

Abstract
Herpes Simplex Virus thymidine kinase (HSVTK) with ganciclovir (GCV) is currently the most widely used suicide gene/prodrug system in cancer gene therapy. A major limitation in this therapy is the inefficient activation of GCV by HSVTK to its active antimetabolites. We previously described two strategies to overcome this limitation: 1) generation of HSVTK mutants with improved GCV activation potential and 2) construction of a fusion protein encoding HSVTK and mouse guanylate kinase (MGMK), the second enzyme in the GCV activation pathway. As a means to further enhance GCV activation, two MGMK/HSVTK constructs containing the HSVTK mutants (mutant 30 and SR39) were generated and evaluated for their tumor and bystander killing effects in vitro and in vivo. One fusion mutant, MGMK/30, demonstrates significant reduction in IC50 values of approximately 12,500-fold, 100-fold, and 125-fold compared to HSVTK, mutant 30 or MGMK/HSVTK, respectively. In vitro bystander analyses reveal that 5% of MGMK/30-expressing cells are sufficient to induce 75% of tumor cell killing. In an xenograft tumor model, MGMK/30 displays the greatest inhibition of tumor growth at a GCV concentration (1mg/kg) that has no effect on wild type HSVTK-, MGMK/HSVTK-, or mutant 30-transfected cells. Another fusion construct, MGMK/SR39, sensitizes rat C6 glioma cells to GCV by 2,500-fold or 25-fold compared to HSVTK or MGMK/HSVTK, respectively. In vitro analyses show similar IC50 values between cells harboring SR39 and MGMK/SR39, although MGMK/SR39 appears to elicit stronger bystander killing effects where 1% of MGMK/SR39-transfected cells result in 60% cell death. In a xenograft tumor model, despite observable tumor growth inhibition, no statistical significance in tumor volume was detected between mice harboring SR39- and MGMK/SR39-transfected cells when dosed with 1mg/kg GCV. However, at a lower dose of GCV (0.1mg/kg), MGMK/SR39 appears to have slightly greater tumor growth inhibition properties compared to SR39 (P≤0.05). In vivo studies indicate that both mutant fusion proteins display substantial improvements in bystander killing in the presence of 1mg/kg GCV, even when only 5% of the tumor cells are transfected. Such fusion mutants with exceptional prodrug converting properties will allow administration of lower and non-myelosuppressive doses of GCV concomitant with improved tumor killing and as such are promising candidates for translational gene therapy studies.
Keywords: thymidine kinase, guanylate kinase, fusion gene, suicide gene therapy, ganciclovir
Introduction
Thymidine kinase (TK) (EC 2.7.1.21) is an essential enzyme in the pyrimidine salvage pathway, catalyzing the transfer of the γ-phosphate from ATP to thymidine to produce dTMP1. Unlike human TK, Herpes Simplex Virus type 1 (HSV-1) thymidine kinase (HSVTK) has a broad substrate specificity and is able to phosphorylate pyrimidines, pyrimidine analogues (thymidine, deoxycytidine, and azidothymidine), and guanosine analogues (ganciclovir, acyclovir, buciclovir, and penciclovir). Originally suggested by Moolten, HSVTK has become widely used as a suicide gene in combination with the guanosine analogue ganciclovir (GCV) for the treatment of a variety of cancers2, 3. Upon successful delivery of HSVTK to cancer cells followed by systemic administration of GCV, transfected cancer cells become sensitized to the prodrug. Inside the cell, GCV is initially phosphorylated by HSVTK to form GCV-monophosphate (GCV-MP), which is then further phosphorylated to GCV-diphosphate (GCV-DP) by cellular guanylate kinase (GMK) and to the active antimetabolite GCV-triphosphate (GCV-TP) by cellular nucleoside diphosphokinase4, 5. Cytotoxicity is mainly due to the action of GCV-TP after its incorporation into nascent DNA, which results in inhibition of DNA synthesis and ultimately leads to cell death5, 6. Since human TK has a narrow substrate specificity and is not active towards GCV, prodrug-associated cytotoxicity is limited to the site of transfection. Because low gene transfer is a key limitation in gene therapy, the success of suicide gene therapy relies heavily on a phenomenon known as the bystander effect, in which untransfected neighboring cancer cells are eradicated by the transfer of antimetabolites via gap junctions or apoptotic vesicles2, 7. Previous studies reported that complete tumor ablation in vivo could be achieved in the presence of GCV even when only a small percentage of the tumor was transfected with retroviral vectors carrying the HSVTK gene8. Implicit in the bystander effect is that sufficient phosphorylated GCV is transferred to neighboring cells to elicit cell killing. In addition, an immune-related response also contributes to the BE although in a delayed fashion.
Although the bystander effect promotes tumor cell killing in HSVTK/GCV gene therapy, inefficient activation of GCV by HSVTK is a major limitation. The Km value of HSVTK toward GCV (Km = 47μM) is approximately 10-fold higher than its Km value for thymidine (Km = 0.4μM), its natural substrate9. This lower GCV binding ability is exacerbated by a 5-fold lower kcat (turnover rate) resulting in more than a 500-fold lower overall enzyme catalytic efficiency (kcat/Km). Due to the combination of low gene transfer and inadequate GCV activation by HSVTK, myelosuppressive doses of GCV are required to achieve significant or complete tumor ablation9. The second step in the activation of GCV is performed by guanylate kinase (GMK, ATP:GMP phosphotransferase, EC 2.7.4.8). GMK is an essential enzyme involved in purine biosynthesis and is responsible for the phosphorylation of GMP and dGMP, as well as GCV-MP10. Previous reports have shown the Km value of GMK for GCV-MP is 2-fold higher (Km = 42-54μM) than its Km toward its natural substrate GMP (∼25μM)10, 11. This suggests that endogenous GMK may be a second rate limiting step during the production of sufficient antimetabolites for complete tumor ablation 12, 13.
One approach to improve prodrug activation, which may consequently reduce the dose of GCV required to kill tumor cells and thereby limit the prodrug-associated negative side effects, is to use HSVTK mutants with increased activity and/or substrate specificity toward GCV9, 14. We have previously reported the construction and characterization of two such HSVTK mutants, mutant 30 and SR39, that show not only an improvement in kinetic properties towards GCV, but also in vitro tumor killing and in vivo tumor growth inhibition compared to HSVTK alone9, 15. As a means to overcome the bottleneck of prodrug activation from the monophosphate to the diphosphate form, a fusion protein expressing both functional GMK and HSVTK (GMK/HSVTK) was generated16. Mouse GMK (MGMK) was used because in our hands, the mouse enzyme was soluble in E. coli whereas the human enzyme was not10. In vitro cytotoxic assays using this fusion protein demonstrated that cells expressing MGMK/HSVTK confer a ∼175-fold decrease in IC50 value compared to HSVTK alone in response to GCV treatment in stably transfected rat C6 glioma cells16.
In this study, we evaluated tumor toxicity and bystander killing activity of the previously created fusion MGMK/HSVTK in vitro and in an in vivo tumor model. Results from in vivo studies suggest that the fusion protein inhibits tumor growth at a GCV concentration (1mg/kg) that has no affect on wild type HSVTK-bearing tumors. Moreover, the fusion also elicits a stronger bystander effect in vitro and in vivo. To further augment its GCV-mediated tumor killing activity, we have taken a further step by engineering the improved HSVTK mutants, mutant 30 and SR39, into the fusion protein construct. Here, we report that the mutant fusion proteins created in this study exhibit a superior GCV-mediated toxicity and bystander killing activity at very low doses of prodrug in vitro and in an in vivo xenograft tumor model compared to MGMK/HSVTK and their respective single gene constructs. The results reported here demonstrate that the mutant chimeric proteins offer significant advantages to the suicide gene therapy approach to that of the widely used wild type HSVTK gene, previously characterized HSVTK mutants, or the previously created MGMK/HSVTK fusion construct.
