

Abstract
Objectives
To investigate the effect of location, coding type, and topology of KCNH2(hERG) mutations on clinical phenotype in Type-2 long-QT syndrome.
Backgrounds
Previous studies were limited by population size in their ability to examine phenotypic effect of location, type and topology.
Methods
Study subjects included 858 Type-2 long-QT syndrome patients with 162 different KCNH2 mutations in 213 proband-identified families. The Cox proportional-hazards survivorship model was used to evaluate independent contribution of clinical and genetic factors to the first cardiac events.
Results
For patients with missense mutations, the transmembrane pore (S5-loop-S6) and N-terminus regions were a significantly greater risk than the C-terminus region (HR=2.87 and 1.86, respectively), but the transmembrane non-pore (S1-S4) region was not (HR=1.19). Additionally, the transmembrane pore region was significantly riskier than the N-terminus or transmembrane non-pore regions (HR=1.54, 2.42). However, for non-missense mutations, these other regions were no longer riskier than the C-terminus (HR=1.13, 0.77 and 0.46, respectively). Likewise, subjects with non-missense mutations were at significantly higher risk than those with missense mutations in the C-terminus region (HR=2.00), but this was not the case in other regions. This mutation location-type interaction was significant (p-value=0.008). A significantly higher risk was found in subjects with mutations located in α-helical domains than in those with mutations in β-sheet domains or other locations (HR=1.74 and 1.33, respectively). Time-dependent β-blocker use was associated with a significant 63% reduction in the risk of first cardiac events (p<0.001).
Conclusions
KCNH2 missense mutations located in the transmembrane S5-loop-S6 region are associated with the greatest risk.
Keywords: arrhythmia, electrocardiography, long-QT syndrome, genetics, syncope
Introduction
Long QT syndrome (LQTS) is a congenital disorder caused by mutations of several cardiac ion channel genes and is diagnosed clinically by a prolonged QT interval on the electrocardiogram (ECG) and variable clinical outcomes including arrhythmia-related syncope and sudden death.1,2 Mutations involving the KCNH2 gene (hERG, human ether-a-go-go-related gene), which codes for the pore forming α-subunit of a cardiac K+ channel, have been linked to the type-2 LQTS, the second most common variant of LQTS.3 KCNH2 mutations lead to a reduction in the rapid component of the delayed rectifier repolarizing current (IKr), which contributes to lengthening of the QT interval.4 KCNH2 subunits oligomerize to form a tetramer that inserts into the cell membrane to form the functional K+ channel. Each subunit is comprised of six α-helical transmembrane segments (S1-S6), where the K+-selective pore is found between S5-S6. The transmembrane segments are flanked by amino (N)- and carboxyl (C)- terminus regions.5-8 In a previous study of patients with type-2 LQTS, mutations in the pore region were associated with an increased risk for arrhythmia-related cardiac events when compared to patients with non-pore mutations.9 However, this study was limited by population size in its ability to examine the phenotypic effect of mutations within distinct domains of the non-pore region.
There are several coding types of mutations in genes that form the functional K+ channel; missense, nonsense, splice site, in-frame deletion, and frameshift mutations.10 Missense mutations are point mutations that result in a single amino acid change within the protein, nonsense mutations generate a stop codon and can truncate the protein. Insertion and deletion mutations cause in-frame or frameshift mutations, the latter of which change the grouping of nucleotide bases into codons. Splice site mutations may alter splicing of mRNA. In our recent cohort of type-1 LQTS,11 a missense mutation accounted for 81 percent of all the mutations, and the type of mutation (missense vs. non-missense) was not an independent risk factor. On the other hand, non-missense mutations such as frameshift and nonsense mutations have been reported to be more frequently identified in the type-2 LQTS patients.11,12
Moreover, topology of mutations (α-helical domain, β-sheet domain, and other uncategorized location) has been recently reported to relate to the function of mutated channel in the type-2 LQTS patients.8
We hypothesized that the distinct location, coding type and topology of the channel mutation would have important influence on the phenotypic manifestations and clinical course of patients with type-2 LQTS. To test this hypothesis, we investigated the clinical aspects of 858 subjects having a spectrum of KCNH2 mutations categorized by the distinct location, coding type, and topology of the channel mutations.
Methods
Study Population
The study population of 858 subjects was derived from 213 proband-identified families with genetically confirmed KCNH2 mutations. The proband in each family had corrected QT (QTc) prolongation not due to a known cause. The subjects were drawn from the U.S. portion of the International LQTS (Rochester) Registry (n=456), The Netherlands' (Amsterdam) LQTS Registry (n=214), the Japanese (National Cardiovascular Center) LQTS Registry (n=95), and the Mayo Clinic LQTS Registry (n=93). All subjects or their guardians provided informed consent for the genetic and clinical studies. Not included in the study population were 58 subjects with evidence of two or more LQTS mutations and an additional 18 who had polymorphisms (p.R176W or p.R1047L) that the authors felt might reduce IKr current. Two hundred one of the 456 patients enrolled from the U.S. portion of the International LQTS Registry and 61 of the 95 patients from the Japanese LQTS Registry were reported in our prior reports.9,12
Phenotype Characterization
Routine clinical and electrocardiographic parameters were acquired at the time of enrollment in each of the registries. Follow-up was censored at age 41 to minimize the influence of coronary disease on cardiac events. Measured parameters on the first recorded ECG included QT and R-R intervals in milliseconds, with QT corrected for heart rate by Bazett's formula. The QTc interval was expressed in its continuous form and categorized into four levels: <460msec, 460-499msec, 500-530msec, and >530msec. The QTc interval was categorized into three levels: <500msec, 500-530msec, and >530msec for the endpoint of lethal cardiac events (aborted cardiac arrest or LQTS-related sudden cardiac death), because there were few lethal cardiac events in the lowest QTc group (<460msec). Clinical data were collected on prospectively designed forms with information on demographic characteristics, personal and family medical history, electrocardiographic findings, therapy, and endpoints during long-term follow-up. Data common to all 4 LQTS registries involving genetically identified patients with type-2 LQTS genotype were electronically merged into a common database for this study.
Genotype Characterization
The KCNH2 mutations were identified using standard genetic tests performed in molecular-genetic laboratories in the participating academic centers. From the Rochester registry there were 60 subjects who died of sudden cardiac death at a young age and were not genotyped. These 60 subjects were assumed to have the same KCNH2 mutation as other affected close members of their respective family.
