

Abstract
Protein S-acylation (palmitoylation), a reversible post-translational modification, is critically involved in regulating protein subcellular localization, activity, stability, and multimeric complex assembly. However, proteome scale characterization of S-acylation has lagged far behind that of phosphorylation, and global analysis of the localization of S-acylated proteins within different membrane domains has not been reported. Here we describe a novel proteomics approach, designated palmitoyl protein identification and site characterization (PalmPISC), for proteome scale enrichment and characterization of S-acylated proteins extracted from lipid raft-enriched and non-raft membranes. In combination with label-free spectral counting quantitation, PalmPISC led to the identification of 67 known and 331 novel candidate S-acylated proteins as well as the localization of 25 known and 143 novel candidate S-acylation sites. Palmitoyl acyltransferases DHHC5, DHHC6, and DHHC8 appear to be S-acylated on three cysteine residues within a novel CCX7–13C(S/T) motif downstream of a conserved Asp-His-His-Cys cysteine-rich domain, which may be a potential mechanism for regulating acyltransferase specificity and/or activity. S-Acylation may tether cytoplasmic acyl-protein thioesterase-1 to membranes, thus facilitating its interaction with and deacylation of membrane-associated S-acylated proteins. Our findings also suggest that certain ribosomal proteins may be targeted to lipid rafts via S-acylation, possibly to facilitate regulation of ribosomal protein activity and/or dynamic synthesis of lipid raft proteins in situ. In addition, bioinformatics analysis suggested that S-acylated proteins are highly enriched within core complexes of caveolae and tetraspanin-enriched microdomains, both cholesterol-rich membrane structures. The PalmPISC approach and the large scale human S-acylated protein data set are expected to provide powerful tools to facilitate our understanding of the functions and mechanisms of protein S-acylation.
Protein S-acylation, commonly but somewhat inaccurately known as protein palmitoylation, is a post-translational lipid modification involving the covalent addition of long-chain fatty acids (predominantly the 16-carbon palmitic acid) to protein cysteine thiols via thioester linkages (1). Like other lipid modifications such as myristoylation and prenylation, S-acylation increases the hydrophobicity of cytoplasmic proteins, including many signaling molecules, thereby increasing their affinity for cytosolic membrane surfaces. However, compared with myristoylation and prenylation, S-acylation is more frequently detected on transmembrane proteins such as G protein-coupled receptors, immune cell receptors, and ion channels, all of which are already tightly associated with membranes (2). Moreover, protein S-acylation, which may either occur spontaneously or be catalyzed by palmitoyl acyltransferases (PATs),1 can be reversed by protein thioesterases such as acyl-protein thioesterase-1 (APT1) and protein palmitoylthioesterase-1 (1, 2). In this respect, protein S-acylation, as a dynamic and reversible modification, can be regarded as potentially analogous to protein phosphorylation. Notably, reversibility is not shared by any other lipid modification, making S-acylation an attractive mechanism for modulation of protein activity and stability, protein-protein interactions, and shuttling of proteins between subcellular compartments in response to changes in signal transduction (1, 3).
Evidence suggests that protein S-acylation may play important roles in a wide range of biological processes such as cell signaling, apoptosis, and carcinogenesis (4, 5). However, understanding of the mechanisms and functions of reversible S-acylation has progressed at a slow pace because of several challenges. First, S-acylated proteins are generally present in low abundance. Second, long-chain fatty acids attached to proteins via thioester bonds turn over rapidly. Third, although a class of PATs sharing a conserved Asp-His-His-Cys (DHHC) motif within a cysteine-rich domain (CRD), as well as several deacylating enzymes, has been recognized for several years, the enzymology of protein S-acylation and deacylation remains poorly understood (6). Fourth, there is no general consensus motif for S-acylation site prediction. Although two software tools, CSS-PALM (7) and NBA-PALM (8), have recently been developed to predict S-acylation sites on proteins, their reliability is uncertain. Last and important, no convenient method (e.g. S-acylation-specific antibody) is available to detect and purify S-acylated proteins. The commonly used [3H]palmitate in vivo labeling method is hazardous, sometimes insufficiently sensitive, and time-consuming, typically requiring several weeks or months for autoradiographic exposure (9).
To gain a more comprehensive view of protein S-acylation in vivo, two independent and complementary methods have been developed to detect and/or purify S-acylated proteins at the proteome scale. One method involves the metabolic incorporation of an azide- or alkyne-containing palmitate analogue into proteins (10–12). The azido/alkynyl groups, which are metabolically inert in cellular environments, can be specifically and efficiently conjugated with a tag (e.g. biotin, Myc, and fluorescein) by chemoselective ligations (e.g. Staudinger reaction or azide-alkyne [3 + 2] cycloaddition reaction) in vitro. Consequently, fatty acylated proteins containing the azide/alkyne moiety can be detected and/or purified with minimal contamination. Using this method, Martin and Cravatt (12) have recently purified 17-octadecynoic acid (17-ODYA)-modified proteins from immortalized Jurkat T cells. Multidimensional protein identification technology analysis and spectral counting quantification led to the identification of 125 high confidence and about 200 medium confidence predicted palmitoylated proteins (12). However, the metabolic labeling method cannot be readily applied to analyze tissue samples. It is also potentially difficult to use this method to study protein S-acylation in cancer cells, in which overexpression and hyperactivity of fatty-acid synthase dramatically decrease the intake of exogenous long-chain fatty acids (13).
A second method, named acyl-biotinyl exchange (ABE) (14–16), has been more widely used than the palmitate analogue labeling method. In this approach, thioester bonds are selectively cleaved by neutral hydroxylamine (HA), and S-acyl moieties are replaced by biotinyl groups. Consequently, S-acylated proteins can be detected and/or purified via streptavidin-biotin interactions. Given that no metabolic labeling is required, the ABE approach can be readily applied to analyze tissue samples and cancer cells. In the first ever global analysis of protein S-acylation, Roth et al. (15) enriched S-acylated proteins from yeast and validated the S-acylation of 35 proteins from 58 novel candidate S-acylated proteins. More recently, Kang et al. (16) identified 68 known and >200 candidate S-acylated proteins from whole rat brain, purified rat synaptosomes, and cultured embryonic rat neurons. Among these S-acylated protein candidates, 21 were confirmed by [3H]palmitate labeling, immunoprecipitation/ABE, and/or ABE/immunoblotting (16).
Although both proteomics methods have been demonstrated to be powerful for global analysis of protein S-acylation, the procedures reported in the above studies are not suitable for S-acylation site localization. Recently, Zhang et al. (17) applied their adaptation of the ABE method, called palmitoyl-cysteine isolation capture and analysis, to screen substrates of human DHHC2 in HeLa cells. A total of 57 sites were identified from 50 proteins, but the majority of the identified proteins are abundant cytoplasmic proteins, and half of the sites were identified only once in four runs, indicating that this method is technically limited. In addition, no controls were used to distinguish putative S-acylated peptides from contaminating peptides; thus, the actual S-acylation sites were not evaluated.
All the above mentioned large scale studies were performed on total membranes. However, biological membranes are not homogeneous but instead are compartmentalized into microdomains that exhibit particular lipid and protein compositions. Lipid rafts, a designation that includes vesicular caveolae, are liquid-ordered membrane microdomains enriched in cholesterol and sphingolipids (18, 19). Because of the saturated nature of palmitate, protein S-palmitoylation (the highly predominant form of S-acylation) has been proposed to target proteins to lipid rafts, a claim supported by studies of a limited number of proteins (20). However, no proteome scale localization of palmitoylated/S-acylated proteins in different membrane domains has been reported to date. In the present study, we isolated lipid raft-enriched and non-raft membrane fractions using a recently described procedure shown to be suitable for proteomics studies (21). With this approach, S-acylated proteins and peptides were purified and then analyzed using a novel ABE-based method designated palmitoyl protein identification and site characterization (PalmPISC). In combination with the widely used label-free spectral counting method of quantitation (22), PalmPISC led to the identification of 67 known and 331 novel candidate S-acylated proteins as well as the localization of 25 known and 143 novel candidate S-acylation sites. The PalmPISC approach, in combination with other tools such as stable isotope labeling with amino acids in cell culture, RNA interference, and cell imaging techniques, will greatly expand our understanding of protein S-acylation as a regulatory post-translational modification.
EXPERIMENTAL PROCEDURES
Cell Culture
The human prostate cancer cell line DU145 was obtained from the American Type Culture Collection. DU145 cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal bovine serum (Valley Biomedical) supplemented with 2 mm l-glutamine (Invitrogen), 100 units/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen) in a water-saturated 5% CO2 atmosphere at 37 °C. Cells were dissociated with 0.05% trypsin plus 0.02% EDTA (Invitrogen) and subcultured with 1:7 split ratios every 3–4 days.
