

Abstract
The transcriptional co-activators CBP/p300 and PCAF participate in transcriptional activation by many factors. We have shown that both CBP/p300 and PCAF stimulate the transcriptional activation by KLF13, a member of the KLF/Sp1 family, either individually or cooperatively. Here we further investigated how CBP and PCAF acetylation regulate KLF13 activity, and how these two co-activators functionally interplay in the regulation of KLF13 activity. We found that CBP and PCAF acetylated KLF13 at specific lysine residues in the zinc finger domain of KLF13. The acetylation by CBP, however, resulted in disruption of KLF13 DNA binding. Although the acetyltransferase activity of CBP is not required for stimulating the DNA binding activity of all of the transcription factors that we have examined, the disruption of factor DNA binding by CBP acetylation is factor-specific. We further showed that PCAF and CBP act synergistically and antagonistically to regulate KLF13 DNA binding depending on the status of acetylation. PCAF blocked CBP acetylation and disruption of KLF13 DNA binding. Conversely, acetylation of KLF13 by CBP prevented PCAF stimulation of KLF13 DNA binding. PCAF blocked CBP disruption of KLF13 DNA binding by preventing CBP acetylation of KLF13. These results demonstrate that acetylation by CBP has distinct effects on transcription factor DNA binding, and that CBP and PCAF regulate each other functionally in their regulation of transcription factor DNA binding.
Keywords: CBP/p300, PCAF, KLF13, acetylation, regulation of DNA-binding
Introduction
Sequence-specific DNA-binding factors regulate transcription by directing the assembly of promoter-specific and functional complexes containing co-activators/chromatin modifiers and the general transcription apparatus at the target gene promoter.1 The Sp1/KLF family of transcription factors regulates the expression of a variety of genes by binding to the GC/GT box through three highly conserved Cys2His2 type zinc fingers.2,3 Members of this family have been shown to regulate target gene expression through recruiting co-activators and co-repressor complexes.3 The protein acetylation and chromatin remodeling activities associated with these co-activator/chromatin modifier complexes play important roles in gene regulation.4–9 Several transcriptional co-activators with intrinsic acetyltransferase activity have been identified including GCN5,10,11 PCAF,12 CBP/p300,13,14 TAFII 250,15 ACTR16 and SRC-1.17 The transcriptional co-activators CBP/p300 and PCAF/GCN5 are among the best-studied co-activators. CBP/p300 proteins play important roles in cell growth and development.18 CBP/p300 and PCAF exist in multi-molecular complexes in vivo and function as co-activators for a variety of transcription factors.19,20 Current models suggest that these co-activators are brought to target gene promoters through interaction with sequence-specific DNA binding proteins where they may function: (1) as bridging molecules to mediate the interaction of DNA binding activators with the general transcription machinery; (2) as docking sites for multiple proteins to mediate synergistic activation; and (3) as acetylases to acetylate transcription factors and histones to regulate transcription. At the transcriptional factor level, these co-activators have been reported to regulate transcription factor activity at multiple levels including DNA binding, protein–protein interaction, stability and nucleocytoplasmic shuttling.21–24 Studies have shown that both CBP/p300 and PCAF/GCN5 transcriptional co-activators often act synergistically in stimulating transcription: Lee et al.,25 and references therein). In spite of the progress, several fundamental questions on the mechanisms of their action remain to be answered. For example, why different acetyltransferase co-activators are involved in transcriptional activation? Are they functionally redundant? If not, what is the exact role of each individual acetyltransferase and how they functionally interact in regulation of transcriptional activation?
We have shown that both CBP/p300 and PCAF physically interact with KLF13, a member of the KLF family of transcription factors, and stimulate its transcriptional activation individually and cooperatively using transient reporter assays.26 Here we report our studies on the role of acetylation by CBP and PCAF in the regulation of KLF13 activity, and the molecular mechanisms by which these two acetyltransferase co-activators functionally interact in regulation of KLF13 acetylation and DNA binding. Our results demonstrate that acetylation by CBP has distinct effects on the DNA binding activity of transcription factors. CBP and PCAF act cooperatively and antagonistically to regulate KLF13 DNA binding. These results demonstrate a dynamic functional interplay between CBP/p300 and PCAF acetyltransferase co-activators in the regulation of transcription.
