigned weight will be.
For example, when processing a peptide in the ID database, given that the peptide is identified in some LC-MS/MS runs, say, {X1, X2, …, XN} at elution time {x1, x2, …, xN} and unidentified in a specific LC-MS/MS run r, we construct N prediction models (f(x) + F(x)) for run r versus each run of Xi (Equations 1 and 2) to calculate the predicted elution time y of the peptide by Equation 3.
SCI: Three-dimensional Peptide Peak Cluster Validation
When quantifying a specific peptide in an LC-MS/MS run, i.e. given the elution time (predicted by IDEAL for an unidentified peptide or derived from the search results for an identified peptide) and m/z, IDEAL-Q first extracts the MS1 data within the range of the elution time ±3 min and the precursor m/z ± 3.5 Da. (Note that the range of the elution time can be adjusted for different instruments.) Next, peak cluster detection is performed on the extracted MS1 data after which the detected peak cluster, which contains three isotopic peaks, will be validated by the SCI process. SCI stands for the three criteria used for peptide validation: 1) signal-to-noise (S/N) ratio, 2) charge state, and 3) isotope pattern. These three criteria are checked sequentially unless violation of a criterion is detected.
The S/N criterion checks whether the precursor peak, i.e. the monoisotopic peak, has a valid S/N ratio (≥2). The charge state criterion is used to eliminate peak clusters that have an incorrect charge state by examining whether the distance between adjacent peaks is equal to 1/z. Finally, the isotope pattern criterion examines the correlation between the isotopic distribution of the observed peak intensities and the theoretical isotopic distribution (32) of the peptide. The correlation is evaluated by the χ2 goodness of fit test. Any peptide that does not satisfy this criterion is eliminated because of the occurrence of possible co-eluting peptides with close m/z values.
Peptide Abundance Determination and Normalization at LC-MS/MS Run Level
For each peptide in an LC-MS/MS run that passes SCI validation, we construct the XIC of the selected peak cluster by summing the MS signals within the m/z width and elution time range of the precursor peak. (Both the m/z width and elution time range are determined by peak detection.) Then, we use the B-spline algorithm (33) for curve smoothing. The area under the XIC curve is used to determine the peptide abundance in the LC-MS/MS run, called the peptide run abundance. Note that when a peptide fails SCI validation it is regarded as absent from the LC-MS/MS run. In this case, the peptide run abundance is reported as zero, so it will not be involved in the subsequent normalization and peptide ratio calculation procedures.
After determining all peptide run abundances in the LC-MS/MS run, we normalize them to eliminate systematic errors. IDEAL-Q supports four normalization strategies in an LC-MS/MS run: 1) the XIC areas of spiked internal standard proteins used to support both single and multiple spiked internal standards, 2) the median of all peptide run abundances in the LC-MS/MS run, 3) the mean of all peptide run abundances in the LC-MS/MS run, and 4) a user-defined normalization factor.
Determination of Peptide Ratios and Protein Ratios at Sample Level
In addition to peptide run abundance, based on different experiment designs, IDEAL-Q defines the following levels of peptide abundance: peptide abundance in the fraction level (called peptide fraction abundance) and peptide abundance in the sample level (called peptide sample abundance). A number of fractionation strategies, such as strong cation exchange and SDS-PAGE, have been proposed to reduce the complexity of a sample mixture before LC-MS/MS analysis. If a prefractionation strategy is not adopted, the peptide sample abundance is retrieved directly from the peptide fraction abundance. For experiments with multiple fractions and where each fraction is used to conduct multiple LC-MS/MS runs, peptide fraction abundance is defined as the average of the peptide run abundances of the LC-MS/MS runs in the fraction. Then, the peptide abundances in all the fractions are summed to represent the peptide sample abundance. After determining peptide sample abundances, the peptide ratio of any two samples can be calculated.
Before determining the protein ratio, IDEAL-Q supports the following mechanisms to normalize the peptide ratios of sample i versus sample j: central tendency normalization, linear regression normalization, and quantile normalization (34), which can be used to further reduce systematic biases. For protein ratios, IDEAL-Q provides the flexibility to calculate each ratio by only the non-degenerate peptides of the protein (the default setting) or by all detected peptides. It is noted that the degeneracy of peptide/protein identification was based on the database search results. It also provides an option to further eliminate the outlier peptide ratios of a protein by using Dixon's Q-test (35). The protein ratio Ri, j of sample i to sample j is determined by a weighted average of the peptide ratios where the weight of each peptide ratio is determined by the corresponding peptide sample abundance.