Isolation of Lipid Raft-enriched and Non-raft Membrane Fractions
DU145 cells were harvested when they reached 90% confluence. Cells were rinsed three times with cold PBS (Invitrogen), scraped off 150-mm culture dishes (Falcon), and pelleted at 500 × g for 5 min. Lipid raft-enriched and non-raft membrane fractions were isolated using a modified successive detergent extraction method as described previously (21). Briefly, DU145 cells were homogenized and centrifuged at 500 × g for 5 min to pellet nuclei and intact cells. The resulting supernatant was centrifuged at 16,000 × g for 20 min to pellet membranes. Non-raft fractions were extracted with 1% Triton X-100, and lipid raft-enriched fractions were subsequently isolated with 60 mm β-octyl glucoside. Immunoblotting analysis was performed to confirm the enrichment of lipid raft markers caveolin-1 (Cav-1) and Giα2 as well as the cytosol/non-raft marker β-tubulin in lipid raft-enriched fractions and non-raft fractions, respectively.
ABE Chemistry
ABE was performed as described previously (15, 23) with some modifications. Protein extracts from lipid raft-enriched and non-raft membrane fractions were precipitated using the chloroform/methanol (CM) precipitation method. Protein pellets were redissolved with 4% SDS Buffer (50 mm Tris-HCl, 4% SDS, 5 mm EDTA, pH7.4) at 37 °C for 10 min. Protein concentration was determined using the Micro BCA protein assay (Pierce) according to the manufacturer's instructions. Following dilution with 3 volumes of Dilution Buffer (50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 0.2% Triton X-100, 1 mm PMSF, protease inhibitor mixture, pH 7.4), samples were reduced with 10 mm tris(2-carboxyethyl)phosphine (TCEP) (Pierce) for 30 min and alkylated with 50 mm N-ethylmaleimide (NEM) (Fluka) for 2.5 h at room temperature (RT) with end-over-end rotation. Excess NEM was removed with five sequential CM precipitations. Protein pellets were redissolved with 4% SDS Buffer and divided equally into two portions. To each portion was added 3 volumes of freshly prepared 1.33 mm N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide (biotin-HPDP) (Pierce), 0.27% Triton X-100, 33.3% N,N-dimethylformamide, 1.33 mm PMSF, protease inhibitor mixture, pH 7.4 containing 1 m HA (experimental (EXP) group) or 50 mm Tris-HCl (control (CON) group). Samples were incubated at RT for 60 min with end-over-end rotation, and excess biotin-HPDP was removed by three sequential CM precipitations.
S-Acylated Protein Purification, Separation, and Tryptic Digestion
Protein pellets were redissolved with 2% SDS Buffer (50 mm Tris-HCl, 2% SDS, 5 mm EDTA, pH 7.4) at 37 °C for 10 min, and then 19 volumes of Dilution Buffer was added to decrease the concentration of SDS to 0.1%. Following incubation at RT for 30 min with end-over-end rotation, EXP and CON samples were centrifuged at 16,000 × g for 5 min. The supernatants were incubated with streptavidin-agarose beads (GE Healthcare) pre-equilibrated with 50 volumes of Equilibration Buffer (50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 0.2% Triton X-100, 0.1% SDS, pH 7.4). After 1-h incubation at RT with end-over-end rotation, streptavidin-agarose beads were washed with 50 volumes of Equilibration Buffer six times. Bound proteins were eluted by incubating beads with 10 volumes of 20 mm TCEP in Equilibration Buffer for 30 min at RT with end-over-end rotation. Samples were centrifuged at 200 × g for 1 min to pellet beads. The supernatants were moved to new Eppendorf tubes and centrifuged again. Enriched proteins were recovered by a CM precipitation, resolved by 12.5% SDS-PAGE, and stained with Coomassie Brilliant Blue solution (Bio-Rad). In-gel digestion was performed essentially as described (24, 25). Each lane was cut into four gel slices, reduced with 10 mm DTT in 50 mm NH4HCO3 for 45 min, and alkylated with 55 mm iodoacetamide in 50 mm NH4HCO3 for 45 min in the dark. Proteins were in-gel digested with MS grade trypsin (Promega) and incubated at 58 °C for 30 min. Tryptic peptides were successively extracted with 100 μl of 5% acetic acid, 100 μl of 2.5% acetic acid and 50% acetonitrile, and 100 μl of 100% acetonitrile. Samples were dried down in vacuo using a SpeedVac concentrator (Thermo Scientific) and stored at −80 °C until mass spectrometric analysis.
S-Acylated Peptide Purification
Protein pellets obtained after ABE chemistry were redissolved with 2% SDS Buffer, diluted with 19 volumes of Dilution Buffer, and digested in solution with trypsin by incubating at 58 °C for 60 min. After centrifugation at 16,000 × g for 5 min, supernatants were combined with pre-equilibrated streptavidin-agarose beads and incubated at RT for 60 min with end-over-end rotation. Beads were successively washed with 50 volumes of Equilibrating Buffer five times and 20% acetonitrile in 10 mm NH4HCO3 buffer twice and then incubated with 2.5 volumes of 5 mm TCEP, 10 mm NH4HCO3, 20% acetonitrile solution at 37 °C for 30 min. Samples were centrifuged at 200 × g for 1 min, and the supernatants were centrifuged again. Finally, the supernatants were moved to LoBind tubes (Eppendorf), dried down in a SpeedVac concentrator, and stored at −80 °C until mass spectrometric analysis.
Mass Spectrometry
Peptides derived from in-gel digested proteins were analyzed by on-line C18 nanoflow reversed-phase HPLC (Eksigent nanoLC·2DTM) connected to an LTQ Orbitrap mass spectrometer (Thermo Scientific). Samples were loaded onto an in-house packed 100-μm-inner diameter × 15-cm C18 column (Magic C18, 5 μm, 200 Å, Michrom Bioresources Inc.) and separated at 200 nl/min with 80-min linear gradients from 5 to 35% acetonitrile in 0.4% formic acid. Survey spectra were acquired in the Orbitrap with the resolution set to a value of 30,000. Up to five of the most intense ions per cycle were fragmented and analyzed in the linear trap.
Peptides derived from in-solution digested proteins were analyzed on an LTQ ProteomeX mass spectrometer connected to a Surveyor HPLC pump and a microsampler (all from Thermo Scientific). Peptides were separated at about 400 nl/min with 80-min linear gradients from 5 to 35% acetonitrile in 0.4% formic acid. The ion trap was operated in data-dependent acquisition mode, fragmenting up to six of the most intensive ions after each survey scan.
Database Searching
The Thermo .raw files were converted into complete peak lists and entered into a relational database. The data conversion was carried out using in-house written software (26) utilizing the XRawfile2.dll library (version 2.1.1.0), which was provided by Thermo Scientific as part of the Xcalibur software package. The library is also used by MSQuant, Trans-Proteomic Pipeline, and the Sashimi project to convert the proprietary .raw file format into generic peak lists. For each MS/MS spectrum, the 200 most intense fragment ions were converted into an .mgf file without any further data processing such as smoothing, deisotoping, and filtering. Comparisons between the .mgf files generated by our approach and those generated by DTASuperCharge showed that our approach performed favorably (26).
All MS data sets were searched against the IPI_Human database (v3.36; 69,012 sequences) using the MASCOT search engine (Matrix Science, v2.1.04). For Orbitrap data, no fixed modification was set for any of the amino acids; variable protein modifications were selected as carbamidomethyl (Cys), deamidation (Asn and Gln), N-acetyl (protein N terminus), NEM (Cys), and oxidation (Met). The mass tolerance was set as ±20 ppm for MS spectra and ±0.8 Da for MS/MS spectra. Peptides were identified with an ion score no less than 33 (p < 0.05), and finally proteins were identified based on at least two unique peptides. For ion trap data, no fixed modification was set for any of the amino acids; variable protein modifications were selected as N-acetyl (protein N terminus), NEM (Cys), and oxidation (Met). The mass tolerance was set as ±1.5 Da for MS spectra and ±1.5 Da for MS/MS spectra. Peptides were identified with an ion score above 41 (p < 0.05). For both data sets, up to one missed tryptic cleavage was allowed, and probable contaminants (e.g. keratins and albumin) were removed from the identified protein/peptide lists.