Genetic alterations of the amino acid sequence were characterized by location in the channel protein, by the type of mutation (missense, splice site, in-frame insertions/deletions, nonsense [stop codon], and frameshift), and by the topology of mutation (α-helical domain, β-sheet domain, and other uncategorized location) (Figure 1). The transmembrane region of the KCNH2 encoded channel was defined as the coding sequence involving amino acid residues from 398 through 657 (S5-loop-S6 region: 552-657), with the N-terminus region defined before residue 398, and the C-terminus region after residue 657 (Figure 1).13,14
Figure 1. Diagramatic location of 162 different mutations in the KCNH2 potassium channel involving 858 subjects.
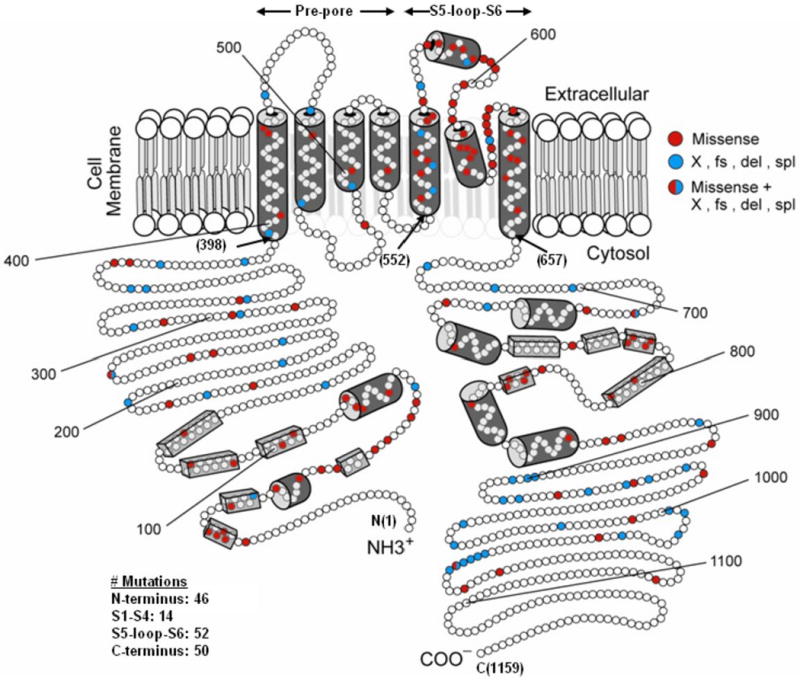
The α subunit involves the N-terminus (NH3+), six membrane-spanning segments, and the C-terminus portion (COO-). The numbers in the parentheses refer to the position of the amino acid beginning at the N-term position (1), the beginning of the transmembrane non-pore S1-S4 sequence (398), the beginning of the transmembrane S5-loop-S6 sequence (552), the end of the transmembrane S6 sequence (657), and at the C-term end position (1159). The open circles represent individual amino acids, the red circles indicate the missense mutations, and the blue circles indicate non-missense mutations. The cylinders represent putative α-helical segments, and the bars represent putative β-sheets.
We evaluated the risk associated with 4 main prespecified regions: 1) N-terminus, 2) transmembrane “non-pore” region (S1-S4), 3) transmembrane “pore” region (S5-loop-S6), and 4) C- terminus. We also evaluated the risk associated with distinct types of mutation and topology of mutation.
Statistical Analysis
Differences in the univariate characteristics by specific groupings were evaluated by standard statistical methods. The primary end point was time to syncope, aborted cardiac arrest, or sudden death, whichever occurred first. The cumulative probability of a first cardiac event was assessed by the Kaplan-Meier method with significance testing by the log-rank statistic. The Cox proportional-hazards survivorship model was used to evaluate the independent contribution of clinical and genetic factors to the first occurrence of time-dependent cardiac events from birth through age 40 years.15 The Cox regression models, stratified by decade of birth year, and allowing for time-dependent covariates, were fit to estimate the adjusted hazard ratio of each factor as a predictor of first cardiac events. We observed that sex was not proportional as a function of age with crossover in risk at age 13 on univariate Kaplan-Meier analysis. In order to fulfill the assumption of proportional hazards for sex over the entire age range, a time-dependent covariate for gender (via an interaction with time) was incorporated, allowing for different hazard ratios by sex before and after age 13 years.
Since almost all the subjects were first- and second- degree relatives of probands, the effect of potential lack of independence between subjects was evaluated by refitting the Cox model using the robust sandwich estimator for family membership.16 All significant predictors of risk maintained significance using this robust measure of variance.
Patients who didn't have an ECG for QTc measurement were identified in the Cox models as “QTc missing”. Pre-specified covariate interactions between mutation location, type and α-helical domains were evaluated. Only the mutation location - missense interaction was significant. In order to test the impact of the interaction between the 4 different mutation locations and missense mutation type, 3 interaction terms were added to the Cox proportional hazards regression model. A 3 degree of freedom likelihood-ratio test was performed to determine their statistical significance. The influence of time-dependent β-blocker therapy (the age at which β-blocker therapy was initiated) on outcome was determined by adding this variable to the final Cox model containing the various covariates.
Results
Total Study Population
The continuum of KCNH2 mutations and their respective number of subjects by location, type, and topology of mutation and contributing registry are presented in the Supplementary Table, and the location, type, and topology of the mutations are diagrammatically presented in Figure 1. A total of 162 different KCNH2 mutations were identified in 858 subjects. The mutations were predominantly found in three regions: the N-terminus (28.4%, n=46), the C-terminus (30.9%, n=50), and the transmembrane domain (40.7%, n=66). Of the 66 mutations within the transmembrane domain, 78.8% (n=52) were located within the S5-loop-S6 region. Missense (single amino acid substitutions) accounted for 61.7% (n=100) of all the mutations, splice site for 1.9% (n=3), in-frame insertions/deletions for 0.6% (n=1), nonsense for 10.5% (n=17), and frameshift for 25.3% (n=41). Sixty six mutations (40.7%) were located in the α-helical domain, 17 (10.5%) in the β-sheet domain, and 79 (48.8%) in other uncategorized location.
The phenotypic characteristics of patients enrolled in each of the 4 registries and by location, type and topology of mutation are presented in Table 1. The age was younger in the Mayo Clinic registry than the other 3 registries. The QTc interval was longer and the cardiac events were more frequent in the U.S. and Japanese registries than the other 2 registries. A pacemaker was more frequently implanted in the U.S. registry, and a defibrillator in the Mayo Clinic registry. LQTS-related death was more frequent in the U.S. registry than the other 3 registries. This seems mainly due to the facts that the U.S. registry included the largest proportion of patients missing ECG data and was the longest-standing registry in which 44 of the 92 deaths occurred before 1980. It is not surprising that the death rate in subjects missing ECG data (i.e. QTc) was very high.
Table 1.