RESULTS AND DISCUSSION
We conducted four experiments to evaluate the performance of IDEAL-Q in terms of the accuracy of elution time prediction, quantitation coverage (i.e. the percentages of quantified peptides/proteins in all identified peptides/proteins), and quantitation accuracy. In the first experiment, the workflow and the quantitation performance of IDEAL-Q were demonstrated on a serially diluted standard protein mixture spiked into E. coli cell lysate. The quantitation coverage and accuracy on the proteome scale were demonstrated on the biological replicates of THP-1 cell lysate. In this experiment, we also manually validated all the quantified peptide ions in a large scale data set for unbiased evaluation of the true quantitation accuracy. To demonstrate the features of robust peptide elution time prediction for alignment and stringent peptide quantitation assurance for unreproducible LC-MS/MS, we generated a special data set from the biological replicate experiment of THP-1 cell lysate using different LC-MS/MS instruments. In the data set, dramatic chromatographic shifts appeared between different LC-MS/MS runs.
For comprehensive quantitation of a proteome profile, an additional fractionation step, such as SDS-PAGE, is usually incorporated to increase the number of identified/quantified peptides. However, label-free quantitation experiments frequently suffer from low resolution and variations in fractionation reproducibility because a peptide or a protein usually appears in two or more consecutive fractions. Therefore, peptide abundance in only one fraction does not necessarily represent the actual peptide expression and could lead to inaccurate peptide ratios. To improve the quantitation accuracy, IDEAL-Q is designed to be compatible with label-free quantitation experiments using different fractionation strategies. The quantitation accuracy is demonstrated by a biological replicate of the THP-1 cell lysate with an additional SDS-PAGE fractionation followed by the shotgun approach. The complete quantitation results are shown in the supplemental figures.
Workflow of IDEAL-Q
As shown in Fig. 1 , quantitation under IDEAL-Q involves four steps. 1) For data preparation, IDEAL-Q is designed to accept search results from major search engines and data in the common mzXML format, which can be converted easily from spectral data files generated by different mass spectrometers. After loading the database search results and mzXML files, an ID database containing the identified peptides and proteins in all LC-MS/MS runs is constructed. 2) For peptide cross-assignment and SCI validation in each LC-MS/MS run, we process all peptides in the ID database and classify them as identified or unidentified. To quantify an identified peptide, i.e. a confidently matched peptide generated by the database search engine, in an LC-MS/MS run, the elution time and precursor m/z of the identified MS/MS spectrum are acquired to extract the spectral data of the peptide that will be processed by SCI validation. Meanwhile, to quantify an unidentified peptide, we first detect the peptide peaks by using the IDEAL algorithm to predict the elution time of the peptide for peptide alignment. Then, we use the predicted elution time and precursor m/z to extract local LC-MS/MS data for detecting the peptide peak cluster; this procedure is called peptide cross-assignment. The detected peptide peak cluster, i.e. the assigned peptide, is processed by SCI validation (described under “Experimental Procedures”). If an identified peptide or an assigned peptide passes SCI validation, its peak cluster is used to construct the XIC to determine peptide abundance; otherwise, it is regarded as unquantifiable. 3) For peptide ratio determination, first, we construct the XIC of the selected peak cluster by summing the MS signals within the m/z width and elution time range of the precursor peak and perform curve smoothing using the B-spline algorithm. Then, the peptide abundance is determined by the XIC area. To correct systematic errors, IDEAL-Q supports four strategies, described under “Peptide Abundance Determination and Normalization at LC-MS/MS Run Level”, to normalize peptide abundance in each LC-MS/MS run. Then, the peptide ratio is calculated accordingly. 4) For protein ratio determination, the protein abundance ratio is determined by the weighted average of non-degenerate peptides. Prior to protein ratio determination, IDEAL-Q allows further normalization based on the peptide ratio distribution. It also provides the option to further eliminate outlier peptide ratios of a protein by using Dixon's Q-test.
Quantitation Performance Evaluation of IDEAL-Q
Highly Accurate Quantitation Evidenced by Standard Protein Mixture
To demonstrate the performance of IDEAL-Q, different concentrations of four standard proteins (glycogen phosphorylase, serum albumin precursor, enolase 1, and alcohol dehydrogenase 1) were spiked into E. coli cell lysate and analyzed by LC-MS/MS. A total of 51 proteins was identified by Mascot and quantified by IDEAL-Q. The protein ratios of 47 E. coli proteins that were unchanged had a mean of 1.05 ± 0.17, as shown in Fig. 2A. The ratios of the other four spiked proteins were consistent with the expected ratio with small deviations (2.9–19.9%), as shown in Table I. Furthermore, the calculated protein ratios for serially diluted BSA (1, 5, 10, 25, and 50 fmol) were highly correlated with the expected ratios and demonstrated good linearity (R2 = 0.996), as shown in Fig. 2B. These results demonstrate that IDEAL-Q can calculate protein expression ratios with high accuracy and precision.
Fig. 2.