Results
CBP and PCAF acetylate specific lysine residues of KLF13
Both CBP and PCAF enhance KLF13 transcriptional activity and acetylate KLF13.26 To further investigate the role of CBP/p300 and PCAF acetylation in KLF13 regulation, we identified the CBP and PCAF target lysine residues in KLF13. There are 13 lysine residues in KLF13 and 11 of them are located in the zinc finger domain (Figure 1(a)). We have shown that CBP and PCAF acetylated primarily the zinc finger domain of KLF13 from amino acid residues 167 to 250. Subdividing the zinc finger into two fragments showed that CBP acetylated both fragments of residues 149–206 (containing five lysine) and of 200–289 (containing six lysine), whereas PCAF acetylated only the fragment from amino acid residues 200 to 289.26 These results indicate that CBP acetylates multiple lysine residues throughout the zinc finger domain of KLF13. To identify the CBP acetylated lysine residues we first generated lysine to alanine mutations using each fragment as the backbone. In this way, we changed the 11 lysine residues in the zinc finger domain to alanine, individually and in combination. Lysine 167 and 169, 206 and 207 or 226 and 227 were mutated together. Acetylation analyses of the lysine to alanine (K–A) mutants identified K167/K169 and K181 in the first zinc finger, and K226/K227 and K235 in the third zinc finger as targets of CBP acetylation (Figure 1(b)). Acetylation assays using the mutant 6A, which contains lysine to alanine mutations in all the CBP target lysine residues in the full KLF13 zinc finger (amino acid residues 149–250), confirmed the results obtained using the two separate fragments. PCAF acetylates KLF13 at K226 and K227 (Figure 1(c)). As shown in Figure 1(a) the CBP target lysine residues are located at the first and the third finger of KLF13. Such extensive acetylation of a transcription factor by CBP is unprecedented as CBP usually acetylates very few lysine residues in its target protein.27 These results show that CBP and PCAF acetylate specific and overlapping lysine residues of KLF13.
Figure 1.

CBP and PCAF acetylate specific lysines in KLF13. (a) Top, the schematic representation of KLF13 domain structure, and the CBP and PCAF acetylated lysine residues. Bottom, the amino acid sequences of the zinc fingers. Zinc coordinating cysteine and histidine residues are underlined. Residues that contact DNA are shaded. CBP and PCAF acetylated lysine residues are in boldface. Unfilled and filled arrowheads denote lysine residues that are acetylated by CBP only and by both CBP and PCAF, respectively. (b) Autoradiography of CBP acetylation of wild-type and lysine to alanine mutants of KLF13. Acetylation assays were carried out using the wild-type or lysine to alanine mutants of KLF13 containing the first finger (residues 149–206), the second and third finger (residues 200–289) and the full zinc finger (residues 149–250). Mutant 6A contains all the K–A mutations in all the six targeted lysine residues in the background of full zinc finger domain (149–250) of KLF13. The Coomassie blue staining of the input GST-KLF13 proteins is shown at the bottom. (c) Identification of PCAF acetylated lysine residues in KLF13. The wild-type and mutant KLF13 in this assay contain full-length zinc finger of KLF13 (amino acid sequences from 149–250). The result shown here is the autoradiography of PCAF acetylation of wild-type and lysine to alanine mutants of KLF13. Asterisks indicate self-acetylated PCAF. The Coomassie blue staining of the input GST–KLF13 proteins is shown at the bottom.
Acetylation by CBP disrupts KLF13 DNA binding
To elucidate the role of CBP acetylation in regulation of KLF13 DNA binding, EMSA assays were performed using acetylated or mock-acetylated KLF13 by wild-type (HATwt) or HAT defective (HAT-) CBP. Consistent with our previous results,26 CBPHATand CBPHATwt stimulated KLF13 DNA binding in the absence of acetyl-CoA (data not shown). Unexpectedly, no stimulation of KLF13 DNA binding by CBPHATwt was observed when acetyl-CoA was added in the acetylation reaction (data not shown). The inability of CBPHATwt to stimulate KLF13 DNA binding with added acetyl-CoA was a result of the disruption of KLF13 DNA binding by CBP acetylation but not a result of the inactivation of CBP by self-acetylation. First, CBP acetylation of KLF13 blocks the stimulation of its DNA binding by PCAF, indicating that the CBP acetylated KLF13 is unable to response to PCAF stimulation (Figure 5(a), below). Second, quantitative EMSA showed that KLF13 is not latent in DNA binding, and PCAF and CBP in the absence of acetyl-CoA act by enhancing its DNA binding but not by converting it from an inactive form to an active form.26 Therefore, by using an increased amount of KLF13 we were able to examine the effect of CBP acetylation on KLF13 DNA binding directly. This assay showed that although the higher basal level of KLF13 DNA binding was further stimulated by CBPHAT or CBPHATwt without added acetyl-CoA (Figure 2(a), lanes 2–6), it was significantly reduced by CBPHATwt with acetyl-CoA (comparing lanes 2 and 3 with lane 7). Taken together, these results lead to the conclusion that acetylation by CBP has disruptive effects on KLF13 DNA binding. We next tested whether the disruption of KLF13 DNA binding is mediated by the acetylation of specific lysine residues by CBP. EMSA assays showed that CBP stimulated the DNA binding by the KLF13 mutants containing individual or double K–A substitutions in the CBP targeted lysine residues in the absence, but not in the presence of added acetyl-CoA, indicating that acetylation of multiple lysine residues by CBP can mediate the disruption of KLF13 DNA binding (Figure 2(b)). Mutant 6A containing K–A substitutions in all the CBP acetylated lysine residues (K167, K169, K181, K226, K227 and K235) is impaired in its DNA binding activity (data not shown). Consequently, we were unable to determine the effect of CBP acetylation on 6A mutant DNA binding directly.