Phenotypic Characteristics by Source of Subjects, Location of Mutation, Type of Mutation, and Topology of Mutation
| Source of Subjects | Location of Mutation | Type of Mutation | Topology of Mutation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics | Rochester | Netherlands | Japan | Mayo | Transmembrane (S1-S4) | Transmembrane (S5- loop -S6) | N-Terminus | C-Terminus | Missense | Frameshift/nonsense | Others | α-helices | β-sheet | Neither |
| # unique mutations | 14 | 52 | 46 | 50 | 100 | 58 | 4 | 66 | 17 | 79 | ||||
| # of Patients | 456 | 214 | 95 | 93 | 44 | 259 | 243 | 312 | 555 | 261 | 42 | 319 | 141 | 398 |
| Female, % | 57 | 59 | 66 | 57 | 57 | 59 | 56 | 60 | 60 | 56 | 55 | 57 | 55 | 61 |
| ECG at enrollment | ||||||||||||||
| Age†‡§, yr | 25±20 | 33±21 | 30±18 | 22±16 | 28±16 | 24±19 | 32±21 | 26±19 | 28±20 | 27±19 | 26±22 | 25±19 | 31±22 | 28±19 |
| QTc†‡¶§, sec | 0.49±0.06 | 0.47±0.05 | 0.49±0.05 | 0.47±0.05 | 0.48±0.05 | 0.50±0.06 | 0.47±0.06 | 0.48±0.05 | 0.49±0.06 | 0.47±0.05 | 0.47±0.06 | 0.49±0.05 | 0.48±0.05 | 0.48±0.06 |
| QTp†‡, sec | 0.34±0.07 | 0.33±0.05 | 0.38±0.06 | 0.34±0.06 | 0.37±0.07 | 0.36±0.07 | 0.34±0.06 | 0.34±0.07 | 0.35±0.07 | 0.34±0.06 | 0.34±0.08 | 0.35±0.07 | 0.33±0.07 | 0.34±0.06 |
| Therapy, % | ||||||||||||||
| β-blockers | 51 | 45 | 45 | 60 | 48 | 53 | 45 | 51 | 49 | 51 | 50 | 51 | 48 | 49 |
| Pacemaker† | 6.8 | 0.5 | 1.1 | 0 | 2.3 | 6.6 | 2.5 | 2.9 | 4.5 | 2.7 | 2.4 | 5.6 | 2.8 | 2.8 |
| Sympathectomy | 2 | 0.9 | 0 | 1.1 | 2.3 | 1.5 | 0 | 2.2 | 1.6 | 0.8 | 2.4 | 1.6 | 2.8 | 0.8 |
| Defibrillator† | 14 | 4.7 | 6.3 | 25 | 18 | 14 | 8.2 | 12 | 11 | 13 | 17 | 12 | 9.9 | 13 |
| 1st Cardiac | 50 | 34 | 52 | 31 | 34 | 58 | 40 | 38 | 45 | 44 | 29 | 54 | 30 | 41 |
| Event*†‡§, % | ||||||||||||||
| Syncope†‡§ | 42 | 31 | 52 | 28 | 32 | 49 | 36 | 34 | 39 | 39 | 29 | 45 | 26 | 38 |
| Aborted Cardiac Arrest | 0.7 | 1.9 | 0 | 3.2 | 2.3 | 1.2 | 1.2 | 1 | 1.3 | 1.1 | 0 | 1.6 | 0.7 | 1 |
| Death†‡§ | 7.7 | 0.5 | 0 | 0 | 0 | 7.7 | 2.9 | 2.9 | 5.0 | 3.1 | 0 | 7.2 | 2.8 | 2.3 |
| Ever Cardiac Event, % | ||||||||||||||
| Syncope†‡§ | 42 | 31 | 55 | 28 | 32 | 49 | 36 | 34 | 39 | 39 | 29 | 45 | 26 | 38 |
| Aborted Cardiac Arrest† | 4.6 | 7.5 | 16 | 7.5 | 11 | 6.9 | 6.2 | 6.7 | 6.1 | 8.4 | 7.1 | 7.8 | 4.3 | 7 |
| Death†‡¶§ | 18 | 3.3 | 0 | 2.2 | 4.5 | 17 | 11 | 6.7 | 14 | 5.7 | 7.1 | 16 | 8.5 | 7.8 |
Plus-minus values are means ±SD. Percentages greater than 10 are rounded to a whole number.
The 858 subjects in this table include 60 subjects from the Rochester-based registry who died suddenly at a young age, were from families with known KCNH2 mutation, and were assumed to have the family mutation. QTp, corrected Q-Tpeak interval.
First cardiac event was syncope, aborted cardiac arrest, or sudden death, whichever occurred first.
P<0.01 for the comparison of characteristics among the four sources of subjects.
P<0.01 for the comparison of characteristics among the four locations of the mutations.
P<0.01 for the comparison of characteristics among the three major type of mutations.
P<0.01 for the comparison of characteristics among the three major topology of mutations.
Location, Type and Topology of Mutation on Clinical Outcome
As to the location of mutation, the QTc interval was longer and cardiac events were more frequent in patients with mutations in the transmembrane pore locations (S5-loop-S6) than those with mutations in either transmembrane non-pore (S1-S4), N-terminus or C-terminus locations. As to the type of mutation, the QTc interval was longer in patients with missense mutations than that in patients with either frameshift/nonsense or other mutations. Sudden death was also more frequent in patients with missense mutations. As to the topology of mutation, the QTc interval was longer and cardiac events were more frequent in patients with mutations located in the α-helical domain than those with mutations in either the β-sheet domain or other uncategorized location.
The cumulative probabilities of first cardiac event by type, location and topology of mutation are presented in Figures 2A, 2B and 2C, respectively. No significant difference in event rates was observed among types of mutation (P=0.68, Figure 2A), although missense mutations were more associated with longer QTc interval and increased risk for sudden death compared with other type of mutations. On the other hand, significantly higher event rates were found in subjects with transmembrane pore mutations than in those with mutations in either transmembrane non-pore, N-terminus, or C-terminus regions, with a gradual increase in event rates occurring during ages 5 to 40 years (Figure 2B). Significantly higher event rates were also observed in subjects with mutations located in the α-helical domains than in those with mutations in either the β-sheet domains or other locations (Figure 2C).
Figure 2. Kaplan-Meier estimate of the cumulative probability of a first cardiac event by type.