Validation of protein quantitation by IDEAL-Q. Serially diluted proteins mixed with E. coli cell lysate were quantified by IDEAL-Q. A, the protein ratios of the 20 proteins in E. coli cell lysate. The result is close to the expected ratio 1. B, given concentrations of BSA quantified by IDEAL-Q show the good linearity of the quantitation and good sensitivity in a highly dynamic range (The error bars indicate the standard deviation of the quantified BSA protein ratio).
Table I. Quantitation results of four proteins with different concentrations.
| Protein name | Molecular mass | No. of identified peptides | No. of quantified peptides | Ratio |
Quantitation error | |
|---|---|---|---|---|---|---|
| IDEAL-Q | Expected | |||||
| Da | % | |||||
| Glycogen phosphorylase | 97,228 | 19 | 19 | 0.53 | 0.5 | 6 |
| Serum albumin precursor | 69,248 | 13 | 11 | 6.40 | 8 | −19.9 |
| Enolase 1 | 46,773 | 7 | 5 | 1.80 | 2 | −9.7 |
| Alcohol dehydrogenase 1 | 36,800 | 11 | 10 | 0.97 | 1 | −2.9 |
Substantially Increased Quantitation Coverage on Proteome Scale with High Quantitation Accuracy
Achieving high quantitation coverage in terms of the percentages of quantified peptides/proteins in all identified peptides/proteins is a challenge in label-free quantitative proteomics. Normally, a peptide is quantified only if it can be identified in all LC-MS/MS runs. Here, we demonstrate the ability of IDEAL-Q to increase the quantitation coverage by cross-assignment of confidently identified peptides in all LC-MS/MS runs. To evaluate the performance of IDEAL-Q on a complex sample, we conducted an experiment on biological duplicate of the THP-1 cell lysate and performed triplicate LC-MS/MS analysis. A total of 1,990 peptides corresponding to 703 proteins was identified (score, >39; false discovery rate, 0.05–2.3%). The elution times of any two LC-MS/MS runs from the two samples exhibited a high correlation; for example, in Fig. 3, there is a high correlation between the first runs of the two samples.
Fig. 3.

Correlation of peptide elution times of THP-1 cell line biological replicate. The elution times of commonly identified peptides in the first runs of two replicate samples are used to show the correlation. The R2 value of 0.9999 and the slope of 1.011 demonstrate that the LC system is stable and reproducible.
In the data set, 1,990 peptides were confidently identified in at least one of the six LC-MS/MS runs in the two biological replicates. Among these peptides, 1,596, 1,289, and 1,107 peptides were identified in at least one, two, and three LC-MS/MS runs for both biological replicates, respectively. The above four peptide sets represent increasingly reliable peptide sets. We use N(m, n) to denote the set of peptides identified in at least m LC-MS/MS runs of one biological replicate and at least n LC-MS/MS runs of the other replicate. Denoting |N(m, n)| as the number of peptides in N(m, n), we have |N(0, 1) U N(1, 0)| = 1,990, |N(1, 1)| = 1,596, |N(2, 2)| = 1,289, and |N(3, 3)| = 1,107. Many identity-based quantitation methods only quantify peptides that are identified in all LC-MS/MS runs; for example, they quantify peptides in the peptide set N(3, 3), which only accounts for 55% of all identified peptides. In contrast, IDEAL-Q tries to quantify all identified peptides, i.e. peptides identified in at least one LC-MS/MS run, by peptide cross-assignment using the predicted elution time.
Although we focused on comparing the quantitation strategies of the conventional identity-based approach and IDEAL-Q, we also used IDEAL-Q to quantify the above four peptide sets to evaluate its performance in terms of the quantitation coverage and quantitation accuracy. In this subsection, we examine how the quantitation coverage is affected by quantifying different peptide sets represented by the above N(m,n) and assess the accuracy based on the coefficients of variation (CVs) of protein ratios. The quantitation accuracy was further examined by manual validation. (We discuss this aspect in the next subsection.) The conventional identity-based approach quantified peptides in N(3, 3). Among them, only 909 peptides corresponded to 353 proteins, i.e. 45.7% of all identified peptides and 50.2% of all identified proteins were quantified. IDEAL-Q quantified N(0, 1) U N(1, 0) by peptide cross-assignment. Among them, 1,672 peptides corresponded to 626 proteins, i.e. 84% of all identified peptides and 89% of all identified proteins were quantified. The protein and peptide quantitation coverage rates for different peptide sets are shown in Fig.4. The results show that peptide cross-assignment can achieve a substantial improvement in the quantitation coverage.
Fig. 4.