Figure 5.

CBP and PCAF interact in the regulation of KLF13 DNA binding activity. The Scheme of the assays were depicted on the top of each panel. (a) EMSA analysis shows that pre-acetylation by CBPHATwt prevents PCAF from stimulating KLF13 DNA binding activity. Purified KLF13 was first subjected to mock acetylation with buffer or acetylation by CBPHATwt or CBPHAT. The mock acetylated or CBPHATwt or CBPHAT acetylated KLF13 was further incubated with different amounts of PCAF and used in EMSA assay. Lanes 3–5, the dose-dependent stimulation of KLF13 DNA binding by PCAF following mock acetylation. Lanes 6–9, the stimulation of KLF13 DNA binding by CBPHAT alone (lane 6) or together with increasing amounts of PCAF (lanes 7–9). Lanes 10 and 11, the acetylation-dependent disruption of KLF13 DNA binding by CBPHAT. Lanes 12–14, that PCAF failed to stimulate KLF13 DNA binding following its pre-acetylation by CBPHAT. The asterisk indicates a band of unknown identity. GST alone has no effect on KLF13 DNA binding or PCAF stimulation of KLF13 DNA binding (data not shown). (b) EMSA assay shows that pre-incubation with PCAF blocked the disruptive effect of CBP acetylation on KLF13 DNA binding. The scheme of the EMSA assay was the same as in (a) except that the pre-incubation was carried out between KLF13 and PCAF. GST–KLF13 is same as in (a). The results show that pre-incubation with GST–PCAFHATwt but not GST alone completely blocked CBP acetylation-mediated disruption of KLF13 DNA binding (lanes 5–7 and 11–13). Pre-incubation with PCAFHAT also partially blocked CBP disruption (lanes 8–10). (c) EMSA assay shows that the acetylation of lysine 226/227 in KLF13 is not required for PCAF protection of disruption of KLF13 DNA binding by CBP acetylation. The assay was essentially the same as in (b) except that PCAFHATwt was first incubated with wild-type KLF13, KLF13 K226A; K227A or KLF13 K167A; K169A.
Figure 2.

CBP acetylation disrupts KLF13 DNA binding activity of factors. (a) EMSA assay shows the basal KLF13 DNA binding is further increased by CBPHAT mutant (L1690K; C1691L) and CBPHAT (1196–1718) wt in the absence of acetyl-CoA, but decreased by CBPHATwt in the presence of acetyl-CoA as denoted. The asterisks indicate fast migrating complexes that appear in EMSA assays (see also Figure 6(a)) using CBPHAT. The identity of this band is unknown. (b) Multiple CBP target lysine residues can mediate the disruption of KLF13 DNA binding by CBP acetylation. The EMSA assay was carried out using purified GST fusion proteins containing the wild-type or mutant KLF13 with lysine to alanine mutations in the CBP acetylated lysine residues (K167A; K169A, K181A, K226A; K227A and K235A) or non-CBP acetylated lysine residues (K207A; K209A). The wild-type and mutant GST–KLF13 were acetylated or mock acetylated by CBPHATwt in the presence or absence of 1 μM unlabeled acetyl-CoA.
We next determined whether the results shown in Figure 2(a) are KLF13 specific. We tested the effect of CBP acetylation on KLF1 and KLF11 binding to the CACCC box. KLF1, the founding member of the KLF family, is regulated by CBP/p300.28 KLF11 was another member of the KLF family. As shown in Figure 3, CBP acetylation disrupted both KLF1 and KLF11 DNA binding. In contrast to KLF1/11/13, CBP acetylation showed no disruptive effect on p53 and E2F DNA binding (data not shown). These results together demonstrate that the HAT activity of CBP is not required for stimulating the DNA binding activity of these factors, and acetylation by CBP disrupts the DNA binding activity in a factor dependent manner.
Figure 3.