Spectral Counting and Statistical Analysis
The relative protein/peptide abundance changes between the paired EXP and CON samples were determined using a label-free spectral counting approach (22). For both data sets generated by the protein-based procedure and by the peptide-based procedure, the spectral counts were merged over all biological/technical replicates and membrane domains. Subsequently, statistical analysis was performed on the merged data sets. For the protein data set, missing values were imputed; all zeros were replaced with 1. The protein-wise natural logarithms (ln) of EXP/CON ratios were calculated and then clustered using a Bayesian information criterion-based Gaussian mixture model (27). The resulting two Gaussian components represent the log ratio distributions of protein sets largely dominated by contaminating proteins and by S-acylated proteins. p values were computed based on the distribution of the contaminating protein-dominated data set. Proteins with p values less than 0.05 (corresponding to EXP/CON > 6.7) and between 0.275 and 0.05 (corresponding to 3.0 ≤ EXP/CON ≤ 6.7) were accepted as S-acylated candidates with high and medium confidence, respectively. For the peptide data set, because of the low peptide signal intensities, no missing value imputation was carried out, and subsequent analysis was identical to the protein analysis.
Immunoblotting Analysis
Immunoblotting analysis was performed essentially as described (28). To confirm the successful isolation of lipid raft-enriched and non-raft membrane fractions or localize ribosomal proteins L10A (RPL10A) and L12 (RPL12), 10 μg of cytoplasmic, non-raft membrane, and lipid raft-enriched membrane fractions was separated by SDS-PAGE and electrotransferred onto nitrocellulose membranes. To validate the S-acylation of RPL10A and RPL12, S-acylated proteins and controls were purified from equal amounts of total proteins extracted from lipid raft-enriched membranes using our adaptation of the ABE method, separated by SDS-PAGE, and electrotransferred onto nitrocellulose membranes. Primary antibodies such as anti-Cav-1 (N-20) pAb, anti-Giα2 pAb, and anti-β-tubulin mAb (clone D10) were purchased from Santa Cruz Biotechnology; anti-RPL12 pAb and anti-RPL10A mAb were obtained from Sigma. Secondary antibodies were from Pierce. All membranes were blocked with PBS, 0.1% Tween 20, 5% milk for 1 h at RT and incubated with primary antibodies (1:1000) overnight at 4 °C followed by incubation with species-specific horseradish peroxidase-conjugated secondary antibodies (1:10,000) for 1 h at RT. Signals were detected using SuperSignal chemiluminescent reagent (Pierce) followed by exposure of blots to x-ray film.
Immunofluorescence Microscopy
DU145 cells seeded in chamber slides were serum-starved for 12 h and treated with 100 μm 2-bromopalmitate (Sigma) or vehicle (0.1% ethanol) for 6 h. Membrane labeling and immunofluorescence were performed essentially as described (28). Briefly, live cells were washed once with ice-cold PBS and incubated on ice with FITC-conjugated cholera toxin B subunit (CTxB) (Sigma) diluted in medium. Cells were fixed in 4% paraformaldehyde and incubated with PBS, 0.1% BSA for 1 h at RT to block nonspecific binding sites prior to 1-h incubation with anti-RPL10A mAb (1:100) at RT. The immune reaction for RPL10A was detected by a Cy3-conjugated anti-mouse secondary antibody (1:250) for 30 min at RT. After washing, slides were mounted in Vectastain medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories) and analyzed using an Axioplan 2 microscope (Carl Zeiss MicroImaging).
Ingenuity Pathway Analysis
High confidence S-acylated proteins identified in this study using protein-based and peptide-based procedures as well as all known human S-acylated proteins were merged and analyzed by the Ingenuity Pathway Analysis (IPA) software (v7.6) (Ingenuity Systems). The IPA software uses an extensive high quality knowledge base to relate gene products to each other according to their interactions and functions. To discover potential S-acylated protein complexes, direct protein-protein interactions, which are contained in the IPA knowledge base, were shown as edges to connect S-acylated proteins. The appearances of the (sub)networks were improved by using Path Designer, a tool included in the IPA package.
RESULTS
Isolation of Lipid Raft-enriched and Non-raft Membrane Fractions
Detergent-resistant membrane (DRM) and non-DRM fractions were isolated using a modified successive detergent extraction method, which generates DRMs that are largely free of nuclear contamination (21). Immunoblotting analysis showed that the DRM fractions were greatly enriched with lipid raft maker proteins Cav-1 and Giα2, whereas the non-DRM fractions were significantly enriched with the cytosol/non-raft marker β-tubulin (Fig.1). Hence, we define DRM and non-DRM fractions hereinafter as lipid raft-enriched and non-raft membrane fractions, respectively.
Fig. 1.
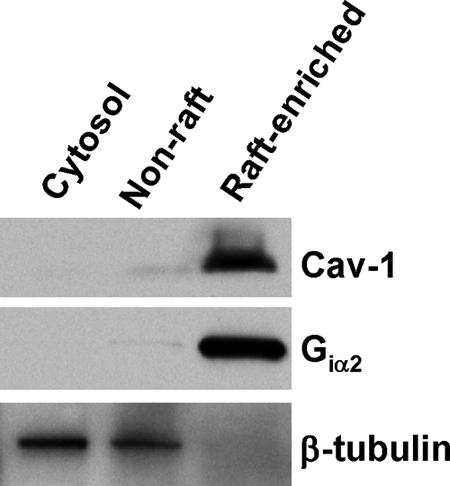
Isolation of lipid raft-enriched and non-raft membranes. DU145 cell lysates were subjected to successive detergent extraction using a method that depletes nuclear proteins from the preparation (21). 10 μg of proteins from cytoplasmic, non-raft, and lipid raft-enriched fractions was separated by SDS-PAGE, electrotransferred, stained with Ponceau S stain to confirm equal loading(supplemental Fig. S7), and immunoblotted with antibodies for the indicated proteins. The data are representative of three independent experiments.
Enrichment and Separation of S-Acylated Proteins
Initially, we optimized the protocols from Davis and co-workers (15,23) for enrichment and separation of S-acylated proteins to minimize background (Fig.2, top and lower left). High concentrations of SDS were used to denature proteins in all samples. TCEP, a potent reducing agent that does not cleave thioester bonds,2 was used to break disulfide bonds. All free thiol groups were irreversibly blocked by NEM, a widely used alkylating agent. Total TCEP-reduced and NEM-alkylated proteins were equally divided into two portions: one was treated with neutral HA (EXP) to specifically cleave off S-acyl moieties (Fig. 2), and the other was treated without HA (CON) (not shown). Subsequently, newly formed thiols were labeled with biotin-HPDP (Fig. 2). The in vitro biotinylated proteins were enriched by streptavidin affinity chromatography and gently eluted by TCEP, leaving endogenous biotinylated proteins and nonspecifically bound proteins on the streptavidin-agarose beads (Fig. 2). Enriched proteins were separated by SDS-PAGE followed by in-gel digestion and LC-MS/MS analysis (Fig. 2). Notably, prior to tryptic digestion, the free thiols (formerly S-acylated) were blocked by iodoacetamide so that the S-acylation sites, which were eventually carbamidomethylated, can be distinguished from non-acylated cysteines, which were blocked by NEM prior to the ABE reaction.
Fig. 2.

Schematic representation of PalmPISC approach for large scale S-acylated protein enrichment and identification as well as S-acylation site characterization. Top, ABE reaction to replace S-acyl groups with biotinyl groups. Lower left, protein-based procedure to enrich and identify S-acylated proteins. Lower right, peptide-based procedure to enrich S-acylated peptides and to localize S-acylation sites.
Fig. 3 shows a representative gel image of EXP and CON groups of proteins, which were purified from equal amounts of total proteins extracted from non-raft (lane 2 and lane 3) and lipid raft-enriched (lane 4 and lane 5) membranes. Optimization of the protocols developed by Davis and co-workers (15, 23) led to low level co-purification of contaminating proteins (lane 2 versus lane 3 and lane 4 versus lane 5). The SDS-PAGE protein patterns were different (lane 2 versus lane 4), suggesting that some S-acylated proteins are in fact concentrated in the lipid raft-enriched fractions, whereas others localize predominantly in the non-raft fractions. In addition, more proteins were found to be purified from the non-raft fractions than from the lipid raft-enriched fractions (lane 2 versus lane 4).
Fig. 3.