Results
Genetic complementation
Two new constructs were generated, MGMK/30 (containing HSVTK mutant 30) and MGMK/SR39 (containing HSVTK mutant SR39). To ascertain whether the constructed mutant fusions displayed functional TK and GMK activity, genetic complementation for both activities was independently evaluated using the previously established thymidine kinase (tdk-) and conditionally gmk deficient E. coli strain TS202A(DE3)17. Upon DNA sequence verification, pET23d, pET23d:mgmk, pET23d:HSVTK, pET23d:30, pET23d:SR39, pET23d:mgmk/HSVTK, pET23d:mgmk/30 and pET23d:mgmk/SR39 were used to transform the E. coli strain, TS202A(DE3) and were plated onto TK and GMK selection media in the presence or absence of arabinose supplemented with kanamycin. As anticipated, cells harboring pET23d and pET23d:mgmk were not able to complement the TK deficiency of TS202A(DE3), whereas cells harboring pET23d:HSVTK, pET23d:30, pET23d:SR39 and their respective fusion constructs were viable (data not shown). Similarly, only pET23d:mgmk, pET23d:mgmk/HSVTK, pET23d:mgmk/30, and pET23d:mgmk/SR39 were viable on GMK selection medium in the absence of arabinose (data not shown). These results indicated that the fusion proteins were expressed and maintained both TK and GMK activities. All cultures grew on control non-selective minimal medium.
In vitro prodrug sensitivity assays
In order to analyze the efficacy of the constructed mutant chimeric proteins, we utilized rat C6 glioma cells. Rat C6 glioma cells have previously served as a model system to evaluate the efficacy of the HSVTK/GCV paradigm18, 19. Mammalian expression vectors (pUB) expressing suicide genes were constructed and used to stably transfect rat C6 glioma cells. Once stable transfection was achieved, expression levels of the suicide proteins were determined by immunoblots using polyclonal serum raised against HSVTK or MGMK. Immunoblots revealed the presence of HSVTK at around 45kDa when anti-HSVTK serum was used, and MGMK at around 23kDa when anti-MGMK serum was used. These results are in accord with the predicted molecular masses for each protein (Figure 1B-C). The fusion proteins cross-reacted with both anti-sera at the predicted molecular mass of approximately 68kDa and relatively equivalent protein levels were observed (Figure 1A). In vitro prodrug sensitivity assays were done as described in the Materials and Methods section and shown as percent survival in Figure 2. This experiment was performed at least three times using different pools of transfectants with similar results. In the presence of GCV, wild type HSVTK expressing cells displayed an IC50 of ∼50μM whereas wild type MGMK/HSVTK-, mutant 30- and fusion MGMK/30-expressing cells displayed an IC50 value of 0.5μM, 0.4μM, and 0.004μM, respectively (Figure 2A and Table 1). A 100-fold decrease in IC50 was seen in cells expressing MGMK/HSVTK compared to cells expressing HSVTK alone. This same trend, a 100-fold reduction in IC50 value, was also noted in cells expressing MGMK/30 when compared to cells expressing mutant 30 or MGMK/HSVTK. Surprisingly, when the mutant construct MGMK/SR39 was analyzed in vitro, no significant difference in tumor cytotoxicity was detected in comparison to SR39. Similar IC50 values of 0.02μM were seen in cells expressing either SR39 or fusion MGMK/SR39 (Figure 2B and Table 1). Microscopically, no cells were visible in the highest concentration of prodrugs with mutant 30, SR39, fusion MGMK/HSVTK, MGMK/30, and MGMK/SR39 despite the artifactual reading of 5% survival.
Figure 1. Immunoblot of stable C6 transfectants.
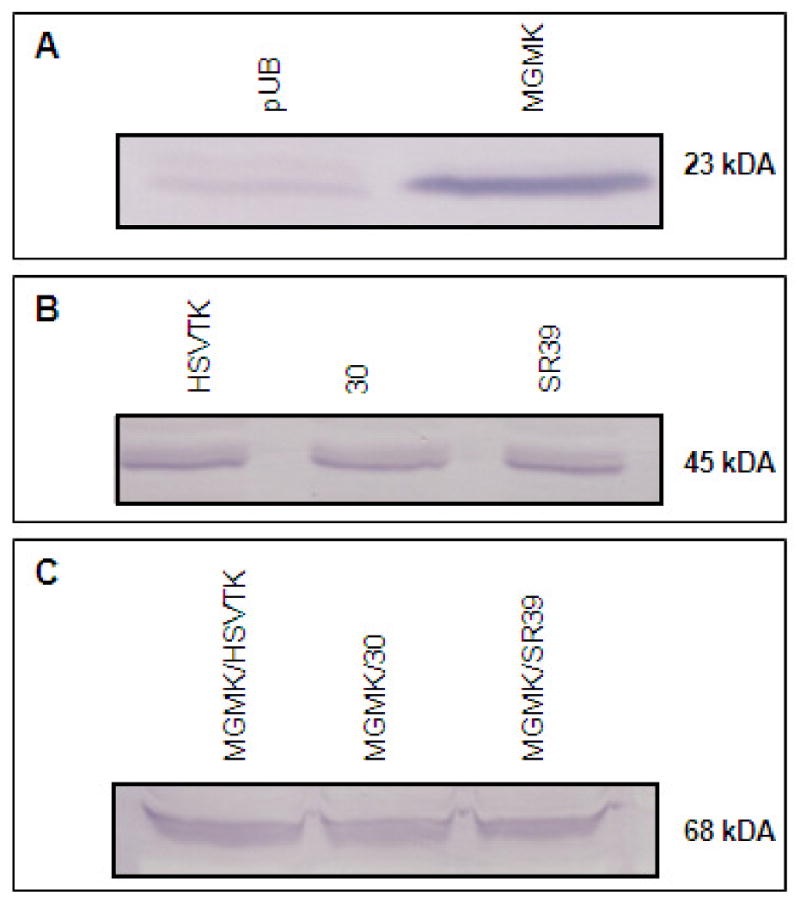
Equivalent amounts of cell lysates (∼1 million cells) were collected from pools of stable transfectants harboring (A) pUB and pUB:mgmk, or (B) pUB:HSVTK, pUB:30, and pUB:SR39, or (C) pUB:mgmk/HSVTK, pUB:mgmk/30, and pUB:mgmk/SR39 were loaded onto an SDS-containing polyacrylamide gel. The proteins were transferred to nitrocellulose and an immunoblot was performed using rabbit anti-HSVTK and anti-MGMK serum. The presence of MGMK, HSVTK (and HSVTK mutants), and MGMK/HSVTK (and the remaining fusion constructs) was detected at ∼23kDA, ∼45kDA, and ∼68kDA, respectively, which was in accord with the expected molecular weight for each sample. Relatively equivalent protein levels were observed.
Figure 2. GCV sensitivity assays of rat C6 transfectants.