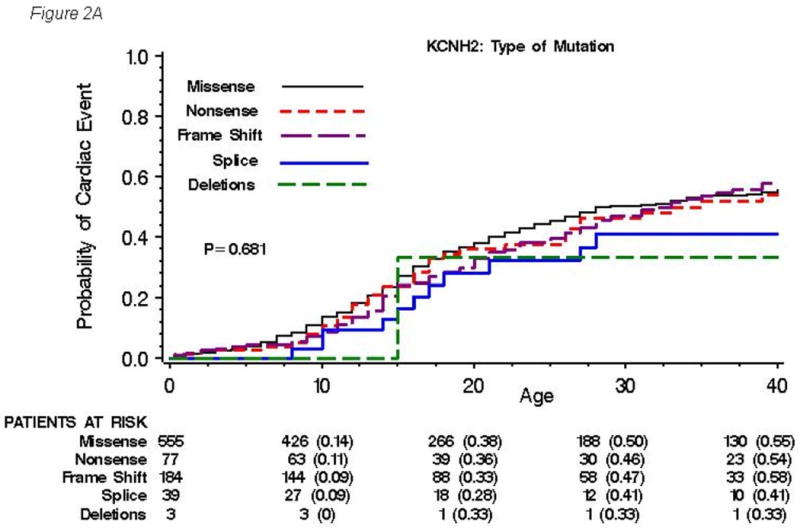
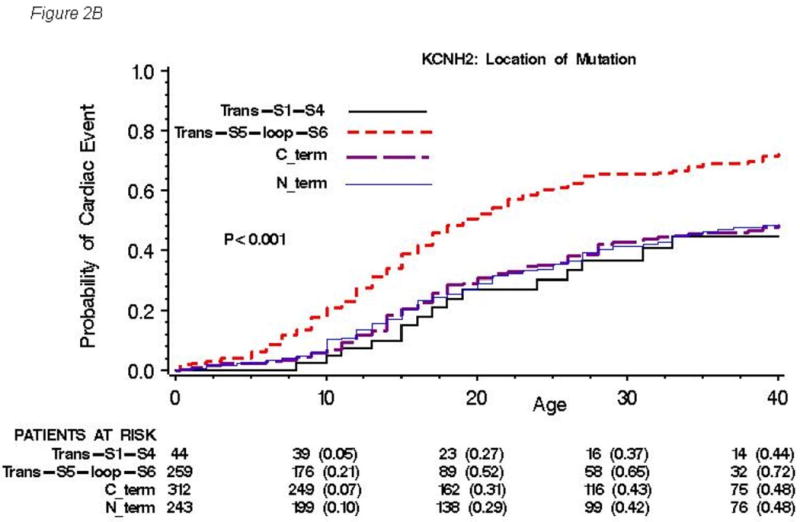
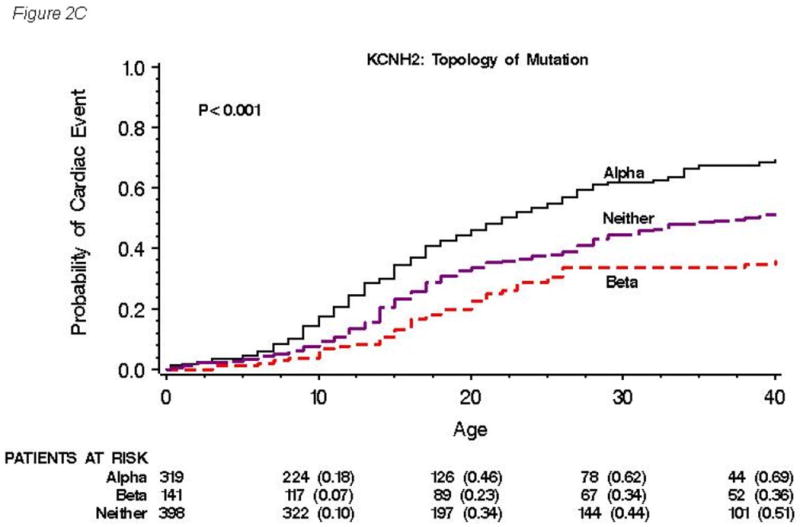
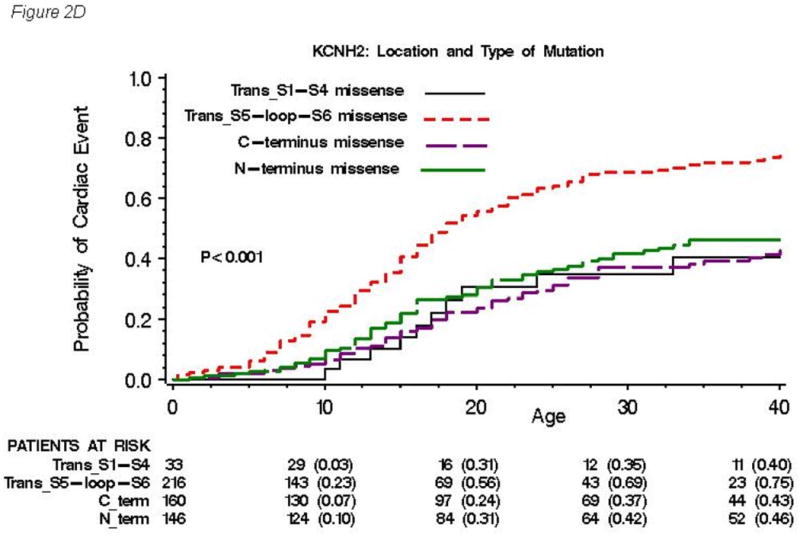
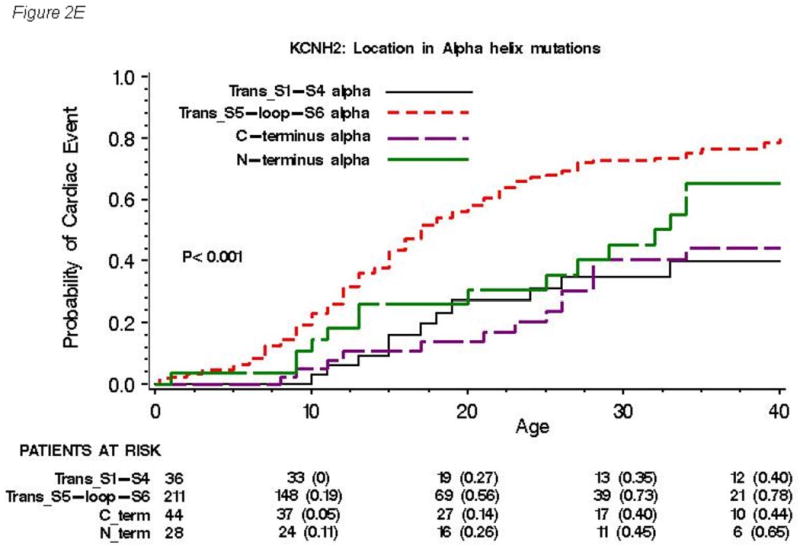
(A), location (B), and topology (C) of the mutation for all 858 subjects with genetically confirmed KCNH2 mutations. Kaplan-Meier estimate of the cumulative probability of a first cardiac event for missense mutations within different locations (D) and for mutations located in the α-helical domains within different locations (E). The numbers in parentheses reflect the cumulative event rate at that point in time.