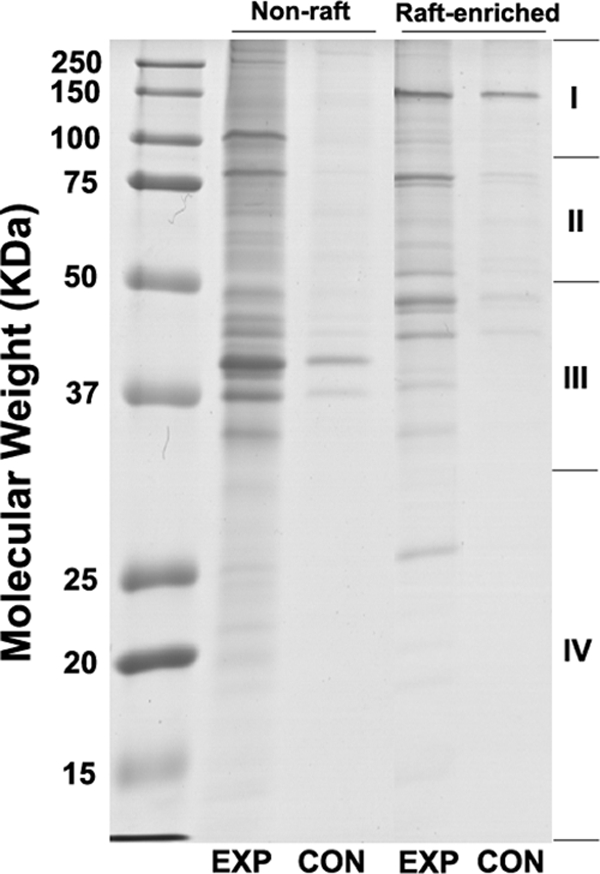
Electrophoretic analysis of +hydroxylamine (EXP) and −hydroxylamine (CON) samples purified from equal amounts of non-raft and lipid raft-enriched membrane fractions using an optimized ABE method. Purified proteins were resolved by 12.5% SDS-PAGE and stained with Coomassie Brilliant Blue R-250 staining solution. Each gel lane was cut into four slices (I, II, III, IV). Data represent at least three independent experiments.
Identification of S-Acylated Proteins
Two biological replicates of enriched proteins (16 groups; non-raft/lipid raft-enriched × EXP/CON × 4 slices) were in-gel digested and analyzed using a nano-LC-LTQ Orbitrap mass spectrometer platform. Fig.4A shows a representative EXP-CON pair of base peak chromatograms of eluted peptide ions, which were derived from the third gel slices of EXP and CON non-raft proteins. Most peptide ions were markedly increased in abundance after HA treatment, suggesting that many peptides derived from proteins containing thioester bonds were greatly enriched in comparison with those derived from the contaminants, which were present in the pair of EXP and CON lanes at the same level. A representative MS/MS spectrum, derived from the analysis of a doubly charged peptide (m/z 965.51) from erythrocyte band 7 integral membrane protein, is depicted in Fig.4B. A total of 928 non-redundant proteins (supplemental Table S1) were identified with at least two unique peptides; each peptide had an ion score no less than 33 (p < 0.05). Using the same criteria, the false discovery rates for proteins and peptides were less than 0.5 and 2%, respectively, when searching the data against a reversed IPI_Human database. Among the proteins identified with high confidence, 793 were from non-raft fractions and 408 were from lipid raft-enriched fractions, with 273 proteins in common (supplemental Table S1).
Fig. 4.

Representative base peak chromatograms (A) and tandem mass spectrum (B) for LC-MS/MS analysis of purified proteins. A, comparison of the EXP and CON chromatograms shows that most peptide ions are much more abundant in EXP than in CON samples. B, MS/MS sequencing of a doubly charged peptide with m/z 965.51 led to the identification of the peptide VQNATLAVANITNADSATR, which is derived from erythrocyte band 7 integral membrane protein.
Some contaminating proteins were repeatedly co-purified with S-acylated proteins. To distinguish S-acylated proteins from contaminants, a widely used label-free quantitative method, spectral counting, was applied (22). For statistical analysis, the spectral counts were merged over all biological replicates and membrane domains. After all missing values in the merged data set were imputed, Bayesian information criterion-based Gaussian mixture modeling (27) suggested that the distribution of the ln(EXP/CON) comprises two Gaussian components (supplemental Fig. S1). The left Gaussian component represents the log ratio distribution of most contaminating proteins and some S-acylated proteins, whereas the right Gaussian component represents the log ratio distribution of most S-acylated proteins and a small number of contaminating proteins. The mean ± S.D. values for the left and the right Gaussian components are 0.64 ± 0.76 and 2.88 ± 0.56, respectively. To distinguish S-acylated proteins from contaminants, the p values were computed based on the distribution of the left Gaussian component. Proteins with p < 0.05 (corresponding to EXP/CON > 6.7) were accepted as high confidence S-acylated protein candidates, whereas those with p values between 0.275 and 0.05 (corresponding to 3.0 ≤ EXP/CON ≤ 6.7) were considered as medium confidence candidates ( supplemental Fig. S1). Among the candidates, the proteins known to use thioester linkages for modifications other than S-acylation (15, 29) are potential false positives. For instance, ubiquitin-activating enzyme E1 links ubiquitin chains, instead of long-chain fatty acids, to Cys residues via thioester linkages (15). Thus, these proteins were removed from the list of S-acylated protein candidates, although S-acylation cannot be completely excluded.
According to these criteria, 169 and 224 proteins were identified to be high confidence and medium confidence candidate S-acylated proteins, respectively (supplemental Tables S2 and S3). Of the 169 high confidence candidates, 34 are known human S-acylated proteins, and 17 are known S-acylated proteins of other mammalian origins, altogether accounting for 30.2% of total high confidence S-acylated protein candidates (supplemental Table S2 and Fig.5). 46 additional proteins were reported previously to be putative S-acylated proteins by using 17-ODYA metabolic labeling and HA treatment (12). In regard to the other 72 proteins, to our knowledge, S-acylation of these proteins has not been reported. Nonetheless, many of these belong to protein families containing well known S-acylated proteins (e.g. Ras, Rho, and G protein-coupled receptor); thus, they are also very likely S-acylated (supplemental Table S2). Among the 224 medium confidence candidate S-acylated proteins, six are known human S-acylated proteins, and five are known S-acylated proteins in other mammalian species (supplemental Table S3). 49 additional proteins have been reported to be S-acylation candidates by using 17-ODYA labeling and HA treatment (12). In addition, many other proteins belong to protein families that contain known S-acylated proteins (e.g. Ras, Rho, syntaxin, tetraspanin, claudin, and casein kinase) or cysteine-rich domains (e.g. DHHC protein), suggesting they are very likely S-acylated. In regard to the other 535 proteins with p > 0.275, albeit many of them are potentially contaminating proteins, five (i.e. GPRIN1, LAT2, LCK, PLD1, and RGS19) are known mammalian S-acylated proteins, and 63 additional proteins were reported to be predicted S-acylated proteins using 17-ODYA labeling and HA treatment (supplemental Table S1).
Fig. 5.

Classification of high confidence candidate S-acylated proteins. A total of 169 high confidence S-acylated protein candidates were classified into four categories: known human S-acylated proteins, known S-acylated proteins of other mammalian origins, putative S-acylated proteins identified by 17-ODYA metabolic labeling and HA treatment (12), and other unknown proteins (see supplemental Table S2 for more details). About 30% of the candidates have been reported to be authentic S-acylated proteins in the peer-reviewed literature.
Fig. 6 shows the comparisons (EXP versus CON) of the total spectral counts of known mammalian S-acylated proteins identified from non-raft (A) and lipid raft-enriched (B) fractions. These data show that S-acylated proteins are highly or exclusively enriched in the EXP groups, strongly suggesting that the PalmPISC approach works as envisioned for S-acylated protein enrichment and identification.
Fig. 6.


Comparison of spectral counts for known mammalian S-acylated proteins identified in non-raft (A) and lipid raft-enriched (B) fractions. All known S-acylated proteins of human and other mammalian origins are highly or exclusively enriched in the EXP samples in comparison with the CON samples.
Localization of S-Acylation Sites
Although the protein-based procedure also provides some information of S-acylation site localization, it is not suitable for large scale characterization of S-acylation sites given that only about 5% of the identified peptides contain carbamidomethylated cysteines (potential S-acylation sites) and that the numbers of spectral counts are low (supplemental Table S4). Therefore, a peptide-based procedure was developed to separate S-acylated peptides from non-acylated peptides (Fig. 2, top and lower right). After ABE chemical reactions, whole protein lysates were digested in solution prior to affinity purification instead of being digested in gel after affinity purification. In vitro biotinylated (potentially formerly S-acylated) peptides were selectively enriched by streptavidin affinity chromatography, and all non-acylated peptides, which may be derived from both S-acylated and non-acylated proteins, were washed away. Bound peptides were gently eluted by TCEP, and originally S-acylated cysteine residues became free cysteines (Fig. 2, lower right). In contrast, non-acylated cysteines were irreversibly blocked by NEM in a previous step.