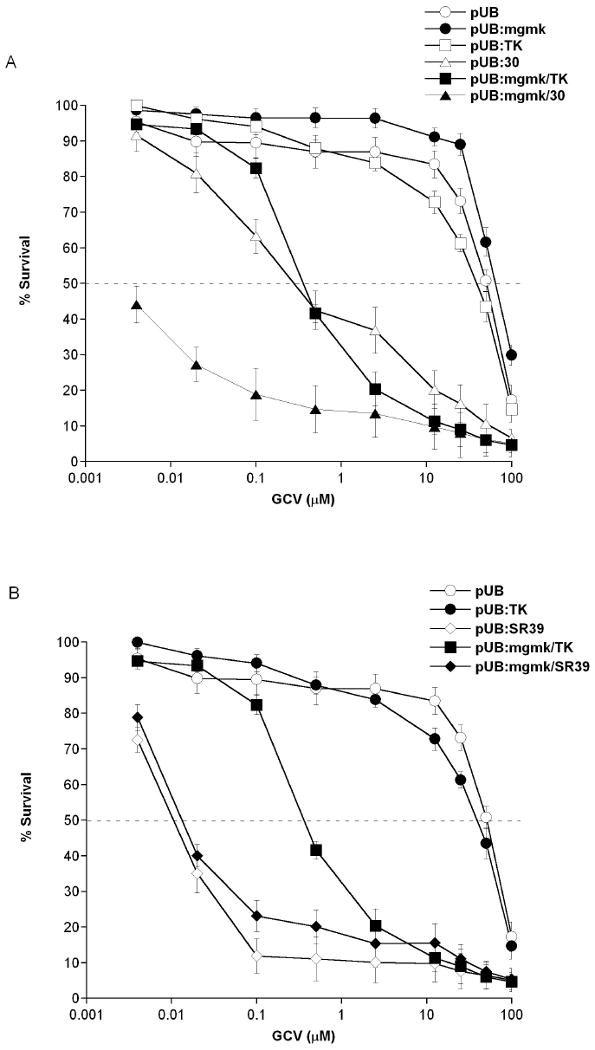
Pools of stably transfectants containing (A) pUB (○), pUB:mgmk (●), pUB:HSVTK (□), pUB:mgmk/HSVTK (■), pUB:30 (∆) and pUB:mgmk.30 (▲), or (B) pUB:SR39 (◇) and pUB:mgmk/SR39 (◆) were evaluated for GCV sensitivity as described in the Materials and Methods section. After 7 days of GCV treatment, cell survival was determined using Alamar Blue according to the manufacturer's instructions. Untreated samples (0μM GCV) were set at 100% cell survival. Each data point (mean ± SEM; n=3; performed with 24 replicates) is expressed as a percentage of the value for control wells with no GCV treatment.
Table 1.
In vitro cytotoxicity and bystander response of rat C6 glioma cells expressing wild type, mutant HSVTKs, wild type fusion or mutant fusion constructs to GCV.
| GCV IC50 (μM) | Relative to HSVTK | Percentage of cell killing achieved with ∼5% cell transfection | |
|---|---|---|---|
| pUB | 50 | 1 | N.A |
| pUB:MGMK | 65 | 0.77 | N.A |
| pUB:HSVTK | 50 | 1 | N.D |
| pUB:30 | 0.4 | 125 | 23% |
| pUB:SR39 | 0.02 | 2,500 | 46% |
| pUB:MGMK/HSVTK | 0.5 | 100 | 45% |
| pUB:MGMK/30 | 0.004 | 12,500 | 72% |
| pUB:MGMK/SR39 | 0.02 | 2,500 | 71% |
N.A = Not Available
N.D = Not Detected
In vitro bystander activity analysis
In vitro bystander analysis was performed as described in the Materials and Methods section. For this experiment, cells carrying empty vector were mixed with cells expressing the tested constructs at different ratios of 99:1, 95:5, 80:20, 60:40, 50:50, 60:40, 70:30, 90:10, and 100:0, respectively. Figure 3 shows results as percent survival from this experiment. This experiment was performed at least three times using different pools of transfectants with similar results. In the presence of GCV, all fusion constructs elicited stronger bystander effects compared to their respective single gene constructs. Results indicate that MGMK/SR39 demonstrates the strongest bystander activity. When only ∼1% of tumor cells expressed MGMK/SR39, 60% tumor killing was achieved. This is a significant improvement over SR39 alone, where approximately 40% of tumor cell transduction was necessary to achieve the same killing effect. MGMK/30 demonstrated the second strongest bystander activity where only ∼5% cells expressing fusion proteins were sufficient to induce about 75% of tumor cell killing. Mutant 30, SR39 and wild type fusion MGMK/HSVTK appeared to have similar bystander activities, where approximately 50% cell transduction resulted in 65% tumor ablation.
Figure 3. Bystander analysis of rat C6 transfectants in the presence of GCV.
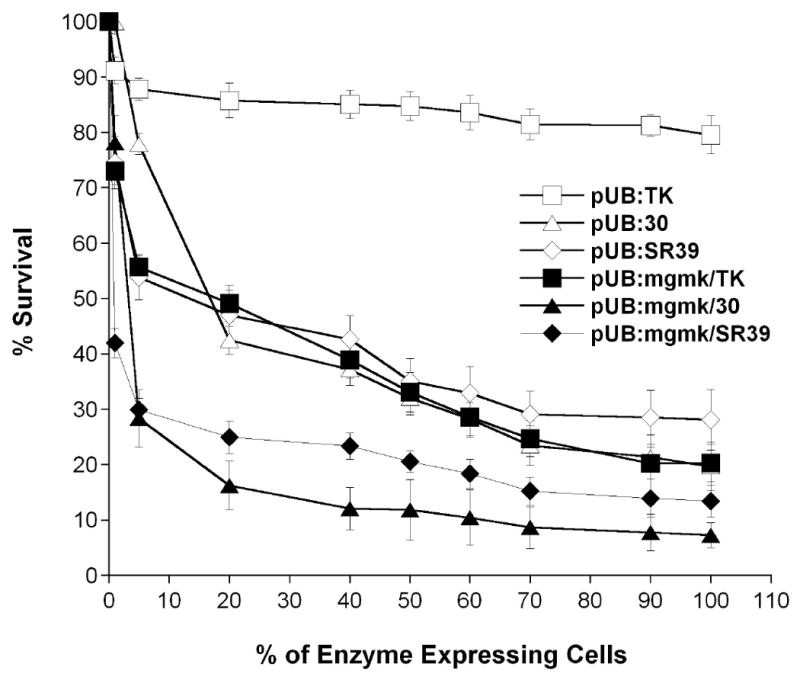
Pools of rat C6 glioma cells containing pUB were mixed with cells harboring either pUB:HSVTK (□), pUB:mgmk/HSVTK (■), pUB:30 (∆), pUB:mgmk.30 (▲), pUB:SR39 (◇) or pUB:mgmk/SR39 (◆) at different ratios and were subjected to 80 μM GCV for a period of 7 days and cell survival was determined using Alamar Blue as described in Materials and Methods. Each data point (mean ± SEM; n=3; performed with 24 replicates) is expressed as a percentage of the value for control wells with without HSVTK-expressing cells (pUB).
Xenograft tumor model
In vivo analyses of all constructs were conducted as described in the Materials and Methods section. Figure 4 displays the time course of tumor growth in mice harboring stable transfected cells treated with GCV or PBS. Tumor cells transfected with empty vector (pUB) showed no statistical difference in tumor size when mice were treated with PBS or GCV (Figures 4A and 4D) (P ≥ 0.05). Similarly, no statistical difference was observed in tumor volume in mice seeded with cells transfected with pUB:HSVTK that were injected with PBS or GCV (Figures 4A and 4D) or in mice seeded with cells transfected with empty vector (pUB) or pUB:HSVTK receiving GCV treatment (Figure 4A) (P values ≥ 0.05). The lack of difference in tumor volume is likely a reflection of the low GCV dose administered. The prodrug-treated mice bearing mutant 30- or fusion MGMK/HSVTK-expressing tumors elicited a similar anti-tumor response (Figure 4A). However, once GCV injections were stopped, rapid tumor growth was observed in these groups of mice. In contrast, mice bearing MGMK/30-expressing tumors and treated with GCV displayed the greatest restriction in tumor growth (day 16 mean tumor volume: 50.8mm3) (Table 2). This is an improvement of approximately 33-fold over fusion MGMK/HSVTK or mutant 30 (P values ≤ 0.05). In line with in vitro data, similar anti-tumor responses were observed between SR39 and MGMK/SR39. Although both are efficient in inhibiting tumor growth, no statistical difference in tumor volume was observed in mice seeded with cells transfected with SR39 or MGMK/SR39 and treated with 1mg/kg GCV (Figure 4B and Table 3) (P ≥ 0.05).
Figure 4. Tumor growth course during and after GCV treatment in an xenograft tumor model.