The findings from the Cox regression analysis by location and by topology of KCNH2 mutations for first cardiac events are presented in Table 2A and those for aborted cardiac arrest or LQTS-related sudden cardiac death are presented in Table 2B. The clinical risk factors associated with first cardiac events involved males before age 13 years (HR=1.54 vs. females), females after age 13 (HR=3.29 vs. males), and longer QTc intervals (HR=3.33, QTc>530msec (n=112) vs. QTc<460msec (n=239); HR=2.09, QTc 500-530msec (n=146) vs. QTc<460msec; HR=1.56, QTc 460-499msec (n=251) vs. QTc<460msec). Mutations located in the transmembrane pore region made significant and independent contributions to the risk model with C-terminus region as reference (HR=1.56). Mutations located in the α-helical domains made significant contributions to the risk model with the β-sheet domains as reference (HR=1.74). A mutation in the α-helical domain located in the transmembrane pore region would have a risk equal to the multiplicative product the two hazard ratios, i.e., 1.56 × 1.74 = 2.71. On the other hand, a mutation in the α-helical domain located in the non-pore transmembrane S1-S4 region would have a risk of 0.61 × 1.74 = 1.06, and this value was very similar to 1. Time-dependent β-blocker use was associated with a significant 63% reduction in the risk of first cardiac events (p<0.001). The clinical risk factors associated with lethal cardiac events showed similar tendency to those with cardiac events, and involved females after age 13 (HR=2.38 vs. males), longer QTc intervals (HR=4.97, QTc>530msec vs. QTc<500msec; HR=2.57, QTc 500-530msec vs. QTc<500msec). History of prior syncope was a significant risk for lethal cardiac events (HR=3.42). Time-dependent β-blocker use showed a reduction in the risk of lethal cardiac events by 26%, but this did not reach statistical significance.
Table 2. Table 2A. Cox Regression with Multiple Predictor Variables Including Location of Mutations for First Cardiac Event.
| Variable | Hazard Ratio | 95% Confidence Interval | P Value | |
|---|---|---|---|---|
| Enrolling Sites with Netherlands as Reference | ||||
| Rochester | 1.38 | 1.03 | 1.85 | 0.030 |
| Japan | 1.10 | 0.74 | 1.63 | 0.644 |
| Mayo | 0.94 | 0.60 | 1.47 | 0.786 |
| Gender | ||||
| Male <13 years:Females<13 years | 1.54 | 1.08 | 2.20 | 0.018 |
| Female 13-40 years:Males 13-40 years | 3.29 | 2.36 | 4.60 | <0.001 |
| QTc Categories with QTc<460msec as Reference | ||||
| QTc >530msec | 3.33 | 2.30 | 4.83 | <0.001 |
| QTc 500-530msec | 2.09 | 1.45 | 3.02 | <0.001 |
| QTc 460-499msec | 1.56 | 1.11 | 2.21 | 0.011 |
| QTc missing* | 3.33 | 2.25 | 4.92 | <0.001 |
| C-terminus region as Reference | ||||
| Transmembrane S5-loop-S6 region | 1.56 | 1.14 | 2.14 | 0.006 |
| Transmembrane S1-S4 region | 0.61 | 0.34 | 1.09 | 0.095 |
| N-terminus region | 1.22 | 0.92 | 1.62 | 0.173 |
| Subunit Topology with β-Sheets as Reference | ||||
| α-Helical Domains | 1.74 | 1.15 | 2.63 | 0.009 |
| Uncategorized Locations | 1.33 | 0.93 | 1.90 | 0.115 |
| Time-dependent β-blocker use | 0.37 | 0.22 | 0.63 | <0.001 |
| Table 2B. Cox Regression with Multiple Predictor Variables Including Location of Mutations for Aborted Cardiac Arrest / LQT-Death | ||||
| Variable | Hazard Ratio | 95% Confidence Interval | P Value | |
| Enrolling Sites with Netherlands as Reference | ||||
| Rochester | 1.38 | 0.79 | 2.41 | 0.257 |
| Japan | 1.87 | 0.89 | 3.94 | 0.099 |
| Mayo | 1.37 | 0.53 | 3.52 | 0.520 |
| Gender | ||||
| Male <13 years:Females<13 years | 1.55 | 0.52 | 4.64 | 0.433 |
| Female 13-40 years:Males 13-40 years | 2.38 | 1.50 | 3.79 | <0.001 |
| QTc Categories with QTc<500msec as Reference | ||||
| QTc >530msec | 4.97 | 2.72 | 9.07 | <0.001 |
| QTc 500-530msec | 2.57 | 1.39 | 4.76 | 0.003 |
| QTc missing* | 25.58 | 14.46 | 45.26 | <0.001 |
| History of prior syncope | 3.42 | 2.25 | 5.20 | <0.001 |
| C-terminus region as Reference | ||||
| Transmembrane S5-loop-S6 region | 1.00 | 0.56 | 1.80 | 0.993 |
| Transmembrane S1-S4 region | 1.00 | 0.39 | 2.61 | 0.994 |
| N-terminus region | 1.33 | 0.80 | 2.22 | 0.271 |
| Subunit Topology with β-Sheets as Reference | ||||
| α-Helical Domains | 1.47 | 0.72 | 2.98 | 0.269 |
| Uncategorized Locations | 1.12 | 0.60 | 2.10 | 0.658 |
| Time-dependent β-blocker use | 0.74 | 0.42 | 1.31 | 0.300 |
The Cox analysis involved 858 subjects with 259 transmembrane S5-loop-S6, 44 transmembrane S1-S4, 243 N-terminus, and 312 C-terminus mutations.
QTc missing category involves 110 subjects, 69 of whom died suddenly at a young age without a prior ECG.
Combination of Location and Type of Mutation on Clinical Outcome
The inter-relation between location, type and topology of mutation is presented in Table 3. Among 52 mutations within the transmembrane pore region, 46 mutations (88.5%) were missense mutations, and only 6 mutations (11.5%) were frameshift/nonsense mutations. On the other hand, frameshift/nonsense mutations were more frequently located in the C-terminus region (31/50 mutations, 62.0%); 17 mutations (34.0%) were missense mutation, and the remaining 2 mutations (4.0%) were from any other type (splice mutation). Since transmembrane pore mutations are more risky than mutations in the transmembrane non-pore, C-terminus or N-terminus regions (Figure 2B), and there is no significant difference in event rates among the types of mutation (Figure 2A), then non-missense mutations, mainly frameshift/nonsense mutations in the C-terminus region may be an independent risk. Therefore, we further investigated the risk associated with a combination of location and type of mutation.