Two biological replicates of purified peptides were analyzed twice using an LTQ ion trap mass spectrometer for the sake of higher scan speed and sensitivity. A total of 527 unique peptides containing at least one free cysteine residue were identified with ion scores above 41 (p < 0.05). A representative MS/MS spectrum is shown in Fig.7 , from which Cys17 was identified to be one potential S-acylation site of flotillin-1, whereas Cys5 was not. Because a maximum of one miscleavage may give rise to a maximum of three unique peptides, only 475 peptides, which are representative of the 527 peptides, are shown in supplemental Table S5. For statistical analysis, the spectral counts were merged over all biological and technical replicates as well as membrane domains, but no imputation was carried out on the peptide level for two reasons. First, with relatively low spectral counts for the peptides, the effect of imputation was found to exceed the interpeptide differences. Second, imputation would have caused an intense artificial increase in the number of contaminating peptides, distorting the true underlying distribution. Consequently, we estimated the distributions of S-acylated peptides and contaminating peptides based on 102 peptides, for which EXP and CON measurements were available (i.e. spectral count ≥1). p value estimates for this set of peptides were calculated using the aforementioned statistical method for S-acylated protein analysis (supplemental Fig. S2 and Table S5). The peptides with p < 0.05 (EXP/CON > 2.4) were treated as significant and accepted as high confidence S-acylated peptide candidates (supplemental Table S6). Given that peptides with three EXP and zero CON measurements are more likely to be S-acylated than those with three EXP and one CON measurements (i.e. EXP/CON = 3), the peptides not identified in the CON groups but with no less than three EXP measurements were also accepted as high confidence S-acylated peptide candidates (supplemental Table S6). Free cysteines (potentially S-acylated prior to ABE reaction; see Fig. 2) on these high confidence S-acylated peptide candidates were accepted as high confidence candidate S-acylation sites. In addition, the peptides with two EXP and zero CON measurements were considered as medium confidence S-acylated peptide candidates (supplemental Table S7), because although it is uncertain whether they are as likely to be S-acylated as those peptides with an EXP/CON ratio of 2.4 (p = 0.05), they are more likely to be S-acylated than those with two EXP and one CON measurements (EXP/CON = 2, p = 0.095).
Fig. 7.

Representative tandem mass spectrum for S-acylation site characterization. MS/MS analysis of a doubly charged peptide with m/z 1069.2 identified the peptide MOxFFTCNEMGPNEAMVVSGFCR (where MOx is oxidized methionine) derived from flotillin-1. The candidate S-acylation site Cys17 (underlined in the sequence and indicated by ▴ in the figure) can be easily distinguished from non-acylated Cys5, which was previously blocked by NEM.
Using these criteria, a total of 127 non-redundant sites were identified to be high confidence candidate S-acylation sites from 85 proteins, of which 22 and 16 were previously identified to be high confidence and medium confidence S-acylated protein candidates by the protein-based procedure, respectively (supplemental Table S6). Among the 127 high confidence candidate S-acylation sites, 21 have been reported in the peer-reviewed literature and/or annotated in the UniProt database (supplemental Tables S6, S8, and S9). In addition, 39 non-redundant sites were identified as medium confidence S-acylation site candidates (supplemental Tables S5 and S7). These sites were identified from 30 proteins, of which seven and two were identified to be high confidence and medium confidence S-acylated protein candidates using the protein-based procedure, respectively (supplemental Table S7). Among these medium confidence candidate S-acylation sites, two have been reported previously (supplemental Tables S7 and S8). To ensure the accuracy of single peptide-based identification, the MS/MS spectra of all candidate S-acylated peptides were manually verified (supplemental Fig. S3). In regard to the other free cysteines with p > 0.05, although many of them may be non-acylated, at least two (C240C241 on PLD1) have been annotated in the UniProt database (supplemental Tables S5 and S9).
S-Acylation Targeting Certain Ribosomal Proteins to Lipid Rafts
Ribosomal proteins are a major component of ribosomes and play key roles in cellular protein biosynthesis. They are generally localized in both the cytoplasm and nucleus, but are also associated with rough endoplasmic reticulum membranes through the formation of complexes comprising ribosomes, nascent polypeptide chains, signal recognition particles, and rough endoplasmic reticulum-associated signal recognition particle receptors. Surprisingly, the global analysis of S-acylated proteins performed here indicates that certain ribosomal subunits are targeted to lipid raft-enriched membranes through protein S-acylation. To confirm this finding, immunoblotting and immunofluorescence analyses were performed. Localization of a pool of RPL10A and RPL12 proteins in lipid raft-enriched membranes was validated by Western blotting (Fig.8 A). ABE purification followed by Western blotting confirmed that both ribosomal proteins contain thioester bonds, suggesting that they are very likely S-acylated (Fig. 8B). An independent study recently showed that RPL10A and RPL12 can be metabolically labeled by a palmitate analogue, 17-ODYA (12), supporting our conclusion of the likely S-acylation of the ribosomal proteins. Immunofluorescence analysis showed that a fraction of RPL10A proteins co-localize with CTxB, which has been widely used to visualize lipid rafts because it specifically binds to the lipid raft-enriched GM1 ganglioside (Fig.9 , upper panels). Treatment with the protein S-acylation inhibitor 2-bromopalmitate (2-BP) disrupted the localization of RPL10A in lipid rafts and resulted in the accumulation of the ribosomal protein in nuclei (probably nucleoli) (Fig. 9, lower panels). Collectively, these data suggest that protein S-acylation targets a fraction of certain ribosomal proteins to lipid rafts.
Fig. 8.
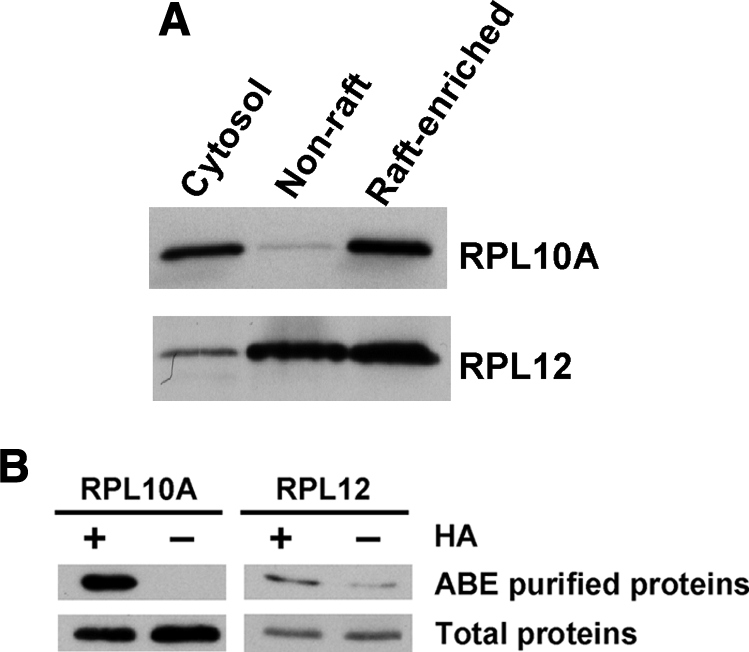
Ribosomal proteins RPL10A and RPL12 exist in lipid raft-enriched membranes and contain thioester bonds. A, 10 μg of proteins from the cytoplasmic, non-raft, and lipid raft-enriched fractions was separated by SDS-PAGE, electrotransferred, and immunoblotted with anti-RPL10A and anti-RPL12 antibodies. B, validation of the existence of thioester bonds in RPL10A and RPL12 proteins using ABE/immunoblotting. Proteins containing thioester bonds and controls were ABE-purified from equal amounts of lipid raft-enriched fractions in the presence and absence of HA, respectively. RPL10A and RPL12 proteins were detected by Western blotting. As a control, portions of the unpurified samples (i.e. total proteins) were also blotted. Data represent at least three independent experiments.
Fig. 9.

Pool of RPL10A proteins localizes in lipid rafts and localization can be disrupted by the general palmitoylation (S-acylation) inhibitor 2-BP. DU145 cells treated with 100 μm 2-BP (lower panels) or vehicle (0.1% ethanol; upper panels) were stained with 0.5 μg/ml FITC-CTxB for 5 min followed by staining with anti-RPL10A mAb (1:100) and Cy3-conjugated secondary antibody (1:250). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Original magnification, 63×.
S-Acylated Protein Network Construction
Increasing evidence shows that reversible protein S-acylation modulates protein-protein interactions (30) and provides a mechanism for regulating the formation of multiprotein complexes. To investigate whether S-acylated proteins interact with each other and form dynamic higher order assemblies, we used IPA software to analyze the potential for direct interactions among high confidence candidate S-acylated proteins identified using protein-based and peptide-based procedures (supplemental Tables S2 and S6) as well as currently known human S-acylated proteins (supplemental Table S8). This analysis revealed that, although about half of the proteins do not directly interact with any other proteins (supplemental Fig. S4, bottom), 151 proteins formed a large network via direct protein-protein interactions, in which two subnetworks emerged (supplemental Fig. S5). One subnetwork, centered around Cav-1, involves 23 other S-acylated proteins (Fig.10 A). The other subnetwork involves a number of tetraspanins, including CD9, CD37, CD53, CD63, CD81, CD82, and CD151 (Fig.10B). This analysis suggests that S-acylated proteins are greatly enriched in the core complexes of caveolae and tetraspanin-enriched microdomains.