Comparison of results derived by conventional ID-based strategy and IDEAL-Q in THP-1 cell line biological replicate experiment. A, the number of quantifiable peptides, the CVs of peptide sample abundance, and the CVs of the protein ratios on the four peptide sets, two of which correspond to the ID-based strategy and two of which correspond to IDEAL-Q quantitation strategy. B, the peptide ratio distributions on the four peptide sets. Compared with the identity-based quantitation strategy, IDEAL-Q achieves high quantitation accuracy and increases the number of quantifiable peptides substantially.
Next, we investigated whether using peptide cross-assignment to increase the quantitation coverage affects the quantitation accuracy. The average CVs of the protein ratios calculated for the four peptide sets are shown in Fig. 4A. The CV of the protein ratios in the peptide set N(0, 1) U N(1, 0) is 3.4%, which is comparable to 2.6% in N(3, 3). In addition, as shown in Fig. 4B, the protein ratio distributions in log2 scale of the four peptide sets are all close to 0 (i.e. close to 1 in non-log scale) with standard deviations of 7–11%. The narrow log2 ratio distributions of the four sets of data revealed that the improved quantitation coverage did not reduce the quantitation accuracy.
Large Scale Manual Validation Demonstrates Highly Reliable Quantitation Performance of IDEAL-Q
We also manually validated all the quantitation results to evaluate the overall performance of IDEAL-Q. The quantitation result of a manually validated peptide ion is considered correct if the following three conditions are satisfied. 1) The detected peptide peak cluster has the correct m/z as the assigned peptide, i.e. correct peptide alignment. 2) The peptide peak cluster does not contain noise or co-eluting peptides and can be quantified, i.e. correct SCI validation. 3) The ratios determined by IDEAL-Q and manual inspection are consistent, i.e. correct ratio determination.
Of 11,940 peptide ions detected by IDEAL-Q in all six LC-MS/MS runs from the two biological replicates, 8,806 peptide ions (1,990 peptides) were identified by the Mascot search. For those peptide ions, we only needed to check condition (2) for manual validation, and as high as 95% of them were correctly quantified by IDEAL-Q, as shown in Fig. 5B. Of the remaining 3,134 unidentified peptide ions, which were assigned by IDEAL-Q, 90% were properly aligned and correctly quantified, 2% were misaligned, and 8% were correctly aligned, but SCI validated them incorrectly, as shown in Fig. 5B. After manual validation, the overall accuracy for all the peptide ions was 93.3%, demonstrating the effectiveness of the data processing capability of IDEAL-Q. In addition, the large scale manual validation results revealed that 1,240 peptide ions had poor spectral quality, which was mainly because of peptide co-elution. However, SCI validation successfully detected and filtered out 1,114 (89.8%) of them. The results demonstrate that the high accuracy achieved by IDEAL-Q can be attributed to correct peptide alignment and the SCI validation process used to filter out noisy unquantifiable data. Thus, fully automated quantitation can be performed without time-consuming manual inspection.
Fig. 5.

Manually validated results of THP-1 cell line biological replicate experiment. A total of 11,940 peptide ion quantitations were manually validated. The results demonstrate the high accuracy rates achieved by SCI validation and peptide alignment. A, 11,940 peptide ions detected by IDEAL-Q in all six LC-MS/MS runs from the two biological replicates. 8,806 peptide ions (1,990 peptides) were identified by the Mascot search, and there were 3,134 unidentified peptide ions. B, Of 8,806 identified peptide ions, SCI correctly validated 8,339 peptide ions and failed on 467 identified peptide ions. C, Of all unidentified peptide ions, 69 were misaligned, 2,808 were correctly validated by SCI, and SCI failed on 257 unidentified peptide ions.
Using IDEAL Elution Time Prediction and SCI Validation to Rectify Chromatographic Shift
In addition to the demonstrated quantitation coverage and accuracy, IDEAL-Q provides a unique fragmental elution time prediction method (see “Experimental Procedures” for details) that enables robust quantitation of data sets with inconsistent elution profiles. Thus, we conducted a biological replicate experiment on the THP-1 cell line with triplicate LC-MS/MS analysis on two different LC systems to derive huge chromatographic shifts. Fig. 6 compares the elution time prediction results of IDEAL with those of two other prediction methods, namely, the ID-time method and the linear regression method. Under the ID-time method, the elution time of an unidentified peptide was determined by the experimental elution time of the same peptide identified in the other LC-MS/MS run. In the linear regression method, the elution time was predicted by Equation 1.
Fig. 6.

Correlation of peptide elution times in heterogeneous LC system experiment. The commonly identified peptides are plotted as gray circles based on the identified elution times of the first runs of the two biological replicates obtained from two different LC systems. The figure shows the prediction curves of three methods. The correlations (R2) between the plotted points and the prediction curves show that the IDEAL curve has the best data fit; therefore, it can predict the most accurate peptide elution times.