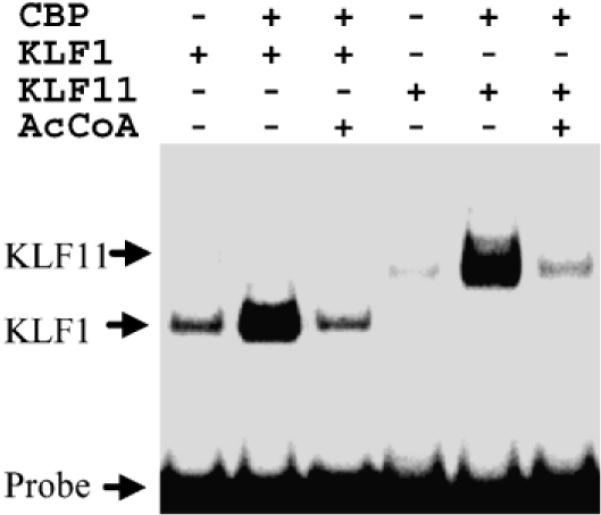
Regulation of KLF1 and KLF11 DNA binding by CBP acetylation. EMSA analysis of stimulation of KLF1 and KLF11 DNA binding by purified GST–CBPHAT (1196–1718) wt or GST–CBP HAT mutant (L1690K; C1691L) in the presence or absence of acetyl-CoA as denoted. Purified GST fusion proteins containing the full-length zinc finger domains of KLF1 and KLF11 were used in the EMSA assay as indicated.
Acetylation of lysine residues 226 and 227 is required for CBP/p300 and PCAF stimulation of KLF13 transcriptional activation
Lysine 226 and 227 are specific targets of both CBP and PCAF acetylation. We next tested the requirement of their acetylation for PCAF and CBP/p300 regulation of KLF13 transcriptional activity. Transient reporter assays showed that KLF13 (K226R; K227R) containing lysine to arginine mutations at residues 226 and 227 was significantly impaired in co-activation with PCAF indicating acetylation of these lysine residues are required for PCAF stimulation of KLF13 transcriptional activity (Figure 4(a)). Since CBP/p300 also acetylates these two lysine residues, we also examined their role in p300 co-activation. As shown in Figure 4(b), acetylation of lysine 226 and 227 is also required for p300 co-activation of KLF13 transcriptional activity. Since the endogenous PCAF may be required for p300 co-activation, these results could not distinguish whether the inability of p300 to activate KLF13 (K226R; K227R) is primarily due to its defect in acetylation and activation by PCAF or p300. Similar results were observed using KLF13 (K226A; K227A), CBP and PCAF (data not shown). Since the side-chain and charge of arginine is more similar to lysine, the results using KLF13 (K226R; K227R were shown to emphasize the defect is due to a loss of acetylation, but not due to changes in side-chain structure or charge, which may also affect the conformation and activity of FKLF13.
Figure 4.

Acetylation of lysines226 and 227 is required for PCAF and CBP/p300 stimulation of KLF13 transcriptional activation. (a) COS cells were transfected with 10 ng of pγluc reporter plasmid, 100 ng of PCAF expression plasmid and 5 ng of wild-type or K226R; K227R mutant KLF13. The KLF13 K226R/K227R contains lysine to arginine mutations at the PCAF target lysine 226 and 227. Results are presented as the mean ± SD (n = 3) of the relative light unit. (b) The same as (a) except 100 ng of p300 was used in co-transfection.
CBP and PCAF functionally interplay to regulate KLF13 DNA binding
Our previous studies have shown that CBP/p300 and PCAF act cooperatively to stimulate KLF13 transcriptional activity in transient reporter assays, indicating that both co-activators participate in KLF13 transcription.26 Therefore, we next sought to determine how these two acetyltransferases interact functionally to regulate KLF13 DNA binding. EMSA assays using purified recombinant CBP and PCAF demonstrate that pre-acetylation of KLF13 by CBP blocks the stimulation of KLF13 DNA binding by PCAF (Figure 5(a), lanes 12–14). Acetyl-CoA by itself has no effect on KLF13 DNA binding (Figures 2(a) and 5(a)). The blocking effect of CBP on PCAF stimulation of KLF13 DNA binding is CBP acetylation-dependent, since it is not observed with CBPHAT or CBPHATwt in the absence of acetyl-CoA (Figure 5(a), lanes 7–9, and data not shown). Instead, a certain level of cooperative stimulation of KLF13 DNA binding by CBPHAT and PCAF was observed (Figure 5(a), comparing lane 6 with lanes 7–9, and lanes 3–4 with 7–8). GST alone also showed no effect on PCAF stimulation of KLF13 DNA binding (data not shown). Therefore, the blocking of PCAF stimulation of KLF13 DNA binding by CBP is dependent on both CBP HAT activity (Figure 5(a), lanes 6–9 and lanes 10–14) and acetyl-CoA (Figure 5(a), lane 10 and lanes 11–14). On the other hand, PCAFHATwt completely prevented the disruption of KLF13 DNA binding by CBP acetylation (Figure 5(b), comparing lane 4 with lanes 5–7). PCAFHAT also partially prevented the disruption of KLF13 DNA binding by CBP acetylation (comparing lane 4 with lanes 8–10), indicating that the acetylase activity of PCAF is not absolutely indispensable for its blocking activity. This blocking effect is specific since GST alone has on effect (lanes 11–13).