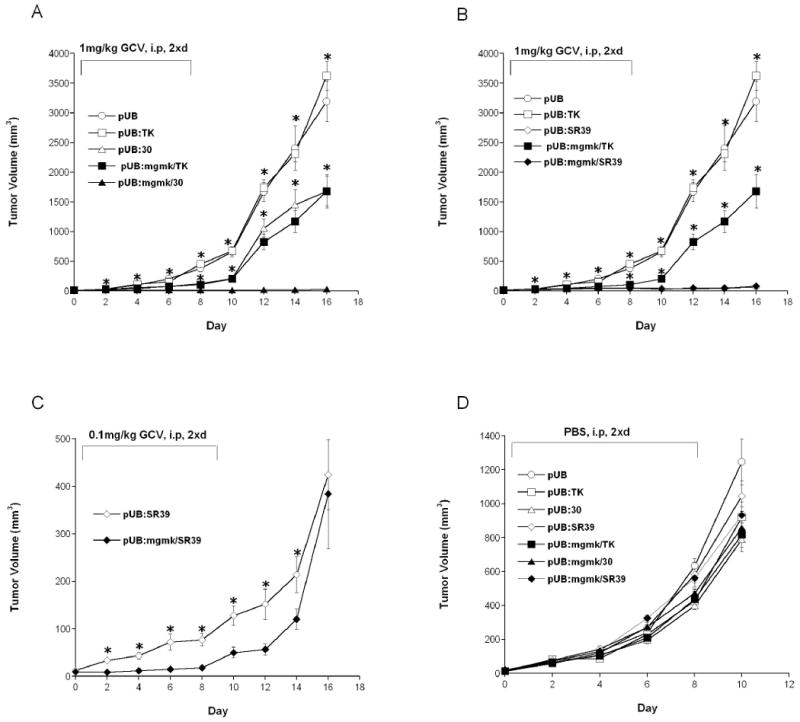
Pools of rat C6 glioma cells transfected with (A) pUB (○), pUB:HSVTK (□), pUB:mgmk/HSVTK (■), pUB:30 (∆) and pUB:mgmk/30 (▲); or (B) pUB (○), pUB:HSVTK (□), pUB:mgmk/HSVTK (■), pUB:SR39 (◇) and pUB:mgmk/SR39 (◆); or (C) pUB:SR39 (◇) and pUB:mgmk/SR39 (◆); or (D) pUB (○), pUB:HSVTK (□), pUB:mgmk/HSVTK (■), pUB:30 (∆), pUB:mgmk/30 (▲), pUB:SR39 (◇) and pUB:mgmk/SR39 (◆) were used to seed tumors in nude mice (n=5 for each group). When tumor size reached 3-4mm (day 0), (A & B) GCV (1mg/kg), (C) GCV (0.1mg/kg) or (D) PBS was intraperitoneally administered twice a day for 8 days. During this period, tumor growth was measured every other day. Tumor volume was calculated using the formula 4/3π ((Width × Length × Height)/2), plotted and analyzed for statistical significance using Student's t-test. Asterisks denote statistical significance (P ≤ 0.05) in tumor sizes between: (A) mice harboring MGMK/30-expressing tumor cells and those that received either vector- or HSVTK-, or mutant 30- or MGMK/HSVTK-expressing tumor cells in the presence of 1mg/kg GCV; (B) mice harboring SR39- or MGMK/SR39-expressing cells (no statistical difference between the two) and those that received either vector- or HSVTK- or MGMK/HSVTK-expressing cell in the presence of 1mg/kg GCV; (C) mice harboring SR39 and those that received MGMK/SR39-expressing cells in the presence of 0.1mg/kg GCV.
Table 2.
In vivo response of rat C6 tumors expressing wild type HSVTK, mutant 30, MGMK/HSVTK or MGMK/30 to PBS or GCV treatment.
| Mean Tumor Volume (mm3) | |||
|---|---|---|---|
| Day 8 | Day 16 | ||
| PBS | GCV (1mg/kg) | GCV (1mg/kg) | |
| HSVTK | 428.29 (39.27)a | 453.42 (43.17)** | 3622.50 (242.60)** |
| Mutant 30 | 399.30 (23.58) | 126.19 (20.03)** | 1681.80 (248.88)** |
| MGMK/HSVTK | 438.51 (47.73) | 109.85 (14.94)** | 1679.10 (284.62)** |
| MGMK/30 | 473.76 (39.49) | 16.60 (3.06) | 33.39 (5.55) |
Standard error mean
P values ≤ 0.0001, this P values reflect a comparison between MGMK/30 and HSVTK, or between MGMK/30 and mutant 30, or between MGMK/30 and MGMK/HSVTK tumor sizes treated with GCV (1mg/kg) at two different time points (day 8 and day 16) (n=5).
Since no difference in tumor growth inhibition was observable between SR39 and MGMK/SR39, another in vivo xenograft tumor studies using one tenth the dose of GCV or 0.1mg/kg was performed (Figure 4C). When 0.1mg/kg GCV was used to treat nude mice bearing SR39- or MGMK/SR39-expressing tumor cells, a difference in tumor growth was observed starting on day 4 and continued up to day 14 (GCV treatment was stopped on day 8) (Figure 4C). At the lower dose of GCV, the efficacy to inhibit tumor growth was discernable and results show that MGMK/SR39 generated a more potent GCV-mediated tumor growth impairment compared to SR39 alone (Figure 4C). On day 14, the mean tumor size for the MGMK/SR39-transfected tumor cells was 120.46 mm3, compared to the mean tumor size of SR39-transfected tumor cells at 214.04 mm3 (P ≤ 0.05) (Table 3). Figure 4D displays the results obtained from nude mice receiving PBS from each group and the data indicate that all tumors grew at approximately the same rate with no statistical difference. Mice receiving PBS from all groups were sacrificed on day 10 due to heavy tumor burden.
Bystander xenograft tumor model
Following protocols described in the Materials and Methods section, a mixture of cells expressing empty vector and cells harboring the tested constructs at a ratio 95:5, respectively, was injected into nude mice. The bystander activity of these constructs was analyzed in an in vivo xenograft tumor model and results are shown in Figures 5 and 6. In the presence of GCV, MGMK/30 exhibited substantial bystander killing activity as indicated by the greatest inhibition in tumor growth. On the other hand, HSVTK, MGMK/HSVTK, and mutant 30 did not exhibit sufficient bystander activity able to suppress tumor growth (Figure 5A). Tumors from mice receiving PBS treatment all grew at approximately the same rate (Figure 5B). In a separate series of experiments, the bystander activity of SR39 and MGMK/SR39 was evaluated. In the presence of 1mg/kg GCV, MGMK/SR39 generated a slightly stronger bystander effect compared to SR39 alone as indicated by a more restricted tumor growth in mice bearing MGMK/SR39-expressing cells (Figure 6A). When PBS was used, tumors from all mice group grew at approximately the same rate with no statistical difference (Figure 6B).
Figure 5. Bystander xenograft tumor model of mutant 30 and MGMK/30.