We used 699 commonly identified peptides from the first LC-MS/MS run of each LC system to compare the prediction curves of IDEAL, linear regression (y = 0.76x − 16.7), and ID-time (y = x). As shown in Fig. 6, the ID-time prediction deviated significantly from the actual elution times of the peptides. IDEAL achieved the highest R2 score of 0.998 followed by 0.982 for the linear regression method. The results demonstrate that IDEAL-Q is capable of robust quantitation compatible with huge chromatographic shifts, which are not unusual in practice.
Next, we used the entire data sets of the two LC systems to compare IDEAL with the other two methods on a large scale by performing k-fold cross-validation (6), which is widely used for performance evaluation in machine learning. We used a data set consisting of all identified peptides in any two of six LC-MS/MS runs to perform a k-fold cross-validation test and repeated it 15 times (the number of combinations of choosing two from six). On average, 693 peptides were identified in any two of the six LC-MS/MS runs. In a k-fold cross-validation test, the data set is randomly divided into k partitions of approximately equal size. One of the k partitions is used as the test set, and the remaining k − 1 partitions are used as the training data set, i.e. to produce corresponding IDEAL prediction models for predicting the elution time of peptides in the test set. In each iteration, the average prediction error is used to evaluate the performance as shown in Equation 4.
After performing k iterations by selecting each partition in turn as the test set, the final result of a k-fold cross-validation test is the average accuracy of the k iterations. The overall prediction errors of all k-fold tests using the three methods are shown in Table II. Clearly, the overall accuracy of the IDEAL algorithm across different k-fold tests was consistently better than that of the linear regression and ID-time methods. Notably, the prediction performance of IDEAL was good even on 2-fold cross-validation, which has the smallest training data set in all k-fold tests (i.e. the training data set and test data set are equal in size). The results show that the performance of IDEAL is very stable in large scale proteomics experiments. Furthermore, its predictions were accurate even on two different LC systems with chromatographic shifts.
Table II. Performance of elution time prediction by IDEAL, tested by k-fold cross-validation.
| k-Fold validation | ID-time |
Linear regression |
IDEAL |
|||
|---|---|---|---|---|---|---|
| Average error | S.D. | Average error | S.D. | Average error | S.D. | |
| 2 | 34.02 | 5.49 | 1.24 | 1.70 | 0.39 | 0.54 |
| 3 | 34.02 | 5.49 | 1.24 | 1.70 | 0.39 | 0.55 |
| 4 | 34.02 | 5.49 | 1.24 | 1.70 | 0.39 | 0.54 |
| 5 | 34.02 | 5.49 | 1.24 | 1.69 | 0.39 | 0.53 |
To further compare the quantitation accuracy achieved by different elution time prediction methods, the data set was quantified based on the elution time predicted by the ID-time, linear regression, and IDEAL methods. Furthermore, SCI validation criteria were applied incrementally to filter out noisy data. The quantitation performance in terms of peptide/protein quantitation coverage and quantitation accuracy are reported in Table III. The quantitation accuracy is measured by the mean and S.D. of the peptide and protein ratios, respectively. The accuracy of elution time prediction obviously affects the quantitation accuracy and quantitation coverage, as evidenced by the results in Table III. The worst performance was the elution time predicted by the ID-time method, which yielded the lowest quantitation accuracy as well as the lowest quantitation coverage (after SCI validation). However, the mean of the protein ratio based on the ID-time method improved substantially from 14.59 (without SCI validation) to 1.18 (with SCI validation). A similar improvement was also evident on the data based on the linear regression method. In summary, the results show that SCI validation can effectively filter out noisy data, especially in very noisy data sets, leading to improved quantitation accuracy.
Table III. The effect of SCI validation on quantitation.
CS, charge state; IP, isotope pattern.
| Prediction method and validation strategy | Protein quantitation coverage | Peptide quantitation coverage | Protein ratio |
Peptide ratio |
||
|---|---|---|---|---|---|---|
| Mean | S.D. | Mean | S.D. | |||
| % | % | |||||
| ID-time | ||||||
| No validation | 87.3 | 90.8 | 14.59 | 60.75 | 17.84 | 137.08 |
| S/N | 74.2 | 74.1 | 4.77 | 21.11 | 7.25 | 36.98 |
| S/N, CS | 60.6 | 55.3 | 4.74 | 25.03 | 5.26 | 22.88 |
| S/N, CS, IP | 43.1 | 34.2 | 1.18 | 1.23 | 1.41 | 3.2 |
| Linear regression | ||||||
| No validation | 87.3 | 79.0 | 1.34 | 3.07 | 1.45 | 3.07 |
| S/N | 85.5 | 75.0 | 1.28 | 2.64 | 1.28 | 2.32 |
| S/N, CS | 74.5 | 61.9 | 1.31 | 2.79 | 1.23 | 2.3 |
| S/N, CS, IP | 61.5 | 46.7 | 1 | 0.64 | 0.98 | 0.55 |
| IDEAL | ||||||
| No validation | 95.2 | 99.7 | 1.18 | 1.16 | 1.28 | 1.64 |
| S/N | 95.2 | 99.6 | 1.18 | 1.16 | 1.29 | 1.64 |
| S/N, CS | 95.0 | 98.7 | 1.19 | 1.16 | 1.28 | 1.59 |
| S/N, CS, IP | 76.8 | 66.3 | 1.08 | 0.55 | 1.08 | 0.61 |
After SCI validation, the linear regression method and the IDEAL method generated comparable results in terms of the mean and S.D. of protein ratios. However, using the elution time prediction of IDEAL achieved much better protein quantitation coverage than the linear regression method (76.8% compared with 61.5%).