Fig. 10.

Virtual S-acylated protein complexes. A number of S-acylated proteins directly interact with each other and form a caveolin-1 complex (A) and a tetraspanin complex (B), suggesting that S-acylated proteins are greatly enriched in the core complexes of caveolae and tetraspanin-enriched microdomains. Cyan, green, and yellow indicate high confidence S-acylated protein candidates identified only by the protein-based procedure, only by the peptide-based procedure, and by both procedures, respectively. White indicates known human S-acylated proteins that were not identified in the present study.
DISCUSSION
In this study, we developed a novel proteomics approach, which we designated PalmPISC, for purification and identification of S-acylated (“palmitoylated”) proteins and peptides. We used this new method to perform the first ever proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membrane domains. This approach allowed us to purify and identify S-acylated proteins and peptides with minimal contamination and to localize S-acylation sites on a large scale. In addition to the technical advance we describe, our study resulted in several important biological findings.
S-Acylated proteins have been hypothesized to be enriched in lipid raft microdomains, which are liquid-ordered membrane patches that reside primarily in the plasma membrane. However, this study revealed that non-raft membranes also contain a considerable amount of S-acylated proteins and that a number of S-acylated proteins show greater enrichment in the non-raft fractions than in the lipid raft-enriched fractions. There may be several reasons for this interesting finding. First, tetraspanin-enriched microdomains, which can be disrupted by Triton X-100 at 4 °C, contain a number of S-acylated proteins (31). Second, some S-acylated proteins (e.g. Ras family proteins) are also prenylated, and prenylation may exclude modified proteins from lipid rafts (32). Indeed, only a few Ras-related proteins were identified in the present study in the lipid raft-enriched fractions (supplemental Table S1). Third, although most S-acylated proteins are acylated with C16:0 palmitate, some S-acylated proteins are acylated with other long-chain fatty acids (e.g. C14:0 myristate, C16:1 palmitoleate, C18:0 stearate, C18:1 oleate, and C20:4 arachidonate) (33, 34), and unsaturated fatty acids may limit association with raft membranes (33, 35). Finally, some S-acylated transmembrane proteins have been found almost exclusively outside raft microdomains. For example, S-acylation of Isoform 1 of tumor endothelial marker TEM8 excludes the protein from lipid rafts, whereas mutations of the cysteine residues target the protein to rafts (36).
DHHC proteins are integral membrane proteins in which PAT activities are conserved from Saccharomyces cerevisiae to mammals (4). Although other PATs do exist (37), most protein S-acylation appears to be catalyzed by the DHHC proteins (15). Mutations in the DHHC box abolish the PAT activities of the proteins, indicating that the cysteine residue in the DHHC box is potentially the catalytic site (38, 39). In our study, DHHC5, a plasma membrane DHHC protein (40), was identified to be a high confidence S-acylated protein candidate that appears to be S-acylated on three cysteine residues downstream of the DHHC-CRD domain (supplemental Tables S1 and S5). But what are the functions of S-acylation on a PAT? One possibility is that protein S-acylation may target DHHC5 to specific membrane microdomains. Another possibility is that S-acylation protects the protein from degradation by preventing its ubiquitination (36). When the protein structure is considered, a far more attractive possibility is that a short loop (NNVSRVL), formed upon S-acylation of DHHC5, may act as a scaffold to recruit specific substrates to DHHC5 and contribute to the specificity of this PAT enzyme (Fig.11 A). Alternatively, the short loop may recruit specific regulators to DHHC5 and block the interactions between DHHC5 and its substrates, consequently inhibiting the S-acylation of the substrates (Fig. 11B). Deacylation of DHHC5 may change the structure of this enzyme and regulate its PAT specificity and/or activity. Interestingly, DHHC6 and DHHC8, which are mainly localized in the endoplasmic reticulum and the Golgi apparatus, respectively (40), were also found to be likely S-acylated on the three cysteines downstream of the DHHC-CRD domain (supplemental Tables S1 and S5). Alignment analysis revealed that the three cysteine residues are embedded in a CCX7–13C(S/T) motif, which may be a conserved mechanism for regulating PAT specificity and/or activity shared by these DHHC proteins (supplemental Fig. S6). Although the other 20 human DHHC proteins do not contain this motif, it is worthwhile to identify all the S-acylation sites of the DHHC proteins and find out whether other types of motifs exist and whether different motifs result in different substrate specificities.
Fig. 11.

Two models for regulating PAT specificity and/or activity by protein S-acylation. DHHC5 appears to be S-acylated on three cysteine residues downstream of the DHHC-CRD domain. As a result, a seven-amino acid (238NNVSRVL244) loop is formed, which may function as a scaffold to recruit specific substrates (A) or regulators (B) to DHHC5 and contribute to the specificity and/or activity of this PAT.
Currently, only two protein thioesterases (i.e. APT1 and protein palmitoylthioesterase-1) have been characterized as capable of deacylating S-acylated proteins (6). APT1, which is predominantly cytoplasmic, has been shown to deacylate a number of S-acylated proteins such as Ras, Gα subunits, and SNAP-23 (6). However, it remains unclear how cytoplasmic APT1 can efficiently access its substrates, which are all membrane-associated. In the present study, APT1 was identified to be a putative S-acylated protein (supplemental Table S3). Its S-acylation has further been validated by ABE/immunoblotting and 2-BP inhibition studies.3 The S-acylation of APT1 may tether this deacylating enzyme to membranes and facilitate its interaction with and deacylation of its substrates. Moreover, the level of membrane-associated APT1 can be dynamically regulated because protein S-acylation is a reversible lipid modification.
The presence of certain ribosomal proteins in lipid rafts has recently been reported in an unbiased quantitative proteomics study of lipid rafts (41), yet the mechanism(s) of their association with rafts remains unclear. In this study, proteomics analysis revealed that some ribosomal proteins were very likely S-acylated (supplemental Tables S1 and S5). Immunoblotting and immunofluorescence analyses confirmed that RPL10A was targeted to lipid raft microdomains through protein S-acylation. Given that lipid rafts are enriched in kinases and phosphatases, reversible S-acylation of ribosomal proteins may mediate their interactions with lipid raft-resident kinases and phosphatases, thereby providing a mechanism for the regulation of ribosomal protein activity. Another possibility is that a pool of ribosomes may be tethered to or assembled in lipid rafts through S-acylation of ribosomal proteins to facilitate the synthesis of raft proteins in situ. Moreover, the protein synthesis rate might be regulated by different levels of lipid raft-resident ribosomes, which are controlled by dynamic S-acylation of ribosomal proteins.
Tetraspanins are a family of integral transmembrane proteins exhibiting substantial sequence identity and common structural features (42). Increasing evidence shows that tetraspanins affect a broad spectrum of processes such as cell proliferation, apoptosis, and tumor metastasis. Notably, these molecules have an extraordinary capability to associate with each other and with many signaling enzymes, consequently forming the “tetraspanin web,” which may act as a new type of signaling platform distinct from lipid rafts (42, 43). IPA analysis of all known and high confidence candidate human S-acylated proteins revealed that S-acylated proteins were disproportionately represented in the core complexes of tetraspanin-enriched microdomains (Fig.10B) as well as caveolae (Fig. 10A), both cholesterol-rich membrane structures. It would be interesting to know whether certain PATs (e.g. DHHC5) and acyl-protein thioesterases are also included in or close to the core complexes and regulate their compositions. Because the long-chain fatty acids on S-acylated proteins can turn over quickly and can be regulated by exogenous stimulation, high levels of S-acylation may play a crucial role in dynamic formation and disassembly of multiprotein signaling complexes that localize to discrete subcellular locations (44). Interestingly, 2-BP treatment of DU145 cells led to the disruption of lipid rafts (Fig. 9, lower left panel), probably through the inhibition of S-acylation of a variety of S-acylated proteins, especially those in the core complexes of caveolae.