Table 3. Mutation Group by Location, Type and Topology.
| Location | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Transmembrane (S1-S4) | Transmem-brane (S5-loop-S6) | N-Terminus | C-Terminus | Total | |||||||
| # | % | # | % | # | % | # | % | # | % | ||
| Type | Topology | ||||||||||
| Missense | α-helices | 31 | 12 | 170 | 63 | 25 | 9 | 44 | 16 | 270 | 100 |
| β-sheet | - | - | - | - | 56 | 43 | 74 | 57 | 130 | 100 | |
| Neither | 2 | 1 | 46 | 30 | 65 | 42 | 42 | 27 | 155 | 100 | |
| Total | 33 | 6 | 216 | 39 | 146 | 26 | 160 | 29 | 555 | 100 | |
| Frameshift/Nonsense | Topology | ||||||||||
| α-helices | 5 | 10 | 41 | 84 | 3 | 6 | - | - | 49 | 100 | |
| β-sheet | - | - | - | - | 4 | 100 | - | - | 4 | 100 | |
| Neither | 6 | 3 | 2 | 1 | 56 | 27 | 144 | 69 | 208 | 100 | |
| Total | 11 | 4 | 43 | 17 | 63 | 24 | 144 | 55 | 261 | 100 | |
| Others | Topology | ||||||||||
| α-helices | - | - | - | - | - | - | - | - | - | - | |
| β-sheet | - | - | - | - | - | - | 7 | 100 | 7 | 100 | |
| Neither | - | - | - | - | 34 | 97 | 1 | 3 | 35 | 100 | |
| Total | - | - | - | - | 34 | 81 | 8 | 19 | 42 | 100 | |
| Total | Topology | ||||||||||
| α-helices | 36 | 11 | 211 | 66 | 28 | 9 | 44 | 14 | 319 | 100 | |
| β-sheet | - | - | - | - | 60 | 43 | 81 | 57 | 141 | 100 | |
| Neither | 8 | 2 | 48 | 12 | 155 | 39 | 187 | 47 | 398 | 100 | |
| Total | 44 | 5 | 259 | 30 | 243 | 28 | 312 | 36 | 858 | 100 | |
Number of patients
The cumulative probabilities of first cardiac event for missense mutations within different locations are presented in Figure 2D, and that for mutations located in the α-helical domains within different locations are presented in Figure 2E. Significantly higher event rates were found in subjects with missense mutations (Figure 2D) and mutations in the α-helical domains (Figure 2E) located in transmembrane pore region than in those located in any other regions. Among 261 patients with frameshift/nonsense mutations, the event rates were not different by location of mutations (data not shown).
The Cox regression analysis by a combination of location and type of mutations for first cardiac events is presented in Table 4A and that for aborted cardiac arrest or LQTS-related sudden cardiac death is presented in Table 4B. For patients with missense mutations, the transmembrane pore (S5-loop-S6) and N-terminus regions were a significantly greater risk than the C-terminus region (HR=2.87 and 1.86, respectively), but the transmembrane non-pore (S1-S4) region was not (HR=1.19). However, for non-missense mutations, these other regions were no longer riskier than the C-terminus (HR=1.13, 0.77 and 0.46, respectively). Likewise, subjects with non-missense mutations, mainly frameshift/nonsense mutations, were at significantly higher risk than those with missense mutations in the C-terminus region (HR=2.00), but this was not the case in other regions. This mutation location-type interaction was significant (p-value=0.008). However, a mutation topology-type analysis did not reveal a significant interaction (p=0.11). Also, the mutation location-type interaction was not seen for the aborted cardiac arrest or LQTS-related sudden cardiac death endpoint reported in Table 4B.
Table 4. Table 4A. Cox Regression with Multiple Predictor Variables Including Location and Type of Mutations and their Interaction for First Cardiac Event.
| Hazard Ratio | 95% Confidence Interval | P Value | ||
|---|---|---|---|---|
| 1. Mutation Location by Type | ||||
| Transmembrane pore (S5-loop-S6) / C-terminus (reference) | ||||
| Missense mutations | 2.87 | 2.03 | 4.07 | <0.001 |
| Non-missense mutations | 1.13 | 0.65 | 1.95 | 0.663 |
| N-terminus / C-terminus (reference) | ||||
| Missense mutations | 1.86 | 1.25 | 2.78 | 0.002 |
| Non-missense mutations | 0.77 | 0.50 | 1.17 | 0.220 |
| Transmembrane non-pore (S1-S4) / C-terminus (reference) | ||||
| Missense mutations | 1.19 | 0.59 | 2.39 | 0.632 |
| Non-missense mutations | 0.46 | 0.18 | 1.17 | 0.103 |
| 2. Mutation Type by Location | ||||
| Non-missense / Missense (reference) | ||||
| C-terminus location | 2.00 | 1.33 | 3.00 | 0.001 |
| Other locations (N-terminus, S1-S4, S5-loop-S6) | n.s. | |||
| 3. Interaction Between Mutation Location and Type* | --- | --- | --- | 0.008 |
| Table 4B. Cox Regression with Multiple Predictor Variables Including Location and Type of Mutations and their Interaction for Aborted Cardiac Arrest / LQT-Death | ||||
| Hazard Ratio | 95% Confidence Interval | P Value | ||
| 1. Mutation Location by Type | ||||
| Transmembrane pore (S5-loop-S6) / C-terminus (reference) | ||||
| Missense mutations | 1.65 | 0.93 | 2.91 | 0.085 |
| Non-missense mutations | 0.57 | 0.19 | 1.74 | 0.324 |
| N-terminus / C-terminus (reference) | ||||
| Missense mutations | 1.95 | 0.99 | 3.83 | 0.052 |
| Non-missense mutations | 0.80 | 0.37 | 1.73 | 0.575 |
| Transmembrane non-pore (S1-S4) / C-terminus (reference) | ||||
| Missense mutations | 1.94 | 0.61 | 6.20 | 0.264 |
| Non-missense mutations | 0.55 | 0.12 | 2.56 | 0.446 |
| 2. Mutation Type by Location | ||||
| Non-missense / Missense (reference) | ||||
| C-terminus location | 1.75 | 0.85 | 3.61 | 0.131 |
| Other locations (N-terminus, S1-S4, S5-loop-S6) | n.s. | |||
| 3. Interaction Between Mutation Location and Type* | --- | --- | --- | 0.208 |
Note: Items 1, 2, and 3 identify risk factors from the same model. The model adjusted for enrolling site, gender X age, QTc, and time-dependent beta-blockers as in Table 3. When family members who experienced LQTS-related SCD without being genotyped were omitted from the analyses, the hazard ratios, confidence intervals, and p-values for location and type of mutation were similar to those values in the above table, but the significance for the interaction between mutation location and type was reduced from p=0.008 to p=0.09.