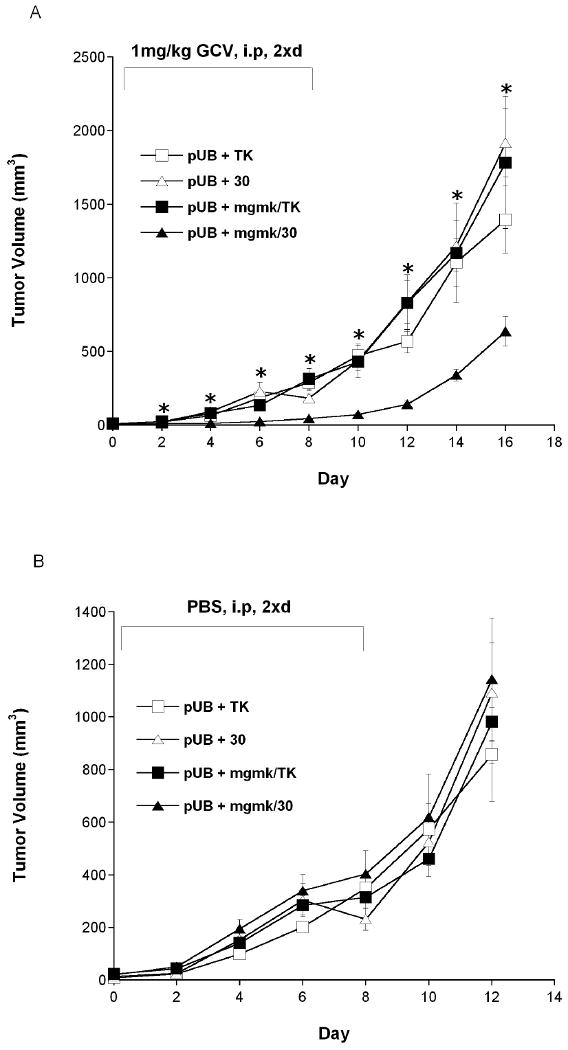
Pools of rat C6 glioma cells transfected with pUB were mixed with cells stably transfected with pUB:HSVTK (□), pUB:mgmk/HSVTK (■), pUB:30 (∆) and pUB:mgmk/30 (▲) at a ratio of 95:5 (vector:suicide genes, respectively). Mixed cells were used to seed tumors in nude mice (n=5 for each group). When tumor size reached 3-4mm (day 0), (A) GCV (1mg/kg) or (B) PBS was administered twice a day for 8 days. During this period, tumor growth was measured every other day. Tumor volume was calculated using the formula 4/3π ((Width × Length × Height)/2), plotted and analyzed for statistical significance using Student's t-test. Asterisks denote statistical significance (P ≤ 0.05) in tumor sizes between mice harboring 5% of MGMK/30-expressing tumor cells and harboring either 5% of HSVTK-, mutant 30-, or MGMK/HSVTK-expressing tumor cells in the presence of 1mg/kg GCV.
Figure 6. Bystander xenograft tumor model of SR39 and MGMK/SR39.
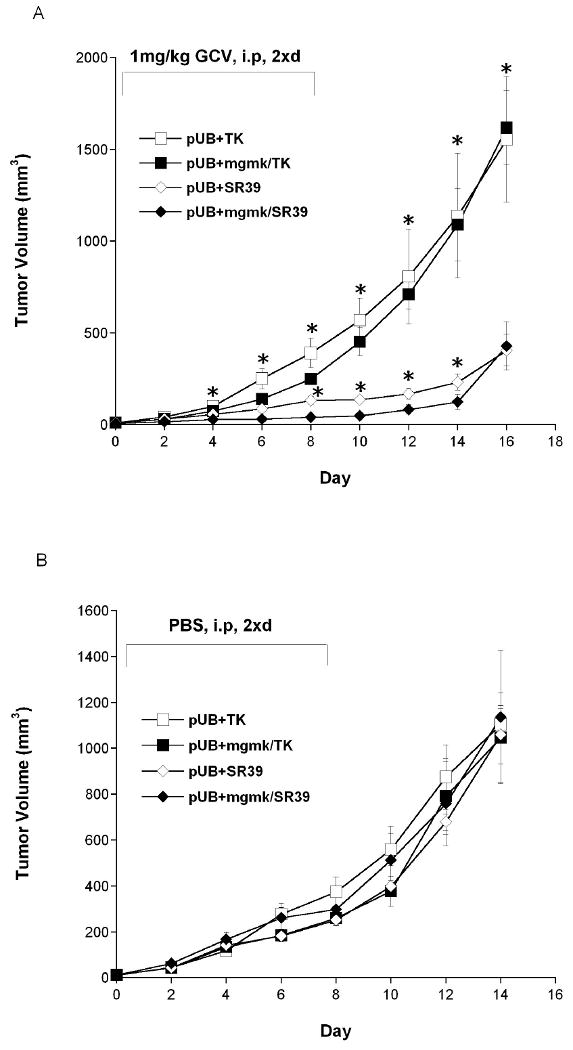
Pools of rat C6 glioma cells transfected with pUB were mixed with cells stably transfected with pUB:HSVTK (□), pUB:mgmk/HSVTK (■), pUB:SR39 (◇) and pUB:mgmk/SR39 (◆) were used to seed tumors in nude mice (n=5 for each group). When tumor size reached 3-4mm (day 0), (A) GCV (1mg/kg) or (B) PBS was administered twice a day for 8 days. During this period, tumor growth was measured every other day. Tumor volume was calculated using the formula 4/3π ((Width × Length × Height)/2), plotted and analyzed for statistical significance using Student's t-test. Asterisks denote statistical significance (P ≤ 0.05) in tumor sizes between mice harboring 5% of MGMK/SR39-expressing tumor cells and those that received 5% of either HSVTK-, MGMK/HSVTK-, or SR39-expressing tumor cells in the presence of 1mg/kg GCV.
Discussion
Despite apparent progress, conventional treatments against cancer such as chemotherapy and radiotherapy still face limitations and place heavy tolls on patients. Systemic administration of chemotherapy not only affects and kills normal tissues, consequently causing unpleasant side effects, but can also lead to the appearance of secondary cancers20, 21. Suicide gene therapy is an attractive alternative treatment for cancer because it offers the prospect of selectively introducing genes into cancer cells, rendering them susceptible to specific antimetabolites, and thereby limits the anti-tumor effect to tumor sites7. HSVTK/GCV is the most extensively studied and described suicide gene/prodrug system in suicide gene therapy. In contrast to human TK, HSVTK is able to phosphorylate select antiviral and anticancer drugs, such as the antiviral drug GCV. The efficacy and anti-tumor effect of HSVTK/GCV has been well documented in vitro and in vivo with more than 70 clinical trials conducted3. As of June 2007, three of these trials have advanced to phase III multicenter programs22, 23.
The selectivity of suicide gene therapy to target cancer cells is primarily due to the vector or delivery vehicle. Unfortunately, successful, selective and effective delivery of suicide genes remains to be one of the most challenging factors in gene therapy. This problem is exacerbated by another major limiting factor: the low efficiency and efficacy of suicide enzymes towards the prodrugs. Because of these limitations, a potent bystander effect is crucial to achieve significant or complete tumor ablation24. The bystander effect associated with suicide gene therapy is achieved via two different mechanisms: local or immune-mediated7. In the local-mediated bystander effect, gap junctions play an important role to transfer phosphorylated-GCV to neighboring cells18. The transfer of toxic drugs through gap junctions has some limitations since certain type of tumors, such as breast tumors have been reported to down-regulate intracellular gap junction communications and thus may have disorganized and/or non-functional gap junctions7, 25. These tumors demonstrate extremely poor bystander effects. Throughout this study, we chose to perform both in vitro and in vivo GCV-sensitivity studies in rat C6 glioma cells due to their low levels of intracellular communication by gap junctions18. By utilizing rat C6 cells, we sought to demonstrate that despite the nature of these cells having poor gap junctions, the fusion constructs are still able to generate broader and stronger bystander killing effects by generating higher levels of antimetabolites compared to their single gene or wild type fusion constructs. Local-mediated bystander effects can also be achieved through the transfer of apoptotic vesicles containing toxic metabolites to neighboring non-transfected cells7. An increase in the amount of cytotoxic products, due to improved prodrug conversion by the suicide genes, will likely lead to more pronounced bystander effects via apoptotic vesicle transfer as well.
One strategy to enhance therapeutic response of HSVTK towards GCV is by improving enzyme activity and efficiency towards to the prodrug. Previously, several HSVTK mutants with significant improvement towards GCV were generated and characterized. Two mutants in particular, mutant 30 and SR39, were analyzed in depth both in vitro and in an in vivo tumor model9, 15. Mutant SR39 contains five amino acid changes, while mutant 30 contains six amino acid substitutions; most of these changes are located at or near the active site. In vitro analyses revealed that both mutants enhanced C6 rat glioma cell sensitivity to GCV by more than 200-fold. When both mutants were evaluated in vivo, significant tumor growth inhibition was observed at a dose that does not affect wild type HSVTK. Clearly, both of these HSVTK mutants have significant advantages over wild type HSVTK.