Demonstration of Quantitation Performance on Large Scale Experiment with Fractionation
To evaluate the performance of IDEAL-Q on large scale quantitation with a fractionation step, we performed quantitation for the biologically duplicate THP-1 cell lysate with an additional SDS-PAGE fractionation step. Each gel was cut into five slices and subjected to trypsin digestion followed by LC-MS/MS analysis. Using the Mascot search engine (score, >32; false discovery rate, 1.5% by decoy database search), five fractions in the two SDS-PAGEs generated 1,438 proteins with 7,247 peptides. Because of the low resolution of SDS-PAGE separation, many proteins inevitably migrate to two or more consecutive fractions. For example, as shown in Fig. 7, the identified peptide DDPSKPVHLTAFLGYK corresponding to the 60 S ribosomal protein L3 was only identified in fraction 1 of both biological replicates. However, IDEAL-Q identified the peptide in fraction 2 of both biological replicates by peptide cross-assignment in addition to that in fraction 1. The conventional strategy only quantifies the peptide that is commonly identified in fraction 1 of the two biological replicates, and the resulting peptide ratio given by peptide abundance in SDS-PAGE A over that in SDS-PAGE B is 0.65 = 338.1/519.2. In contrast, IDEAL-Q detects and quantifies all peptides unidentified in other fractions. By summing the peptide fraction abundances in all fractions, IDEAL-Q calculated the corrected peptide ratio as 1.006 = (338.1 + 593)/(519.2 + 406.2), which corrects the biased ratio calculated by the conventional approach.
Fig. 7.

Example of inaccurate protein quantitation in fractionation experiment. The figure shows a typical problem that arises in quantitation with the fractionation strategy. A protein is separated into two consecutive fractions, leading to quantitation inaccuracy under the conventional quantitative strategy.
Among the 7,247 identified peptides corresponding to 1,438 proteins, 6,829 peptides corresponding to 1,391 proteins were quantified (see supplemental data for further details). The mean and S.D. of the protein ratios (SDS-PAGE A/SDS-PAGE B) is 1.04 ± 0.39, showing the quantitation compatibility of IDEAL-Q on a label-free approach with a fractionation step.
Robust Functionality of IDEAL-Q
Label-free quantitation analysis inevitably involves processing a large number of input files; thus, efficient data analysis is challenging. The most computation-intensive part is the peak alignment procedure. In contrast to pattern-matching alignment algorithms, our fragmental regression algorithm, IDEAL, uses the experimentally identified elution time to predict the elution time of unidentified peptides for peptide cross-assignment. It rectifies the chromatographic shift successfully and also reduces the computation time substantially. In the above four experiments, the input mzXML files were 5, 10.9, 12.8, and 76.5 GB, respectively. IDEAL-Q took 5, 49, 71, and 1,440 min, respectively, to quantify the data sets on a Microsoft Windows Server 2003 R2 (x64 edition service pack 2 with a 64-bit AMD Opteron Processor 2210 CPU 1.8-GHz processors, SATA hard disk (7200 rpm, 500 GB), and 8 GB RAM). It is noteworthy that IDEAL-Q can be executed on a personal computer with the Windows platform. Depending on the user's experiment design, IDEAL-Q provides flexible quantitation mechanisms, including support for various normalization schemes and fractionation strategies. Specifically, IDEAL-Q supports central tendency normalization, linear regression normalization, and quantile normalization (34).
IDEAL-Q also provides a handy user interface for output visualization, validation, and quantitation results, as shown by the screen shot in Fig. 8. This output interface displays protein and peptide lists with the calculated ratios and all identification information, annotated MS/MS spectra, visualization of quantified peptide peaks, and other statistical charts. It also facilitates fast requantitation after the selection or removal of user-selected peptide ions. To enable users to prepare supplemental data from their quantitation results, several output formats, including pdf, csv, and html formats, are supported by IDEAL-Q.
Fig. 8.