To further determine whether acetylation of KLF13 by PCAF is required for preventing CBP acetylation-mediated disruption of KLF13 DNA binding, we tested whether PCAF is able to block CBP acetylation-mediated disruption of KLF13 (K226A; K227A) DNA binding, since this mutant can not be acetylated by PCAF. The results shown in Figure 5(c) demonstrate that PCAF efficiently blocked the disruption of KLF13 (K226A; K227A) DNA binding by CBP acetylation, suggesting that acetylation of KLF13 by PCAF is not required for PCAF to block the disruptive effect of CBP acetylation. In summary, these results demonstrate that CBP acetylation not only disrupts KLF13 DNA binding but it also blocks PCAF stimulation of its DNA binding. PCAF was able to block the disruptive effect of CBP acetylation-mediated disruption of KLF13 DNA binding, and this blocking function does not require PCAF acetylation of KLF13. Therefore, in contrast to CBP, the blocking of CBP acetylation mediated disruption of KLF13 DNA binding by PCAF requires neither the HAT activity (Figure 5(b), lanes 5–7 and lanes 8–10) nor the acetylation of KLF13 by PCAF (Figure 5(c), lanes 3–5 and lanes 7–9). Consistent with the EMSA assays, the data in Figure 6 further demonstrate that PCAF blocking of CBP acetylation of KLF13 is independent of PCAF HAT activity and does not require the acetylation of KLF13 by PCAF.
Figure 6.

PCAF blocks the acetylation of KLF13 by CBP. (a) The acetylation assay was carried out by pre-incubation of wild-type or K226A; K227A mutant KLF13 with PCAF HAT (352–832) for five minutes at room temperature followed by addition of CBPHAT (1196–1718). The reaction was continued for an additional 30 minutes at room temperature. The acetylation of KLF13 was analyzed by SDS-PAGE and PhosphoImage. Input represents SDS-PAGE and Coomassie staining of half of the wild-type or mutant KLF13 proteins from the acetylation assay. (b) The acetylation assay was carried out by first adding GST, PCAFwt or PCAFHAT(Y616A; F617A) to aliquots of a reaction mixture containing purified GST–KLF13 (149–250) (K226A; K227A). CBP acetylation was initiated by adding CBPHAT to the reaction five minutes later. The acetylation was analyzed as for (a). To ensure each reaction receives same amount of GST–KLF13 substrate, GST–KLF13 (149–250) (K226A; K227A) was added to a master reaction mixtures and aliquots of this mixture were used for the assays in each lane. The Coomassie staining of the purified GST, GST–PCAFwt, GST–PCAF HAT and CBP HAT proteins used in the assay are shown to the right with the full-length proteins indicated by an arrowhead.
PCAF prevents CBP acetylation-mediated disruption of KLF13 DNA binding by blocking KLF13 acetylation by CBP
Acetylation of KLF13 by CBP disrupts its DNA binding which can be blocked by PCAF. We next determined the underlying mechanism by examining whether PCAF prevents this disruption by blocking CBP acetylation of KLF13. For this purpose, we used KLF13 (K226A; K227A), since it can not be acetylated by PCAF but can still be regulated by CBP in terms of acetylation and DNA binding disruption (Figure 5(c)). This assay showed that pre-incubation of this mutant with PCAF blocked its acetylation by CBP (Figure 6(a)). The acetyltransferase activity of PCAF is not required for blocking the disruption of KLF13 DNA binding by CBP acetylation. We next tested whether the HAT activity of PCAF is required for blocking CBP acetylation of KLF13 using purified GST, GST–PCAFHATwt and GST–PCAFHAT. This assay revealed that PCAFHAT efficiently blocked CBP acetylation of KLF13 whereas GST alone shows no effect (Figure 6 (b)). This is consistent with the observations that the acetylation of KLF13 by PCAF is not required for PCAF to block CBP acetylation of KLF13 (Figure 6(a)), and that PCAFHAT is able to block CBP acetylation-mediated disruption of KLF13 DNA binding (Figure 5(b)). These data demonstrate that PCAF prevents CBP acetylation-mediated disruption of KLF13 DNA binding by blocking CBP acetylation, and that the HAT activity of PCAF is not required for blocking CBP acetylation of KLF13.