Using the spectral counting method and after statistical analysis, a total of 393 candidate S-acylated proteins and 166 candidate S-acylation sites were identified after in-gel and in-solution digestion, respectively (Table I). A rather different spectrum of S-acylated proteins (see supplemental Tables S6 and S7) was identified by the two different procedures because each procedure has its own merits and weaknesses. The protein-based procedure may distinguish different isoforms (e.g. three isoforms of Gαs based on at least one non-redundant peptide) because higher protein coverage can be achieved by this procedure than by the peptide-based procedure. Likewise, proteins can be identified with higher confidence by the former procedure than by the latter. In contrast, the peptide-based procedure allows the identification of small proteins (e.g. CDC42SE1) that may be lost during SDS-PAGE separation. Another distinct advantage of the peptide-based procedure is that the relative quantification of S-acylated peptides is not subject to interference by abundant contaminating peptides derived from the same protein. For example, actin may be S-acylated on Cys16 of the peptide CPEALFQPSFLGMESCGIHETTFNSIMK (supplemental Table S5). However, the contaminating N-terminal peptides DDDIAALVVDNGSGMCK and EEEIAALVIDNGSGMCK were identified with much higher spectral counts; thus, the actins were classified into the contaminating group according to the criteria for the protein-based procedure (supplemental Table S1). Nonetheless, the peptide-based procedure has its intrinsic defects. Peptides too large or too small may not be identified by mass spectrometry. Moreover, S-acylated peptides containing other modifications such as myristoylation, prenylation, phosphorylation, or glycosylation will not be identified if these modifications are not considered in the database search. Unsurprisingly, few Ras family proteins, whose farnesylation and S-acylation sites are closely spaced in the C terminus, were identified using the peptide-based procedure. Predictably, the use of different endoproteases such as trypsin, Glu-C, and Asp-N and/or removal of myristoyl, prenyl, phosphoryl, and glycosyl groups will facilitate the identification of S-acylation sites.
Table I.
Summary of the identified human S-acylated proteins and S-acylation sites
| Group and category | Known human | Known other species | 17-ODYAa | Others | Total |
|---|---|---|---|---|---|
| Proteins | |||||
| High confidence | 34 | 17 | 46 | 72 | 169 |
| Medium confidence | 6 | 5 | 49 | 164 | 224 |
| Others | 2 | 3 | 63 | 467 | 535 |
| Sites | |||||
| High confidence | 13 | 8 | 106 | 127 | |
| Medium confidence | 2 | 0 | 37 | 39 | |
| Others | 2 | 393 | 395 |
a S-Acylated protein candidates identified by using 17-ODYA metabolic labeling and HA treatment (12).
Unlike other lipid modifications such as myristoylation and farnesylation, which have well defined consensus sequences, S-acylation lacks a universal consensus motif (45). However, cysteine residues adjacent to or near N-myristoylated glycine residues, immediately upstream of prenylated C-terminal cysteine residues, in juxtamembrane regions, or in cysteine-rich domains are often S-acylated, indicating that specificity determinants do exist. However, because of an insufficient number of experimentally validated S-acylation sites, programs developed for prediction of S-acylation sites are not very reliable (7, 8). The PalmPISC approach will accelerate the identification of novel S-acylation sites, thereby improving the accuracy of these programs. In addition, growing evidence shows that DHHC proteins have substrate specificity (4, 15, 46). Systematic knockdown or specific inhibition of the PATs in combination with the PalmPISC approach and quantitative proteomics techniques will establish PAT-substrate relationships on a large scale and may finally reveal DHHC subfamily-specific consensus motifs.
PalmPISC is a robust new tool for global analysis of protein S-acylation; however, it has limitations. First, the ABE-based method is thioester bond-specific but not S-acylation-specific; thus, the false discovery rates derived from other modifications containing thioester bonds are relatively high (∼5–20%). Second, S-acylated proteins can be labeled with fatty acids other than palmitate (33–35). The identities of the long-chain fatty acids attached to S-acylated proteins cannot be determined by PalmPISC because all fatty acids are removed after ABE chemistry. However, neither can the traditional [3H]palmitate in vivo labeling method make this distinction, because [3H]palmitate is often converted into other 3H-fatty acid species during metabolic labeling. The problem can potentially be addressed by gas chromatography-MS, which allows direct identification of lipids attached to proteins. However, gas chromatography-MS is insufficiently sensitive and requires protein purification from large quantities of starting material (47). In contrast, LC-MS is more sensitive and has been successfully applied to rapidly characterize post-translationally modified peptides such as phosphopeptides and glycopeptides. However, S-acylated peptides are generally very hydrophobic and hard to separate and analyze by LC-MS. To address the above mentioned problems, we have developed a novel LC-MS method to validate protein S-acylation and identify the long-chain fatty acids that has been successfully applied to characterize 20 ng of in vitro prepared S-acylated tubulins.3 As might be expected, differential proteomics analysis by PalmPISC followed by detailed characterization of putative S-acylated proteins of interest will facilitate the discovery of authentic S-acylated proteins involved in many biological processes, such as cell growth and differentiation, apoptosis, oncogenesis, and metastasis.
In conclusion, the novel PalmPISC approach in combination with the spectral counting method allowed us to identify a total of 67 known and 331 novel candidate S-acylated proteins as well as 25 known and 143 novel candidate S-acylation sites, comprising a large scale data set of human S-acylated proteins (Table I). S-Acylation of three cysteines within the CCX7–13C(S/T) motif downstream of the DHHC-CRD domain may be a consensus determinant of the specificity and/or the activity of three PATs (i.e. DHHC5, DHHC6, and DHHC8). APT1 may be S-acylated and thus be targeted to membranes to deacylate its S-acylated protein substrates. S-Acylation of certain ribosomal proteins may be involved in lipid raft microdomain targeting, activity modulation, and/or lipid raft protein biosynthesis rate regulation. Finally, S-acylated proteins were found to be highly enriched in the core complexes of caveolae and tetraspanin-enriched microdomains, suggesting that reversible S-acylation plays a pivotal role in regulating the dynamics of the multiprotein assemblies associated with these structures. The PalmPISC approach in combination with other powerful techniques such as quantitative proteomics, RNA interference, and cell imaging techniques will greatly enhance our understanding of the functions and mechanisms of protein S-acylation and facilitate the discovery of disease mechanisms, biomarkers, and novel drug targets.
Supplementary Material
Acknowledgments
We thank Judith A. J. Steen and Rosalyn M. Adam for valuable discussions and Zachary Waldon and Yin Yin Lin for assistance with mass spectrometry. We are also grateful to all the members of the Freeman laboratory and the Steen and Steen laboratory for constructive comments.
Footnotes
* This work was supported, in whole or in part, by National Institutes of Health Grants P50 DK65298, R37 DK47556, and R01CA112303 (to M. R. F.) and Grant K99 CA131472 from the NCI (to D. D. V). This work was also supported by Department of Defense Prostate Cancer Research Program Grant W81XWH-08-1-0139 (to W. Y.).
 The on-line version of this article (available at http://www.mcponline.org) contains supplemental Tables S1–S9 and Figs. S1–S7
The on-line version of this article (available at http://www.mcponline.org) contains supplemental Tables S1–S9 and Figs. S1–S7