User interfaces of IDEAL-Q.A, the protein table, peptide table, and corresponding quantitation results. B, the visualization interface used to inspect data. C, the annotated MS/MS spectra show the status of peptide identification. D, the interface that displays statistical plots. E, comprehensive output results in different formats.
Conclusion
In this study, we present a robust, generic data analysis platform, called IDEAL-Q, for XIC-based label-free quantitative proteomics. It is compatible with different database search engines and mass spectrometers. To avoid the time-consuming computation required to align shifted peptide peaks, we use the fragmental regression method to predict the potential chromatographic shift and use signal processing techniques to detect unidentified peptides in all LC-MS/MS runs for peptide cross-assignment. Because of accurate elution time prediction, peptide/protein quantitation coverage is increased substantially over that achieved by the conventional identity-based approach. Furthermore, applying rigorous SCI validation on detected peptide peak clusters can filter out overlapping peaks or noisy data to ensure high quantitation accuracy. We demonstrated the quantitation performance of IDEAL-Q on a standard protein mixture and a proteome scale by a replicate experiment on the THP-1 cell line and manually validated results to further verify the performance of IDEAL-Q.
The results of triplicate LC-MS/MS analysis of the THP-1 cells on two different instruments show that IDEAL can accurately predict the elution time even when chromatographic shifts occur, and SCI validation can effectively differentiate between good and poor spectral data quality. Because IDEAL-Q is capable of rectifying huge chromatographic shifts and reducing systematic errors via normalization techniques, it could be applied to label-free comparative proteomics across instruments or even laboratories. It would boost the applicability of label-free proteomics significantly and reduce the cost of repetitive data analysis on different instruments. In addition, the robust alignment approach significantly increases the quantitation coverage. In summary, IDEAL-Q is an efficient and robust tool for accurate label-free quantitation of protein expression and compatible for label-free experiments with fractionation steps. It is executable on the Windows platform and available for download.
Supplementary Material
Footnotes
* This work was supported in part by the thematic program of Academia Sinica under Grants AS94B003 and AS95ASIA02 and by the National Science Council of Taiwan under Grants NSC 95-3114-P-002-005-Y, 97-2221-E-001-016-MY2, and 97-3112-B-001-011.
 The on-line version of this article (available at http://www.mcponline.org) contains supplemental data, including all quantitation results and annotated MS/MS spectra of the experiments described in the paper.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental data, including all quantitation results and annotated MS/MS spectra of the experiments described in the paper.
1 The abbreviations used are:
- XIC
- extracted ion chromatogram
- IDEAL
- Identity-based elution time prediction by fragmental regression
- CV
- coefficient of variation
- SCI
- signal-to-noise ratio, charge state, and isotope pattern (it represents a three-dimensional peptide validation procedure)
- S/N
- signal-to-noise ratio
- TEABC
- triethylammonium bicarbonate
- FA
- formic acid
- ID
- identity
REFERENCES
- 1.Gygi S. P., Rist B., Gerber S. A., Turecek F., Gelb M. H., Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 2.Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 3.Yao X., Freas A., Ramirez J., Demirev P. A., Fenselau C. (2001) Proteolytic 18O labeling for comparative proteomics: model studies with two serotypes of adenovirus. Anal. Chem 73, 2836–2842 [DOI] [PubMed] [Google Scholar]
- 4.Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 5.Ong S. E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 6.Stone M. (1974) Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc. Series B Stat. Methodol 36, 111–147 [Google Scholar]
- 7.Chelius D., Bondarenko P. V. (2002) Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J. Proteome Res 1, 317–323 [DOI] [PubMed] [Google Scholar]
- 8.Bondarenko P. V., Chelius D., Shaler T. A. (2002) Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed-phase liquid chromatography-tandem mass spectrometry. Anal. Chem 74, 4741–4749 [DOI] [PubMed] [Google Scholar]
- 9.Wang W., Zhou H., Lin H., Roy S., Shaler T. A., Hill L. R., Norton S., Kumar P., Anderle M., Becker C. H. (2003) Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal. Chem 75, 4818–4826 [DOI] [PubMed] [Google Scholar]
- 10.Cutillas P. R., Geering B., Waterfield M. D., Vanhaesebroeck B. (2005) Quantification of gel-separated proteins and their phosphorylation sites by LC-MS/MS using unlabeled internal standards: analysis of phosphoprotein dynamics in a B cell lymphoma cell line. Mol. Cell. Proteomics 4, 1038–1051 [DOI] [PubMed] [Google Scholar]