Previous reports have suggested that a bottleneck problem arises in the HSVTK/GCV system, leading to accumulation of the ineffective intermediate products, i.e., GCV-MP and GCV-DP4, 16. We hypothesize that this bottleneck problem occurs because endogenous GMK is limited in its ability to convert GCV-MP to GCV-DP. Indeed, the Km value of GMK for GCV-MP is approximately 2-fold higher than its Km value for its natural substrate GMP10, 11. This implies that higher expression of GMK is required to overcome the bottleneck problem in order to produce higher amounts of active antimetabolites. Earlier experiments co-expressing HSVTK and GMK as separate proteins failed to demonstrate notable improvements in GCV sensitivity (data not shown and Akrüyek et al.,26). We hypothesized that the creation of a fusion enzyme may alleviate this problem by creating more efficient and more rapid drug conversion to the active antimetabolites form. We created a fusion chimeric gene, encoding both HSVTK and MGMK (mouse GMK) activities and showed a significant reduction in IC50 value in rat C6 glioma cells compared to HSVTK alone in the presence of GCV16. The current study was aimed to evaluate the anti-tumor and bystander killing activity of the previously described fusion protein MGMK/HSVTK in vitro and in vivo, and to further augment its anti-tumor activity by replacing the wild type HSVTK portion of the fusion construct with the sequences of the well established HSVTK mutants, mutant 30 and SR39. Current in vitro cytotoxic assay data validate the earlier finding that wild type fusion MGMK/HSVTK sensitized rat C6 glioma cells to GCV. However, in our current findings, we observed approximately a 100-fold reduction in IC50 value. This slight discrepancy in IC50 value (less than 2-fold) may be due to different mammalian promoters used in the earlier study (pREP8∆7) compared to the current study (pUB), and therefore protein levels may differ accordingly. Here, the bystander activity of the wild type fusion enzyme was analyzed in vitro and exhibited a stronger bystander effect than HSVTK alone. This suggests that the presence of higher levels of GMK intracellularly in close proximity to HSVTK may be important in producing sufficient antimetabolite levels able to eradicate transfected cancer cells and neighboring non-HSVTK-expressing tumor cells.
When mutant 30 was used to replace wild type HSVTK in the fusion construct, there was an improvement in tumor ablation and bystander activity. In the presence of GCV, a 100-fold decrease in IC50 was observed in MGMK/30-expressing tumor cells compared to mutant 30-expressing cells, translating in a total of 12,500-fold reduction in IC50 compared to HSVTK-expressing cells. When MGMK/30-expressing cells were seeded into nude mice and treated with 1mg/kg GCV, no tumor growth was observed during GCV treatment period (day 0 – 8) up to eight days after GCV treatment ended (day 16). Mice harboring mutant 30- and MGMK/HSVTK-expressing tumor cells demonstrated a delay in tumor growth during the GCV treatment period. Perhaps the most revealing finding was the exceptional improvement in the bystander activity of MGMK/30-expressing cells. The MGMK/30-transfected cells demonstrated a substantial bystander killing effect in vitro and were able to achieve complete tumor cell killing when only ∼25% of cells expressed the fusion enzyme. This was approximately a 3-fold improvement over mutant 30 and MGMK/HSVTK, where about a 75% transfected cell population was necessary to completely eradicate the cells. In a xenograft tumor model, MGMK/30-expressing tumor cells at a vector to suicide gene ratio of 95:5 demonstrated a highly potent GCV-mediated bystander anti-tumor activity compared to mutant 30- or MGMK/HSVTK-expressing tumor cells, where tumors grew similar to those in PBS treated mice. The absence of bystander killing effect could be attributed to the low dose of GCV (1mg/kg), short duration of prodrug treatment, and/or to the low level (5%) of MGMK/30-expressing cells used to evaluate the bystander activity. Slow tumor growth was observed in mice harboring 5% MGMK/30-expressing tumor cells during GCV treatment. Such bystander killing effects in rat C6 glioma cells is in fact impressive given that these cells are notorious for their low levels of gap junctions. This further implies that in rat C6 cells, a high amount of active antimetabolites is required to achieve significant bystander activity. Furthermore, the use of immunocompromised (nude) mice in this study is likely to preclude the full extent of the bystander effect due to the deficiency of the immune-mediated component of this phenomenon.
A 2,500-fold or 25-fold reduction in IC50 was detected between cells expressing either SR39 or MGMK/SR39 and those expressing HSVTK or MGMK/HSVTK, respectively. However, in vitro prodrug sensitivity assays revealed no statistical difference in IC50 value between MGMK/SR39 and SR39-expressing cells. A similar pattern emerged in vivo where both groups of mice harboring either MGMK/SR39- or SR39-expressing tumor cells demonstrated a relatively equal restriction in tumor growth when dosed with 1mg/kg GCV. When the lower dose of GCV (0.1mg/kg) was used, however, a statistically significant difference in GCV-mediated tumor killing could be observed; mice seeded with MGMK/SR39-expressing tumor cells experienced a delay in tumor growth compared to those that received SR39-expressing cells. The impediment in tumor growth seen during the in vivo bystander studies between mice harboring MGMK/SR39-expressing cells and those that received SR39-expressing cells when dosed with 1mg/kg GCV was marginal but statistically significant.
MGMK/30 demonstrates significant improvement in tumor killing and bystander activity compared to mutant 30, whereas only a minor improvement was observed in MGMK/SR39 compared to SR39. One possible explanation for this trend is the difference in kinetic properties toward GCV and thymidine exhibited by mutant 30 and SR39. When GCV is used as substrate, the Km of mutant 30 is 7-fold higher than that of wild type HSVTK. Additionally, the turnover rate (kcat) of mutant 30 for GCV is approximately 6-fold lower than wild type, which subsequently translates to a 41-fold reduction in overall enzyme catalytic efficiency9. Therefore one might predict that mutant 30 would not elicit a greatly enhanced prodrug sensitivity to cells. However, because endogenous thymidine (natural substrate of HSVTK) competes with the prodrug for the active site, it is important to consider the kinetic parameter mutant 30 displays towards thymidine. Indeed, mutant 30 demonstrates much poorer activity towards thymidine when compared to wild type HSVTK. When thymidine is used as a substrate, mutant 30 shows approximately a 3,000-fold reduction in overall enzyme catalytic efficiency compared to wild type HSVTK9.
Mutant SR39, on the other hand, has an advantage over mutant 30 in regards to enzyme activity towards GCV as indicated by a 14-fold improvement in Km value for GCV compared to HSVTK1. Although SR39's activity toward thymidine is slightly impaired, its impairment as shown in the 173-fold lower overall enzyme catalytic efficiency, is not nearly as dramatic as that of mutant 30 (3000-fold). This latter point may provide an explanation for the observed difference in MGMK/30 and MGMK/SR39 tumor killing efficacy. Based on kinetic data, mutant 30-expressing cells are likely to generate lower amounts of GCV-MP than SR39-expressing cells. The current findings with MGMK/30 reveal that the combination of MGMK and mutant 30 demonstrates a much more striking improvement in tumor killing compared to that of SR39. Therefore, taken together that the combination of MGMK and mutant 30 may provide a “balancing” effect to the system, where mutant 30 generates adequate amounts of GCV-MP to be shunted directly to MGMK and efficient phosphorylation of GCV-MP to GCV-DP is achieved. However, because MGMK/SR39 is very efficient at producing GCV-MP, due to the improved actions of SR39 towards GCV, conversion to GCV-DP becomes limited since MSMGK is unable to convert the excessive amounts of GCV-MP to GCV-DP efficiently.