The interaction between mutation location and type measures whether the cardiac event risk for a location relative to the C-terminus varies significantly between missense and non-missense mutations. The hazard ratio and confidence intervals are not provided for this interaction since the p-value is an overall significance level for three interaction terms.
See note in Table 4A. Model adjusted for enrolling site, gender X age, QTc, and time-dependent syncope and beta-blockers as in Table 3.
The interaction between mutation location and type measures whether the cardiac event risk for a location relative to the C-terminus varies significantly between missense and non-missense mutations. The hazard ratio and confidence intervals are not provided for this interaction since the p-value is an overall significance level for three interaction terms.
Among subjects with missense mutations, the transmembrane pore region was significantly higher risk than the transmembrane non-pore, N-terminus or C-terminus regions (not shown HR=2.42, 1.54 and 2.87, respectively). For subjects with non-missense mutations, the transmembrane pore region was not significantly higher risk than transmembrane non-pore, N-terminus or C-terminus regions (HR=2.47, 1.48 and 1.13, respectively). It's interesting to note the while not significant, the effect sizes (HRs) of the pore risk stays relatively constant across mutation type except in the case of the C-terminus, further evidence of the location-type interaction.
Discussion
The major findings of the present study from 858 type-2 LQTS subjects with genetically confirmed KCNH2 mutations derived from 4 LQTS Registries are that 1) there is a significant mutation type-location interaction; specifically that the relative risk between C-terminus and the regions is different for missense versus non-missense locations, 2) patients with missense mutations in the transmembrane pore region have significantly higher cardiac event rates than those with missense mutations in either N-terminus, transmembrane non-pore or C-terminus regions, 3) patients with non-missense mutations were at significantly higher risk than those with missense mutations in the C-terminus region, 4) patients with mutations located in putative α-helical domains have significantly higher cardiac event rates than in those with mutations in either the β-sheet domains or other uncategorized locations, and these higher event rates are independent of traditional clinical risk factors and of β-blocker therapy. Our data indicate that risk stratification and specific management or treatment by distinct location, coding type and topology of the channel mutation in addition to classical risk factors such as QTc, gender or history of prior syncope may be possible in patients with type-2 LQTS, although further studies are definitely required.
A total of 12 forms of congenital LQTS have been reported,2,4,17-20 and clinical studies for genotype-phenotype correlations have been rigorously investigated in the type-1, type-2, and type-3 LQTS, which constitute more than 90 % of genotyped patients with LQTS.2,21-25 More recently, mutation-location specific differences in the severity of clinical phenotype have been investigated in each genotype. 9,11,12,26,27 As to the type-1 LQTS, a large cohort of 600 patients with KCNQ1 mutations has demonstrated that location and biophysical function of mutations were independent risk factors influencing the clinical course.11 However, the distribution of mutation location as well as the frequency of mutation type are reported to be different in each of 3 major genotypes.9,11,12,26,27 More recently, putative secondary structures of α-helices or β-sheet are reported to have an important role on the channel function in the type-2 LQTS.8 Therefore, a larger cohort of patients having a spectrum of KCNH2 mutations is required to test the hypothesis that the location, coding type and topology of mutations would influence the clinical course in the type-2 LQTS.
In contrast to our cohort of 600 type-1 LQTS patients where the majority of mutations were found in the transmembrane region (66.2%),11 in the present study mutations in KCNH2 were more evenly distributed in the N-terminus, the transmembrane domain, and the C-terminus. As to the type of mutation, missense mutations dominated (80.5%), and only 13% of the mutations were frameshift/nonsense mutations in our type-1 LQTS cohort.11 In contrast, missense mutations accounted for 61.7%, and frameshift/nonsense mutations were more frequently observed (35.8%) in this type-2 LQTS cohort. Interestingly, most of the mutations located in the transmembrane pore region were missense mutations (46/52, 88.5%) in the present study, a finding concordant with the previous type-2 LQTS cohort by Moss et al. (13/14, 92.9%).9 This indicated that the severe phenotype in patients with mutations located in the transmembrane pore region was probably due to the fact that missense mutations that are expected to cause dominant negative effects were predominant in this region. However, our type-2 LQTS patients with missense mutations located in the N-terminus, transmembrane non-pore and C-terminus regions were at significantly less risk than those with missense mutations in the transmembrane pore region. These data suggest that location of mutation, i.e. transmembrane pore region, itself was an independent risk in type-2 LQTS patients with KCNH2 missense mutation.
On the other hand, patients with non-missense mutations, mainly frameshift/nonsense mutations were at significantly higher risk than those with missense mutations in the C-terminus region, and the event rates in patients with frameshift/nonsense mutations were not different among the transmembrane pore, transmembrane non-pore, N-terminus, and C-terminus regions. Gong et al. recently suggested that most frameshift/nonsense mutations would cause nonsense mediated decay (NMD) thereby producing less mRNA from the mutant alleles.28 This potentially would allow for the wild type allele to express more normal channels. Therefore, it is expected that the type-2 LQTS patients with frameshift/nonsense mutation causing NMD would have a mild phenotype. In contrast, the type-2 LQTS patients with frameshift/nonsense mutation without NMD would be expected to have a more severe phenotype because a truncated protein would be produced. Thus, the fact that some frameshift/nonsense mutations show NMD, whereas the other mutations do not, makes the clinical phenotype in the type-2 LQTS patients with frameshift/nonsense mutations more complicated, although this scenario is only a speculation. The present study confirmed the higher risk in patients with non-missense mutations than in those with missense mutations in C-terminus region, suggesting that more careful follow-up is required in type-2 LQTS patients with non-missense mutations in the C-terminus region.
With regard to the topology of mutation, Anderson and co-workers recently reported that missense mutations located in a highly ordered structure as α-helices or β-sheet correlated with a class 2 trafficking-deficient phenotype in the type-2 LQTS patients.8 In the present cohort, mutations located in the α-helical domains were associated with a significantly higher risk compared to those with mutations in either the β-sheet domains or other uncategorized locations. It is possible that missense mutations in α-helices, where secondary protein structure is thought to the highly ordered, lead to altered secondary and tertiary channel protein structure and abnormal trafficking. This new analysis considering putative secondary structures of mutated channel would be a useful approach in stratifying risk of cardiac events in patients with LQTS.