2 W. Yang, D. Di Vizio, M. Kirchner, H. Steen, and M. R. Freeman, unpublished observations.
3 W. Yang, D. Di Vizio, M. Kirchner, H. Steen, and M. R. Freeman, manuscript in preparation.
1 The abbreviations used are:
- PAT
- palmitoyl acyltransferase
- ABE
- acyl-biotinyl exchange
- APT1
- acyl-protein thioesterase-1
- biotin-HPDP
- N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide
- Cav-1
- caveolin-1
- CM
- chloroform/methanol
- CON
- control
- CRD
- cysteine-rich domain
- CTxB
- cholera toxin B subunit
- DHHC
- Asp-His-His-Cys
- DRM
- detergent-resistant membrane
- EXP
- experimental
- HA
- hydroxylamine
- IPA
- Ingenuity Pathway Analysis
- NEM
- N-ethylmaleimide
- PalmPISC
- palmitoyl protein identification and site characterization
- RP
- ribosomal protein
- RT
- room temperature
- 17-ODYA
- 17-octadecynoic acid
- TCEP
- tris(2-carboxyethyl)phosphine
- 2-BP
- 2-bromopalmitate
- LTQ
- linear trap quadrupole
- pAb
- polyclonal antibody
- mAb
- monoclonal antibody
- IPI
- International Protein Index
- GM1
- Galβ1–3GalNAcβ1–4Gal(3–2αNeuAc)β1–4Glcβ1–1Cer
REFERENCES
- 1.Linder M. E., Deschenes R. J. (2007) Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol 8, 74–84 [DOI] [PubMed] [Google Scholar]
- 2.Resh M. D. (2006) Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci. STKE 2006, re14. [DOI] [PubMed] [Google Scholar]
- 3.Greaves J., Chamberlain L. H. (2007) Palmitoylation-dependent protein sorting. J. Cell Biol 176, 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsutsumi R., Fukata Y., Fukata M. (2008) Discovery of protein-palmitoylating enzymes. Pfluegers Arch. Eur. J. Physiol 456, 1199–2206 [DOI] [PubMed] [Google Scholar]
- 5.Baekkeskov S., Kanaani J. (2009) Palmitoylation cycles and regulation of protein function. Mol. Membr. Biol 26, 42–54 [DOI] [PubMed] [Google Scholar]
- 6.Zeidman R., Jackson C. S., Magee A. I. (2009) Protein acyl thioesterases. Mol. Membr. Biol 26, 32–41 [DOI] [PubMed] [Google Scholar]
- 7.Ren J., Wen L., Gao X., Jin C., Xue Y., Yao X. (2008) CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng. Des. Sel 21, 639–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xue Y., Chen H., Jin C., Sun Z., Yao X. (2006) NBA-Palm: prediction of palmitoylation site implemented in naive Bayes algorithm. BMC Bioinformatics 7, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roth A. F., Wan J., Green W. N., Yates J. R., Davis N. G. (2006) Proteomic identification of palmitoylated proteins. Methods 40, 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hang H. C., Geutjes E. J., Grotenbreg G., Pollington A. M., Bijlmakers M. J., Ploegh H. L. (2007) Chemical probes for the rapid detection of fatty-acylated proteins in mammalian cells. J. Am. Chem. Soc 129, 2744–2745 [DOI] [PubMed] [Google Scholar]
- 11.Kostiuk M. A., Corvi M. M., Keller B. O., Plummer G., Prescher J. A., Hangauer M. J., Bertozzi C. R., Rajaiah G., Falck J. R., Berthiaume L. G. (2008) Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J 22, 721–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin B. R., Cravatt B. F. (2009) Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 6, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menendez J. A., Colomer R., Lupu R. (2005) Why does tumor-associated fatty acid synthase (oncogenic antigen-519) ignore dietary fatty acids? Med. Hypotheses 64, 342–349 [DOI] [PubMed] [Google Scholar]
- 14.Drisdel R. C., Green W. N. (2004) Labeling and quantifying sites of protein palmitoylation. BioTechniques 36, 276–285 [DOI] [PubMed] [Google Scholar]
- 15.Roth A. F., Wan J., Bailey A. O., Sun B., Kuchar J. A., Green W. N., Phinney B. S., Yates J. R., 3rd, Davis N. G. (2006) Global analysis of protein palmitoylation in yeast. Cell 125, 1003–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang R., Wan J., Arstikaitis P., Takahashi H., Huang K., Bailey A. O., Thompson J. X., Roth A. F., Drisdel R. C., Mastro R., Green W. N., Yates J. R., 3rd, Davis N. G., El-Husseini A. (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456, 904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J., Planey S. L., Ceballos C., Stevens S. M., Jr., Keay S. K., Zacharias D. A. (2008) Identification of CKAP4/p63 as a major substrate of the palmitoyl acyltransferase DHHC2, a putative tumor suppressor, using a novel proteomics method. Mol. Cell. Proteomics 7, 1378–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobson K., Mouritsen O. G., Anderson R. G. (2007) Lipid rafts: at a crossroad between cell biology and physics. Nat. Cell Biol 9, 7–14 [DOI] [PubMed] [Google Scholar]
- 19.Freeman M. R., Solomon K. R. (2004) Cholesterol and prostate cancer. J. Cell. Biochem 91, 54–69 [DOI] [PubMed] [Google Scholar]
- 20.Brown D. A. (2006) Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 21, 430–439 [DOI] [PubMed] [Google Scholar]
- 21.Adam R. M., Yang W., DiVizio D., Mukhopadhyay N. K., Steen H. (2008) Rapid preparation of nuclei-depleted detergent-resistant membrane fractions suitable for proteomics analysis. BMC Cell Biol 9, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu H., Sadygov R. G., Yates J. R., 3rd (2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 23.Wan J., Roth A. F., Bailey A. O., Davis N. G. (2007) Palmitoylated proteins: purification and identification. Nat. Protoc 2, 1573–1584 [DOI] [PubMed] [Google Scholar]
- 24.Yang W., Liu P., Liu Y., Wang Q., Tong Y., Ji J. (2006) Proteomic analysis of rat pheochromocytoma PC12 cells. Proteomics 6, 2982–2990 [DOI] [PubMed] [Google Scholar]
- 25.Di Vizio D., Kim J., Hager M. H., Morello M., Yang W., Lafargue C. J., True L. D., Rubin M. A., Adam R. M., Beroukhim R., Demichelis F., Freeman M. R. (2009) Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res 69, 5601–5609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Renard B. Y., Kirchner M., Monigatti F., Ivanov A. R., Rappsilber J., Winter D., Steen J. A., Hamprecht F. A., Steen H. (2009) When less can yield more—computational preprocessing of MS/MS spectra for peptide identification. Proteomics 9, 4978–4984 [DOI] [PubMed] [Google Scholar]
- 27.Fraley C., Raftery A. E. (2002) Model-based clustering, discriminant analysis, and density estimation. J. Am. Stat. Assoc 97, 611–631 [Google Scholar]
- 28.Di Vizio D., Adam R. M., Kim J., Kim R., Sotgia F., Williams T., Demichelis F., Solomon K. R., Loda M., Rubin M. A., Lisanti M. P., Freeman M. R. (2008) Caveolin-1 interacts with a lipid raft-associated population of fatty acid synthase. Cell Cycle 7, 2257–2267 [DOI] [PubMed] [Google Scholar]
- 29.Kursula P., Ojala J., Lambeir A. M., Wierenga R. K. (2002) The catalytic cycle of biosynthetic thiolase: a conformational journey of an acetyl group through four binding modes and two oxyanion holes. Biochemistry 41, 15543–15556 [DOI] [PubMed] [Google Scholar]
- 30.Smotrys J. E., Linder M. E. (2004) Palmitoylation of intracellular signaling proteins: regulation and function. Annu. Rev. Biochem 73, 559–587 [DOI] [PubMed] [Google Scholar]
- 31.Le Naour F., André M., Boucheix C., Rubinstein E. (2006) Membrane microdomains and proteomics: lessons from tetraspanin microdomains and comparison with lipid rafts. Proteomics 6, 6447–6454 [DOI] [PubMed] [Google Scholar]
- 32.Melkonian K. A., Ostermeyer A. G., Chen J. Z., Roth M. G., Brown D. A. (1999) Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J. Biol. Chem 274, 3910–3917 [DOI] [PubMed] [Google Scholar]
- 33.Liang X., Nazarian A., Erdjument-Bromage H., Bornmann W., Tempst P., Resh M. D. (2001) Heterogeneous fatty acylation of Src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J. Biol. Chem 276, 30987–30994 [DOI] [PubMed] [Google Scholar]
- 34.Hallak H., Muszbek L., Laposata M., Belmonte E., Brass L. F., Manning D. R. (1994) Covalent binding of arachidonate to G protein alpha subunits of human platelets. J. Biol. Chem 269, 4713–4716 [PubMed] [Google Scholar]
- 35.Casey W. M., Gibson K. J., Parks L. W. (1994) Covalent attachment of palmitoleic acid (C16:1 delta 9) to proteins in Saccharomyces cerevisiae. Evidence for a third class of acylated proteins. J. Biol. Chem 269, 2082–2085 [PubMed] [Google Scholar]
- 36.Abrami L., Leppla S. H., van der Goot F. G. (2006) Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J. Cell Biol 172, 309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xue L., Gollapalli D. R., Maiti P., Jahng W. J., Rando R. R. (2004) A palmitoylation switch mechanism in the regulation of the visual cycle. Cell 117, 761–771 [DOI] [PubMed] [Google Scholar]
- 38.Lobo S., Greentree W. K., Linder M. E., Deschenes R. J. (2002) Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J. Biol. Chem 277, 41268–41273 [DOI] [PubMed] [Google Scholar]
- 39.Roth A. F., Feng Y., Chen L., Davis N. G. (2002) The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J. Cell Biol 159, 23–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohno Y., Kihara A., Sano T., Igarashi Y. (2006) Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim. Biophys. Acta 1761, 474–483 [DOI] [PubMed] [Google Scholar]
- 41.Foster L. J., De Hoog C. L., Mann M. (2003) Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. U.S.A. 100, 5813–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hemler M. E. (2005) Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol 6, 801–811 [DOI] [PubMed] [Google Scholar]
- 43.Boucheix C., Rubinstein E. (2001) Tetraspanins. Cell. Mol. Life Sci 58, 1189–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang W., Steen H., Freeman M. R. (2008) Proteomic approaches to the analysis of multiprotein signaling complexes. Proteomics 8, 832–851 [DOI] [PubMed] [Google Scholar]
- 45.Meinnel T., Giglione C. (2008) Protein lipidation meets proteomics. Front. Biosci 13, 6326–6340 [DOI] [PubMed] [Google Scholar]
- 46.Planey S. L., Zacharias D. A. (2009) Palmitoyl acyltransferases, their substrates, and novel assays to connect them. Mol. Membr. Biol 26, 14–31 [DOI] [PubMed] [Google Scholar]
- 47.Sorek N., Poraty L., Sternberg H., Bar E., Lewinsohn E., Yalovsky S. (2007) Activation status-coupled transient S acylation determines membrane partitioning of a plant Rho-related GTPase. Mol. Cell. Biol 27, 2144–2154 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.