- 11.Cutillas P. R., Vanhaesebroeck B. (2007) Quantitative profile of five murine core proteomes using label-free functional proteomics. Mol. Cell. Proteomics 6, 1560–1573 [DOI] [PubMed] [Google Scholar]
- 12.Park S. K., Venable J. D., Xu T., Yates J. R., 3rd (2008) A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 14.Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 15.Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 16.Jaffe J. D., Mani D. R., Leptos K. C., Church G. M., Gillette M. A., Carr S. A. (2006) PEPPeR, a platform for experimental proteomic pattern recognition. Mol. Cell. Proteomics 5, 1927–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radulovic D., Jelveh S., Ryu S., Hamilton T. G., Foss E., Mao Y., Emili A. (2004) Informatics platform for global proteomic profiling and biomarker discovery using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 3, 984–997 [DOI] [PubMed] [Google Scholar]
- 18.Ono M., Shitashige M., Honda K., Isobe T., Kuwabara H., Matsuzuki H., Hirohashi S., Yamada T. (2006) Label-free quantitative proteomics using large peptide datasets generated by nanoflow liquid chromatography and mass spectrometry. Mol. Cell. Proteomics 5, 1338–1347 [DOI] [PubMed] [Google Scholar]
- 19.Li X. J., Yi E. C., Kemp C. J., Zhang H., Aebersold R. (2005) A software suite for the generation and comparison of peptide arrays from sets of data collected by liquid chromatography-mass spectrometry. Mol. Cell. Proteomics 4, 1328–1340 [DOI] [PubMed] [Google Scholar]
- 20.Palagi P. M., Walther D., Quadroni M., Catherinet S., Burgess J., Zimmermann-Ivol C. G., Sanchez J. C., Binz P. A., Hochstrasser D. F., Appel R. D. (2005) MSight: an image analysis software for liquid chromatography-mass spectrometry. Proteomics 5, 2381–2384 [DOI] [PubMed] [Google Scholar]
- 21.Katajamaa M., Oresic M. (2005) Processing methods for differential analysis of LC/MS profile data. BMC Bioinformatics 6, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katajamaa M., Miettinen J., Oresic M. (2006) MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 22, 634–636 [DOI] [PubMed] [Google Scholar]
- 23.Bellew M., Coram M., Fitzgibbon M., Igra M., Randolph T., Wang P., May D., Eng J., Fang R., Lin C., Chen J., Goodlett D., Whiteaker J., Paulovich A., McIntosh M. (2006) A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics 22, 1902–1909 [DOI] [PubMed] [Google Scholar]
- 24.Sadygov R. G., Maroto F. M., Hühmer A. F. (2006) ChromAlign: a two-step algorithmic procedure for time alignment of three-dimensional LC-MS/MS chromatographic surfaces. Anal. Chem 78, 8207–8217 [DOI] [PubMed] [Google Scholar]
- 25.Prince J. T., Marcotte E. M. (2006) Chromatographic alignment of ESI-LC-MS/MS proteomics data sets by ordered bijective interpolated warping. Anal. Chem 78, 6140–6152 [DOI] [PubMed] [Google Scholar]
- 26.Prakash A., Mallick P., Whiteaker J., Zhang H., Paulovich A., Flory M., Lee H., Aebersold R., Schwikowski B. (2006) Signal maps for mass spectrometry-based comparative proteomics. Mol. Cell. Proteomics 5, 423–432 [DOI] [PubMed] [Google Scholar]
- 27.Zimmer J. S., Monroe M. E., Qian W. J., Smith R. D. (2006) Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev 25, 450–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mueller L. N., Rinner O., Schmidt A., Letarte S., Bodenmiller B., Brusniak M. Y., Vitek O., Aebersold R., Müller M. (2007) SuperHirn—a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 [DOI] [PubMed] [Google Scholar]
- 29.Andreev V. P., Li L., Cao L., Gu Y., Rejtar T., Wu S. L., Karger B. L. (2007) A New Algorithm Using Cross-Assignment for Label-Free Quantitation with LC/LTQ-FT MS. J. Proteome Res 6, 2186–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryu S., Gallis B., Goo Y. A., Shaffer S. A., Radulovic D., Goodlett D. R. (2008) Comparison of a label-free quantitative proteomic method based on peptide ion current area to the isotope coded affinity tag method. Cancer Inform 6, 243–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kubinyi H. (1991) Calculation of isotope distributions in mass spectrometry—a trivial solution for a nontrivial problem. Anal. Chim. Acta 247, 107–119 [Google Scholar]
- 33.De Boor C. (1978) A Practical Guide to Splines, 1st Ed., pp. 114–115, Springer Verlag, New York [Google Scholar]
- 34.Callister S. J., Barry R. C., Adkins J. N., Johnson E. T., Qian W. J., Webb-Robertson B. J., Smith R. D., Lipton M. S. (2006) Normalization approaches for removing systematic biases associated with mass spectrometry and label-free proteomics. J. Proteome Res 5, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rorabacher D. B. (1991) Statistical treatment for rejection of deviant values: critical values of Dixon’s “Q” parameter and related subrange ratios at the 95% confidence level. Anal. Chem 63, 139–146 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.