β-Blockers have long been the first choice of therapy in patients with congenital LQTS.2,29 However, it has been shown in previous studies that the protection that β-blockers provide against cardiac events for type-2, and type-3 LQTS patients is somewhat less effective than for type-1 LQTS patients.23,30 A variety of experimental data also support the genotype-specific efficacy of β-blockers in type-1 LQTS.31 In the present study, time-dependent β-blocker use significantly reduced the risk of first cardiac events by 63% (p<0.001), confirming the efficacy of β-blockers as a first line of therapy in patients with type-2 LQTS as well as suggesting more prophylactic use of β-blockers especially in high risk patients with type-2 LQTS. On the other hand, β-blocker use was associated with less protection (29%) in the prevention of lethal cardiac events compared to first cardiac events (mostly syncope), indicating that additional treatment such as potassium supplement or an ICD implantation may be considered in high risk patients with type-2 LQTS. The patients who have aborted cardiac arrest/sudden death may have a more malignant pathophysiology that is more resistant to β-blockers than are syncopal episodes. We purposely included “ECG missing” in the Cox model so that the β-blocker effect is actually adjusted for subjects with “ECG missing” who probably did not receive β-blockers.
Study Limitation
We did not evaluate the risk associated with distinct type of biophysical ion-channel dysfunction (dominant-negative or haplotype insufficient), since only a small percentage of the mutations present within our patient population have been studied extensively in identical cellular expression experiments. There were 60 patients who were not genotyped, and they had an increased risk for events mainly because their fatal events occurred at a young age before they were genotyped. When these patients were excluded from the analysis, the pattern of risk in the missense and non-missense subgroups remains similar to the total population, but the significance of the effect is attenuated due to the reduced number of events.
Supplementary Material
Acknowledgments
This work was supported in part by: 1) health sciences research grant (H18 - Research on Human Genome - 002) and the Research Grant for the Cardiovascular Diseases (21C-8) from the Ministry of Health, Labour and Welfare, Japan (WS); 2) research grants HL-33843 and HL-51618 (AJM) and HL-60723 (CTJ) from the National Institutes of Health, Bethesda, Maryland, USA; and 3) grant 2000.059 from the Nederlandse Hartstichting, Amsterdam, The Netherlands (AAMW).
Abbreviations and Acronyms
- ECG
electrocardiogram
- IKr
rapid component of the delayed rectifier repolarizing current
- LQTS
long QT syndrome
- NMD
nonsense mediated decay
- QTc
corrected QT
Footnotes
Disclosures: Michael J. Ackerman has a consulting relationship and license agreement/royalty arrangement with PGx Health (FAMILION).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moss AJ, Kass RS. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115:2018–2024. doi: 10.1172/JCI25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimizu W. The long QT syndrome: Therapeutic implications of a genetic diagnosis. Cardiovasc Res. 2005;67:347–356. doi: 10.1016/j.cardiores.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 3.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 4.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 5.January CT, Gong Q, Zhou Z. Long QT syndrome: cellular basis and arrhythmia mechanism in LQT2. J Cardiovasc Electrophysiol. 2000;11:1413–1418. doi: 10.1046/j.1540-8167.2000.01413.x. [DOI] [PubMed] [Google Scholar]
- 6.Ficker E, Dennis AT, Obejero-Paz CA, Castaldo P, Taglialatela M, Brown AM. Retention in the endoplasmic reticulum as a mechanism of dominant-negative current suppression in human long QT syndrome. J Mol Cell Cardiol. 2000;32:2327–2337. doi: 10.1006/jmcc.2000.1263. [DOI] [PubMed] [Google Scholar]
- 7.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 8.Anderson CL, Delisle BP, Anson BD, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 9.Moss AJ, Zareba W, Kaufman ES, et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]
- 10.McConkey EH. Mutation Qualitative Aspects. In: McConkey EH, editor. Human genetics: The molecular revolution. Jones and Bartlett Publishers; Boston, MA: 1993. pp. 139–159. [Google Scholar]
- 11.Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagaoka I, Shimizu W, Itoh H, et al. Mutation site dependent variability of cardiac events in Japanese LQT2 form of congenital long-QT syndrome. Circ J. 2008;72:694–699. doi: 10.1253/circj.72.694. [DOI] [PubMed] [Google Scholar]
- 13.Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 14.http://ca.expasy.org/cgi-bin/ft_viewer.pl?Q12809
- 15.Cox DR. Regression models and life-tables. J Stat Soc [B] 1972;34:187–220. [Google Scholar]
- 16.Lin DY, Wei LJ. The Robust Inference for the Proportional Hazards Model. Journal of the American Statistical Association. 1989;84:1074–1078. [Google Scholar]
- 17.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 18.Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–176. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- 19.Ueda K, Valdivia C, Medeiros-Domingo A, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu G, Ai T, Kim JJ, et al. Alpha-1-Syntrophin Mutation and the Long QT Syndrome: a disease of sodium channel disruption. Circ Arrhythmia Electrophysiol. 2008;1:193–201. doi: 10.1161/CIRCEP.108.769224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zareba W, Moss AJ, Schwartz PJ, et al. Influence of the genotype on the clinical course of the long-QT syndrome. N Engl J Med. 1998;339:960–965. doi: 10.1056/NEJM199810013391404. [DOI] [PubMed] [Google Scholar]
- 22.Wilde AA, Jongbloed RJ, Doevendans PA, et al. Auditory stimuli as a trigger for arrhythmic events differentiate HERG-related (LQT2) patients from KVLQT1-related patients (LQT1) J Am Coll Cardiol. 1999;33:327–332. doi: 10.1016/s0735-1097(98)00578-6. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 24.Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 25.Shimizu W, Noda T, Takaki H, et al. Diagnostic value of epinephrine test for genotyping LQT1, LQT2 and LQT3 forms of congenital long QT syndrome. Heart Rhythm. 2004;1:276–283. doi: 10.1016/j.hrthm.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 26.Zareba W, Moss AJ, Sheu G, et al. Location of mutation in the KCNQ1 and phenotypic presentation of long QT syndrome. J Cardiovasc Electrophysiol. 2003;14:1149–1153. doi: 10.1046/j.1540-8167.2003.03177.x. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu W, Horie M, Ohno S, et al. Mutation site-specific differences in arrhythmic risk and sensitivity to sympathetic stimulation in LQT1 form of congenital long QT syndrome - Multi-center study in Japan - J Am Coll Cardiol. 2004;44:117–125. doi: 10.1016/j.jacc.2004.03.043. [DOI] [PubMed] [Google Scholar]
- 28.Gong Q, Zhang L, Vincent GM, Horne BD, Zhou Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation. 2007;116:17–24. doi: 10.1161/CIRCULATIONAHA.107.708818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–623. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- 30.Priori SG, Napolitano C, Schwartz PJ, et al. JAMA. 2004;292:1341–1344. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.