Discussion
Regulation of transcription factor DNA binding activity by CBP acetylation
We have shown that both the wild-type and acetyltransferase-defective CBP stimulated KLF13 DNA binding in the absence of acetyl-CoA.26 Here we carried out extensive biochemical studies to determine the role of CBP acetylation in KLF13 transcription. We identified six CBP target lysine residues in the first and third zinc fingers of KLF13. We found that acetylation by CBP leads to disruption of KLF13 DNA binding. The combined results using wild-type CBP HAT domain in the presence or absence of acetyl-CoA and acetyltransferase-defective CBP HAT domain suggest that the disruption of KLF13 DNA binding is completely CBP acetylation dependent. Our results provide the first example that acetylation of a sequence-specific DNA binding transcriptional activator by CBP can also disrupt its DNA binding. Therefore, acetylation by CBP could regulate DNA binding activity of transcription factors both positively and negatively. To extend our results to other members of the KLF family, we found that the acetylation of KLF1 and KLF11 by CBP also disrupted their DNA binding. To further examine the effect of CBP acetylation on the DNA binding activity of transcription factors, we tested the effect of CBP acetylation on p53 and E2F-1 DNA binding. EMSA assays showed that the HAT activity of CBP was also not required for stimulating p53 and E2F-1 DNA binding. In contrast to the results from KLF1/11/13, acetylation of p53 and E2F-1 by CBP does not disrupt its DNA binding (Figure 3, and data not shown). One possible explanation for the difference in acetylation by CBP between p53 and KLF13 is that KLF13 is extensively acetylated in its zinc finger DNA binding domain whereas p53 is acetylated in the C-terminal regulatory domain.29 The acetylation at different residues may result in differential changes in the charge and conformation of the acetylated protein. These changes may determine the effects of acetylation on the DNA binding activity of a particular protein. The effect of the HAT-defective mutants of CBP on p53 DNA binding have not been examined directly by using their respective HAT mutants in previous studies. In this study, we show that the CBP acetyltransferase activity is not required for stimulating p53 binding (data not shown).
Functional interaction between CBP and PCAF in regulation of KLF13 DNA binding
Multiple HAT activities including CBP/p300 and PCAF co-activators are involved in transcriptional activation by many DNA binding transcription activators. At present, the exact role of the individual acetyltransferase and how they interact in the regulation of transcription are largely unknown. Our previous results showed that CBP/p300 and PCAF act cooperatively in stimulating KLF13 transcriptional activity indicating both acetyltransferases are involved in KLF13 transcriptional activation.26 To understand how these two acetyltransferase co-activators functionally interact to regulate transcription cooperatively, we carried out biochemical studies to determine the molecular interaction between CBP and PCAF in their regulation of KLF13 DNA binding activity. Our results demonstrate that CBP and PCAF act synergistically and antagonistically in regulation of KLF13 DNA binding depending on the HAT activity and the status of KLF13 acetylation. PCAF blocks the CBP acetylation-mediated disruption of KLF13 DNA binding by preventing its acetylation by CBP. Since the GST–PCAF (352–832) used in our experiments does not contain the CBP interacting region of PCAF,30 this blocking activity is probably mediated through PCAF interaction with KLF13 but not with CBP.
The results that acetylation by CBP has disruptive effects on KLF1, KLF11 and KLF13 DNA binding, and that CBP and PCAF functionally antagonize each other in their regulation of KLF13 DNA binding raise the question of what is the biological significance. As both CBP and PCAF act cooperatively in stimulating KLF13 transcriptional activation, another obvious question is how these observations fit into the transcriptional process and lead to cooperative stimulation. Transcription is a very dynamic process involving the assembly and disassembly of pre-initiation, initiation or elongation complexes. Therefore, in the context of the complex process of transcriptional regulation, CBP and PCAF may function at distinct steps of the transcription process to facilitate the assembly or disassembly of the complex through acetylation of target proteins in the complex. Several previous studies support this possibility. For example, it has been shown that PCAF interacts with the elongation-competent, phosphorylated form of RNA polymerase II, whereas p300 interacts specifically with the non-phosphorylated, initiation-competent RNA polymerase II.31 Functional studies have shown that p300 functions at the step of transcriptional initiation but not re-initiation in its co-activation with ligand-activated estrogen receptor.32 Studies have revealed a dynamic and ordered recruitment of these co-activators to the target gene promote in vitro and in vivo.33–35 Recent studies further support this possibility. Acetylation of the architectural protein HMGI(Y) by GCN5/PCAF and CBP has been shown to have distinct effects on HMGI(Y) DNA binding and PCAF acetylation of HMGI(Y) prevents the disruption of its DNA binding by CBP acetylation.36 Acetylation of ACTR, an acetylase itself, by p300/CBP disrupts its association with nuclear receptor.33 The CBP/p300 acetylation-mediated disruption of ACTR interaction with the nuclear receptor and disassembly of enhanceosome have been suggested to play a role in the switching from transcriptional activation to attenuation. Therefore, the distinct effects of CBP and PCAF on KLF13, HMGI(Y) and ACTR DNA binding and protein–protein interaction, and their antagonistic action are integral parts of the dynamic process of RNA synthesis.