The lack of complete tumor eradication seen in these studies is likely due the short duration of GCV treatment (compared to other reports of 14-21 days of treatment). Furthermore, the GCV doses used in this study (0.2mg/kg to 2mg/kg/day) is between 150- to 1500-fold lower than the doses used in most animal experiments (up to 300mg/kg/day)7. A higher dose of GCV combined with a longer GCV treatment is likely to result in complete tumor eradication when used in concert with MGMK/30 and MGMK/SR39. In the end, enzymes with high catalytic activity can impact cell killing efficacy directly by generating a more active prodrug or indirectly through a broader bystander effect. The use of superior fusion enzymes offers significant advantages over wild type HSVTK, HSVTK mutants, or MGMK/HSVTK in at least two important ways: (a) by enhancing GCV-mediated cell killing and bystander killing effects and (b) by further reducing the amount of myelosuppressive GCV required for effective cell killing. Ultimately, the application of such novel fusion enzymes will likely improve the clinical outcome of suicide gene therapy in cancer patients and compensate for the inefficient and ineffective problem of current delivery methods.
Materials and Methods
Materials
Oligonucleotides used to mutate and sequence HSVTK were obtained from Integrated DNA Technologies (Coralville, IA). Restriction endonucleases and T4 DNA ligase were purchased from New England Biolabs (Beverly, MA). DNA purification was done using several kits: Wizard PCR prep kits from Promega (Madison, WI), HiSpeed Plasmid Mini Kit from Qiagen (Valencia, CA), and StrataPrep EF Plasmid Midikit from Stratagene (La Jolla, CA). Alamar Blue was purchased from Serotec Limited (Oxford, UK). All cell culture reagents were purchased from Gibco (Carlsbad, CA). All other reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Bacterial strains
Escherichia coli strain NM522 [F+ lacIqΔ(lacZ)-M15proA+B+/supE thiΔ(lac-proAB)Δ(hsdMS-mcrB)5(rk-mk-McrBC-)] and E. coli strain XL1-Blue [F′∷Tn10 proA+B+ lacIq D(lacZ) M15/recA1 endA1 gyrA96 (Nalr) thi hsdR17 (rk-mk+) supE44 relA1 lac] were used as a recipient for certain cloning procedures. E. coli strain TS202A(DE3) (LAM-, tdk-1, IN(rrnD-rrnE)1, ilv-276 kanR ara-), a thymidine kinase deficient and a conditional guanylate kinase deficient strain, was used in genetic complementation assays to assess TK and GMK activity17.
Construction of mgmk/TK fusion mutant constructs and genetic complementation
Construction of pET23d:mgmk/TK fusion mutant constructs was done as previously described by Willmon et al. (2006)16. Briefly, the mouse gmk (mgmk) gene was isolated from pET23d:mgmk as an BglII/MscI fragment and ligated to BglII/MluI (blunt ended)-digested pET23d:30 or pET23d:SR39. The resulting plasmids, designated pET23d:mgmk/30 and pET23d:mgmk/SR39, were confirmed by DNA sequencing. For complementation studies, E. coli strain TS202A(DE3) harboring pET23d, pET23d:mgmk, pET23d:HSVTK, pET23d:mgmk/TK pET23d:mgmk/30 and pET23d:mgmk/SR39 were grown at 37°C on either GMK complementation media or TK complementation media as previously described14, 17.
Construction of mammalian expression vectors
The pET23d:mgmk, pET23d:SR39, pET23d:mgmk/HSVTK, pET23d:mgmk/30, and pET23d:mgmk/SR39 were subcloned into pUB6/V5-His B (Invitrogen, Carlsband, CA) digested with EcoRV as NcoI (blunt-ended) fragments. After restriction enzyme verification, DNA sequencing analysis confirmed the presence of the mgmk, SR39, mgmk/HSVTK, mgmk/30, and mgmk/SR39 genes. The resulting plasmids were designated pUB:mgmk, pUB: SR39, pUB:mgmk/HSVTK, pUB:mgmk/30, and pUB:mgmk/SR39, respectively. In order to expedite the construction of mammalian vectors, site-directed mutagenesis was performed on plasmid pUB:SR39 to introduce mutations derived from mutant 30 or to revert the sequence back to wild type HSVTK using the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) according to the manufacturer's protocol. Two pairs of oligonucleotides containing wild type sequence at 159-161 amino acid residues (MB428 5′-GCCCTCACCCTCATCTTCGACCGCCATCCC-3′/MB429 5′-GGGATGGCGGTCGAAGATGAGGGTGAGGGC-3′) and wild type sequence at 168-169 amino acid residues (MB432 5′-CCATCCCATCGCCGCCCTCCTGTGCTACCCG-3′/MB433 5′-CGGGTAGCACAGGAGGGCGGCGATGGGATGG-3′) were used to generate wild type sequence of HSVTK. Two pairs of oligonucleotides containing mutant 30's amino acid substitutions at 159-161 amino acid residues (MB432 5′-GCCCTCACCATTTTGGCTGACCGCCATCCC-3′/MB433 5′-GGGATGGCGGTCAGCCAAAATGGTGAGGGC-3′) and amino acid substitutions at 168-169 amino acid residues (MB4345′-CCATCCCATCGCATATTTCTTATGCTACCCG-3′/MB435 5′-CGGGTAGCATAAGAAATATGCGATGGGATGG-3′) were used to generate the mutant 30 sequence. DNA sequencing analysis was performed to confirm the presence of the correct mutation in mutant 30 sequence (pUB:30) or the presence of HSVTK wild type sequence (pUB:HSVTK).
Cell lines
Cell lines were maintained in a humidified incubator at 37°C in 5% CO2. Rat C6 glioma cells (C6) were purchased from ATCC (Manasass. VA) and were grown in Dulbecco's Modified Essential Medium containing 5% fetal bovine serum, 1mM sodium pyruvate, 10mM HEPES, 100μM nonessential amino acids, 100U/mL penicillin G, 10μg/mL streptomycin sulfate, 292μg/mL L-glutamine, 100μM sodium citrate and 0.0014% NaCl. Transfected cells were cultured in media supplemented with blasticidin at a concentration of 4μg/mL for selection of stably transfectants.
In vitro cytotoxicity assay
One μg of each DNA: pUB (empty vector), pUB:mgmk, pUB:HSVTK, pUB:30, pUB:SR39, pUB:mgmk/HSVTK, pUB:mgmk/30, and pUB:mgmk/SR39 was used to transfect 1 × 105 rat C6 glioma cells by lipofection using FuGENE 6 transfection reagent (Roche Diagnostic, Penzberg, Germany) at a 3:1 ratio as described by the manufacturer. Protein expression levels were determined by immunoblot analyses as previously described16. The membrane was probed with rabbit polyclonal HSVTK or mgmk antibody and followed by goat anti-rabbit-AP-conjugated antibody. Color reaction was developed using the AP Conjugate Substrate Kit (Bio-Rad, Hercules, CA). The in vitro cytotoxicity assays were performed using stable transfectants confirmed by immunoblot. Pools of transfectants were transferred to 96-well microtiter plates at an initial density of 500 cells per well. Upon cell adherence overnight, GCV (0-100μM) was added in sets of eight wells for each concentration tested. The cells were subjected to GCV for 7 days, at which time the redox indicator dye Alamar Blue was added. Cell survival was determined by fluorescence recorded at a 530/590nm, using a multi detector microplate reader Biotek Synergy HT, several hours later as described by the manufacturer and data were plotted with the standard deviation. At least three replicates were performed.
In vitro bystander experiment
Rat glioma C6 cells stably transfected with pUB (empty vector) were mixed at different ratios with stable transfectants harboring either pUB:HSVTK, pUB:30, pUB:SR39, pUB:mgmk/HSVTK, pUB:mgmk/30 or pUB:mgmk/SR39. The mixed cells were then transferred to 96-well microtiter plates at a final density of 500 cells per well and following cell adherence overnight, a GCV concentration of 80μM was added into the cells. Cell survival was determined as described above.