Materials and Methods
Plasmids
Plasmids for expression of PCAF, p300 and KLF13 in mammalian cells were as described.26 Plasmid for expression of GST–KLF13 fusion proteins were constructed by inserting the corresponding PCR fragments into pGEX-4T-1 (Pharmacia). Plasmids encoding GST–PCAFwt or GST–PCAFHAT-fusion proteins containing wild-type or HAT defective (Y616A; F617A) were constructed by inserting their corresponding PCAF fragments from amino acid residues 352–832 into pGEX-4T-1. Plasmids encoding GST–CBPHAT wild-type (wt) or acetylase defective (HAT-) mutant (L1690K;C1691L) was constructed by inserting the respective CBP sequences from amino acid residues 1196 to 1718 into pGEX-4T-1. Lysine to alanine or arginine mutation was introduced using Quickchange kit (Stratagene). All the mutants were confirmed by DNA sequencing. GST–p53 was constructed by cloning the full-length p53 into pGEX-4T-1. GST–KLF1 containing the zinc finger domains of KLF1 was as described.37 GST–E2F-1 was constructed by inserting the human E2F-1 (amino acid residues 92–195) into pGEX-4T-1. GST–KLF11 containing the DNA binding domain of KLF11 was constructed by inserting amino acid residues 352–512 of KLF11 into pGEX-4T-1.
Protein acetyltransferase assays
Protein acetylation assays were performed as described.26 Briefly, acetylation assays were carried out in reaction mixtures (30 μl) containing 50 mM Hepes (pH 8.0), 10% (v/v) glycerol, 50 mM KCl, 2 mM DTT, 10 mM sodium butyrate, 1 μl of [14C]acetyl-CoA (50 mCi/mmol; Amersham), 1 μg of purified GST–KLF13 fusion proteins on beads and 50 ng of purified GST–CBP HAT, GST–PCAFwt or GST–PCAF HAT defective mutant. After incubating at room temperature for one hour with gentle mixing, the reaction mixtures were subjected to SDS-PAGE electrophoresis and analyzed using PhosphoImage. We estimated that more than 80% of the protein is acetylated under our acetylation condition.
Electrophoretic mobility shift assay
EMSA was carried out as described.26 The sequence of the oligonucleotide containing the human γ CACCC box is as follows: 5′-GCTAAACTCCACCCATGGGTTGG-3′. The oligonucleotide probe containing p53 binding site29 and E2F-1 binding site21 were as described. For acetylation and EMSA assays, GST–KLF1, GST–KLF11, GST–KLF13, GST–p53 and GST–E2F-1 were acetylated by CBP or PCAF as described in the protein acetyltransferase assays except that 1 μM of unlabeled acetyl-CoA instead of[14C]acetyl-CoA was used. We estimated that more than 80% of the protein is acetylated under our acetylation condition. After the acetylation reaction, the acetylated proteins were used in EMSA assays. The EMSA reaction was incubated with 32P-labeled oligonucleotide probes at room temperature for 40 minutes and protein–DNA complex was separated by electrophoresis on 5% (w/v) gel in 0.5 × TBE at room temperature and analyzed using PhosphorImage.
Cell culture, transfection and reporter assay
COS cells were cultured in DMEM medium containing 10% (v/v) fetal calf serum. For luciferase assays, COS cells cultured in 12-well Plate were transfected with 10 ng of reporter plasmid pγluc containing the human γ globin promoter upstream of the luciferase reporter gene, 5 ng of KLF13 expression plasmid and 100 ng of PCAF or p300 expression plasmid using FuGENE6 (Roche). Cells were harvested at 30–36 hours after transfection. Luciferase activity was measured using the Promega luciferase assay system.
Acknowledgements
We are most grateful to Drs J. Bieker, T. Kouzarides, A. Hecht, Y. Nakatani and I. Talianidis for generously providing the reagents. This research was supported by grant from the National Institutes of Health (to C.-Z. S.).