Xenograft tumor model
Pools of C6 glioma cells stably transfected with pUB, pUB:HSVTK, pUB:30, pUB:SR39, pUB:mgmk/HSVTK, pUB:mgmk/30 or pUB:mgmk/SR39 (0.5 × 106 cells in 200μL of phosphate buffer saline (PBS) at pH 7.2) were injected subcutaneously into 5- to 6-week-old female nude mice (n=5 for each group) (Athymic NCr-nu/nu; National Cancer Institute, Fredrick, MD). Bystander xenograft tumor model experiments were done in a similar fashion except nude mice (n=5) were injected with pools of C6 glioma cells stably transfected with pUB (empty vector) that were mixed with pools of C6 glioma cells stably transfected with either pUB:HSVTK, pUB:30, pUB:SR39, pUB:mgmk/HSVTK, pUB:mgmk/30 or pUB:mgmk/SR39 at a ratio of 95:5 (vector:suicide gene). When the tumors reached 3-4 mm in diameter (day 0), PBS or GCV at 1mg/kg or 0.1mg/kg was administered by intraperitoneal injection twice a day for 8 consecutive days. Starting at day 0, the tumor volume was monitored using caliper measurement (length, width, and height) every other day until day 16 at which time the mice were euthanized. Tumor volume was calculated using the formula: 4/3π ((Width × Length × Height)/2). Tumor volume was plotted and analyzed for statistical significance using Student's T-test.
Acknowledgments
This work was supported by the National Institutes of Health through grants R01CA85939 (to M.E.B) and T32GM008336 (to M.S).
References
- 1.Kokoris MS, Black ME. Characterization of herpes simplex virus type 1 thymidine kinase mutants engineered for improved ganciclovir or acyclovir activity. Protein Sci. 2002;11:2267–2272. doi: 10.1110/ps.2460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moolten FL. Tumor sensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46:5276–5281. [PubMed] [Google Scholar]
- 3.Edelstein ML, Abedi MR, Wixon J, Edelstein RM. Gene Therapy Clinical Trials Worldwide 1989-2004 - An Overview. J Gen Med. 2004;6:597–602. doi: 10.1002/jgm.619. [DOI] [PubMed] [Google Scholar]
- 4.Miller WH, Miller RL. Phosphorylation of acyclovir (acycloguanosine) monophosphate by GMP kinase. J Biol Chem. 1980;255:7204–7207. [PubMed] [Google Scholar]
- 5.Reardon JE. Herpes simplex virus type 1 and human DNA polymerase interactions with 2′-deoxyguanosine 5′-triphosphate analogues. Kinetics of incorporation into DNA and induction of inhibition. J Biol Chem. 1989;264:19039–19044. [PubMed] [Google Scholar]
- 6.Field AK, Davies ME, DeWitt C, Perry HC, Liou R, Germershausen J, et al. 9-([2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine: a selective inhibitor of herpes group virus replication. Proc Natl Acad Sci USA. 1983;80:4139–4143. doi: 10.1073/pnas.80.13.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greco O, Dachs G. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospective. J Cell Physiol. 2001;187:22–36. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1060>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 8.Freeman SM, Ramesh R, Marrogi AJ. Immune system in suicide-gene therapy. The Lancet. 1997;349:2–3. doi: 10.1016/S0140-6736(97)22001-5. [DOI] [PubMed] [Google Scholar]
- 9.Kokoris MS, Sabo P, Black ME. Enhancement of tumor ablation by a selected HSV-thymidine kinase mutant. Gene Ther. 1999;6:1415–1426. doi: 10.1038/sj.gt.3300966. [DOI] [PubMed] [Google Scholar]
- 10.Brady WA, Kokoris MS, Fitzgibbon M, Black M. Cloning, characterization, and modeling of mouse and human guanylate kinases. J Biol Chem. 1996;271:16734–16740. doi: 10.1074/jbc.271.28.16734. [DOI] [PubMed] [Google Scholar]
- 11.Boehme R. Phosphorylation of 9-β-D-arabinofuranosylguanine monophosphate by Drosophila melanogaster guanylate kinase. J Biol Chem. 1984;258:12346–12349. [Google Scholar]
- 12.Ostermann N, Lavie A, Padiyar S, Brundiers R, Veit T, Reinstein J, et al. Potentiating AZT activation: structures of wild-type and mutant human thymidylate kinase suggest reasons for the mutants' improved kinetics with the HIV prodrug metabolite AZTMP. J Mol Biol. 2000;304:43–53. doi: 10.1006/jmbi.2000.4175. [DOI] [PubMed] [Google Scholar]
- 13.Fridland A, Connelly MC, Ashmun R. Relationship of deoxynucleotide changes to inhibition of DNA synthesis induced by the antiretroviral agent 3′-azido-3′-deoxythymidine and release of its monophosphate by human lymphoid cells (CCRF-CEM) Mol Pharmacol. 1990;37:665–670. [PubMed] [Google Scholar]
- 14.Black ME, Newcomb TG, Wilson HM, Loeb LA. Creation of drug-specific herpes simplex virus type 1 thymidine kinase mutants for gene therapy. Proc Natl Acad Sci USA. 1996;93:3525–3529. doi: 10.1073/pnas.93.8.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Black ME, Kokoris MS, Sabo P. Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 2001;61:3022–3026. [PubMed] [Google Scholar]
- 16.Willmon CL, Krabbenhoft E, Black ME. A guanylate kinase//HSV-1 thymidine kinase fusion protein enhances prodrug-mediated cell killing. Gene Ther. 2006;13:1309–1312. doi: 10.1038/sj.gt.3302794. [DOI] [PubMed] [Google Scholar]
- 17.Stolworthy TS, Krabbenhoft E, Black ME. A novel Escherichia coli strain allows functional analysis of guanylate kinase drug resistance and sensitivity. Anal Biochem. 2003;322:40–47. doi: 10.1016/j.ab.2003.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Dilber MS, Abedi MR, Christensson B, Bjorkstrand B, Kidder GM, Naus CCG, et al. Gap junctions promote the bystander effect of herpes simplex virus thymidine kinase in vivo. Cancer Res. 1997;57:1523–1528. [PubMed] [Google Scholar]
- 19.Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 20.Kaldor J, Day N, Pettersson F, Clarke E, Pederson D, Mehnest W, et al. Leukemia following chemotherapy for ovarian cancer. N Engl J Med. 1990;322:1–6. doi: 10.1056/NEJM199001043220101. [DOI] [PubMed] [Google Scholar]
- 21.Relling MV, Rubnitz JE, Rivera GK, Boyett JM, Hancock ML, Felix CA, et al. High incidence of secondary brain tumours after radiotherapy and antimetabolites. The Lancet. 1999;354:34–39. doi: 10.1016/S0140-6736(98)11079-6. [DOI] [PubMed] [Google Scholar]
- 22.Dachs GU, Tupper J, Tozer G. From bench to bedside for gene-directed enzyme prodrug therapy of cancer. Anticancer Drugs. 2005;16:349–359. doi: 10.1097/00001813-200504000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Norris JS, Norris KL, Holman DH, El-Zawahry A, Keane TE, Dong Jy, et al. The present and future for gene and viral therapy of directly accessible prostate and squamous cell cancers of the head and neck. Future Oncol. 2005;1:115–123. doi: 10.1517/14796694.1.1.115. [DOI] [PubMed] [Google Scholar]
- 24.Pope IM, Poston GJ, Kinsella AR. The role of the bystander effect in suicide gene therapy. Eur J Cancer. 1997;33:1005–1016. doi: 10.1016/s0959-8049(96)00483-2. [DOI] [PubMed] [Google Scholar]
- 25.Vrionis FD, Wu JK, Qi P, Waltzman M, Cherington V, Spray DC. The bystander effect exerted by tumor cells expressing the herpes simplex virus thymidine kinase (HSVtk) gene is dependent on connexin expression and cell communication via gap junctions. Gene Ther. 1997;4:577–585. doi: 10.1038/sj.gt.3300438. [DOI] [PubMed] [Google Scholar]
- 26.Akyürek L, Nallamshetty S, Aoki K, San H, Yang Z, Nabel G, et al. Coexpression of guanylate kinase with thymidine kinase enhances prodrug cell killing in vitro and suppresses vascular smooth muscle cell proliferation in vivo. Mol Ther. 2001;3:779–786. doi: 10.1006/mthe.2001.0315. [DOI] [PubMed] [Google Scholar]