Abbreviations used
- EMSA
electrophoretic mobility shift assay
- HAT
histone acetyltransferase
References
- 1.Lemon B, Tjian R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000;14:2169–2551. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- 2.Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucl. Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bieker JJ. Kruppel-like factors: three fingers in many pies. J. Biol. Chem. 2001;276:34355–34358. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- 4.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 5.Wade PA, Wolffe AP. Histone acetyltransferases in control. Curr. Biol. 1997;7:R82–R84. doi: 10.1016/s0960-9822(06)00042-x. [DOI] [PubMed] [Google Scholar]
- 6.Kadonaga JT. Eukaryotic transcription: an interlaced network of transcription factors and chromatin-modifying machines. Cell. 1998;92:307–313. doi: 10.1016/s0092-8674(00)80924-1. [DOI] [PubMed] [Google Scholar]
- 7.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 8.Workman JL, Kingston RE. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 9.Berger SL. Gene activation by histone and factor acetyltransferases. Curr. Opin. Cell Biol. 1999;11:336–341. doi: 10.1016/S0955-0674(99)80046-5. [DOI] [PubMed] [Google Scholar]
- 10.Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 11.Kuo MH, Brownell JE, Sobel RE, Ranalli TA, Cook RG, Edmondson DG, et al. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996;383:269–272. doi: 10.1038/383269a0. [DOI] [PubMed] [Google Scholar]
- 12.Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 13.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 14.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 15.Mizzen CA, Yang XJ, Kokubo T, Brownell JE, Bannister AJ, Owen-Hughes T, et al. The TAF(II)250 subunit of TFIID has histone acetyltransferase activity. Cell. 1996;87:1261–1270. doi: 10.1016/s0092-8674(00)81821-8. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, et al. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 17.Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 18.Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 19.Janknecht R, Hunter T. Transcription. A growing coactivator network. Nature. 1996;383:22–23. doi: 10.1038/383022a0. [DOI] [PubMed] [Google Scholar]
- 20.Nakatani Y. Histone acetylases—versatile players. Genes Cells. 2001;6:79–86. doi: 10.1046/j.1365-2443.2001.00411.x. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. EMBO J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soutoglou E, Katrakili N, Talianidis I. Acetylation regulates transcription factor activity at multiple levels. Mol. Cell. 2000;5:745–751. doi: 10.1016/s1097-2765(00)80253-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W, Kadam S, Emerson BM, Bieker JJ. Site-specific acetylation by p300 or CREB binding protein regulates erythroid Kruppel-like factor transcriptional activity via its interaction with the SWI–SNF complex. Mol. Cell. Biol. 2001;21:2413–2422. doi: 10.1128/MCB.21.7.2413-2422.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munshi N, Agalioti T, Lomvardas S, Merika M, Chen G, Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science. 2001;293:1133–1136. doi: 10.1126/science.293.5532.1133. [DOI] [PubMed] [Google Scholar]
- 25.Lee YH, Koh SS, Zhang X, Cheng X, Stallcup MR. Synergy among nuclear receptor coactivators: selective requirement for protein methyltransferase and acetyltransferase activities. Mol. Cell. Biol. 2002;22:3621–3632. doi: 10.1128/MCB.22.11.3621-3632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song CZ, Keller K, Murata K, Asano H, Stamatoyannopoulos G. Functional interaction between coactivators CBP/p300, PCAF, and transcription factor FKLF2. J. Biol. Chem. 2002;277:7029–7036. doi: 10.1074/jbc.M108826200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Bieker JJ. Acetylation and modulation of erythroid Kruppel-like factor (EKLF) activity by interaction with histone acetyltransferases. Proc. Natl Acad. Sci. USA. 1998;95:9855–9860. doi: 10.1073/pnas.95.17.9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 30.Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McInerney EM, et al. Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science. 1998;279:703–707. doi: 10.1126/science.279.5351.703. [DOI] [PubMed] [Google Scholar]
- 31.Cho H, Orphanides G, Sun X, Yang XJ, Ogryzko V, Lees E, et al. A human RNA polymerase II complex containing factors that modify chromatin structure. Mol. Cell. Biol. 1998;18:5355–5363. doi: 10.1128/mcb.18.9.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraus WL, Kadonaga JT. p300 and estrogen receptor cooperatively activate transcription via differential enhancement of initiation and reinitiation. Genes Dev. 1998;12:331–342. doi: 10.1101/gad.12.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 34.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 35.Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 36.Munshi N, Merika M, Yie J, Senger K, Chen G, Thanos D. Acetylation of HMG I(Y) by CBP turns off IFN beta expression by disrupting the enhanceosome. Mol. Cell. 1998;2:457–467. doi: 10.1016/s1097-2765(00)80145-8. [DOI] [PubMed] [Google Scholar]
- 37.Bieker JJ, Southwood CM. The erythroid Kruppel-like factor transactivation domain is a critical component for cell-specific inducibility of a beta-globin promoter. Mol. Cell. Biol. 1995;15:852–860. doi: 10.1128/mcb.15.2.852. [DOI] [PMC free article] [PubMed] [Google Scholar]