

Conspectus
Antimicrobial peptides (AMPs) provide protection against a variety of pathogenic bacteria and are, therefore, an important part of the innate immune system. Over the last decade, there has been considerable interest in developing AMPs as intravenously administered antibiotics. However, despite extensive efforts in the pharmaceutical and biotechnology industry, it has proven difficult to achieve this goal. While researchers have solved some relatively simple problems such as susceptibility to proteolysis, more severe problems have included the expense of the materials, toxicity, limited efficacy, and limited tissue distribution.
In this Account, we describe our efforts to design and synthesize “foldamers”-- short sequence-specific oligomers based on arylamide and β-amino acid backbones, which fold into well-defined secondary structures-- that could act as antimicrobial agents. We reasoned that small “foldamers” would be less expensive to produce than peptides, and might have better tissue distribution. It should be easier to fine-tune the structures and activities of these molecules to minimize toxicity.
Because the activities of many AMPs depends primarily on their overall physicochemical properties rather than the fine details of their precise amino acid sequences, we have designed and synthesized very small “coarse-grained” molecules, which are far simpler than naturally produced AMPs. The molecular design of these foldamers epitomizes the positively charged amphiphilic structures believed to be responsible for the activity of AMPs. The designed oligomers show greater activity than the parent peptides. They have also provided leads for novel small molecule therapeutics that show excellent potency in animal models for multi-drug resistant bacterial infections. In addition, such molecules can serve as relatively simple experimental systems for investigations aimed at understanding the mechanism of action for this class of antimicrobial agents. The foldamers’ specificity for bacterial membranes relative to mammalian membranes appears to arise from differences in membrane composition and physical properties between these cell types.
Furthermore, because experimental coarse-graining provided such outstanding results, we developed computational coarse-grained models to enable molecular dynamic simulations of these molecules with phospholipid membranes. These simulations allow investigation of larger systems for longer times than conventional molecular dynamics simulations, allowing us to investigate how physiologically relevant surface concentrations of AMP mimics affect the bilayer structure and properties. Finally, we apply the principles discovered through this work to the design of inexpensive antimicrobial polymers and materials.
Keywords: de novo design, antimicrobial peptide, foldamer, antibiotic, membrane-peptide interactions, antimicrobial polymers
Introduction
Biological macromolecules including proteins and RNA generally adopt unique, folded conformations that are responsible for their remarkable properties. Until recently, the process of folding was considered a mystery, but as the fields of protein folding, RNA structure, and molecular organization have advanced it has become increasingly possible to design non-biological molecules that fold into unique structures. To mimic natural proteins, various investigators have synthesized oligomers by sequentially coupling individual monomeric units to provide homogeneous linear molecules of entirely uniform sequence and chain length. An early short list of such oligomers would include poly-pyrol/imidazole DNA-binding oligomers4, pyrrolinone peptide mimetics5, arylamides6-10, N-alkyl-glycines (peptoids11), polyurethanes12, and phenylene-ethynylenes13,14. Oligomers that fold into well-defined secondary structures have come to be known as foldamers (see15-22 for reviews). In the last decade the area of foldamer research has blossomed in manifold directions, providing a rich diversity of backbone structures. The structural simplicity and relative ease of synthesis of many foldamers allow them to be used as three dimensional scaffolds for molecular recognition, which, in iterative steps, can be down-sized to the dimensions of small molecules for various biological applications.
Already there are multiple examples of functional foldamers that target a number of molecular and biological targets including RNA, proteins, membranes and carbohydrates, often with affinities approaching or equaling those of natural α-peptides (for recent reviews see19,23). It is, however, also important to identify applications of foldamers to address problems that cannot be addressed by use of conventional peptides composed of D- or L-α-amino acids. Along these lines, we focused on antimicrobial foldamers that mimic the host defense peptides.
Host defense peptides, innate sources of antibiotics?
Antibiotic resistance is now one of the most pressing global healthcare problems facing society24,25. Although hospital-acquired, or nosocomial, infections in the United States have gradually declined, about 70% of them are resistant to at least one antibiotic and the trend is increasing. For example, of the approximately two million people who will acquire nosocomial infections this year in US hospitals, 99,000 will die from the infection26. AMPs have provided a new approach to developing antibiotics because they play a central role in the innate immunity system of many organisms1,2,27-29. In some primitive organisms that lack a humoral response AMPs and a few enzymes provide the main defense against bacteria, yeast, fungus, and even viruses. Ribosomally encoded AMPs are typically 12 to approximately 80 residues in length, and adopt various active conformations, including α-helices (magainin and cecropin), disulfide-rich β-sheets (bactenecin and defensin) (Figure 1), and other tertiary structures.
Fig 1.

Structure of an α-helical magainins (pdb code 2MAG) and a β-sheet defensin peptide (1BNB) in which the cationic groups are colored dark blue and the nonpolar groups are colored green. The facially amphiphilic nature of the overall conformation is highlighted by the segregation of these groups.
Most AMPs adopt highly amphiphilic conformations in which the cationic hydrophilic and hydrophobic side chains segregate onto distinctly opposing regions or faces of the overall folded conformation. This amphiphilic topology appears to be important for insertion into and disruption of the cytoplasmic membrane, leading to bacteria death1,30. This simple physical mechanism of action has resulted in a relatively low propensity for the development of bacterial resistance. However, AMPs can also interact in multiple ways with other components of the innate immune system (reviewed in31), many AMP’s can act by additional mechanisms32-34, and bacteria are able to respond to AMPs27,35,36 and even evolve novel resistance to their toxic affects37,38.
From natural host defense peptides to antimicrobial β-peptide and peptoids
Previously, antimicrobial peptides have been designed by idealizing the amphiphilicity of natural AMPs2,39-41, introduction of D-amino acids42,43 or long acyl chains44,45, and cyclization42,46. The availability of β-peptides provided another avenue to probe the requirements for activity, as they can adopt distinct secondary structures such as “12-helices” and “14-helices”15,17-20.
Our group47,48 as well as those of Gellman49 and Seebach50 independently showed that β-peptides capable of forming amphiphilic 14- or 12-helices had potent antimicrobial activity. Oligomers with lengths of 10-15 residues that achieved an appropriate hydrophilic/lipophilic balance were selective for killing bacteria vs. mammalian cells. Oligomers that were too short were inactive, and they also failed to adopt the desired conformation, while those that were too long, or hydrophobic, showed unacceptably high toxicities towards human erythrocytes. While these studies were in progress, Gellman and coworkers also described a potent and highly selective antimicrobial peptide based on cyclic β-amino acids. These studies were subsequently extended to a variety of different helical types formed by β-peptides and α/β-peptides51-55. In a similar manner, Patch and Barron designed amphiphilic, helical, antibacterial N-substituted glycine oligomers (peptoids)22, while Guichard and coworkers have synthesized enantiomerically pure antimicrobial foldamers based on a urea backbone56. It was further demonstrated that charge, facial amphiphilicity, and an appropriate hydrophilic/hydrophobic balance were generally important for obtaining selective, non-toxic compounds, although there seems to be no absolute requirement for any one molecular feature57.
Antimicrobial arylamides and arylureas
The diversity of structures and molecular frameworks employed in the above studies led us to ask whether these principles could be extended to design much simpler oligomers, such as aromatic arylamides6-10, which have been examined extensively in fundamental studies of hydrogen bonding and molecular recognition. Given their relative ease of synthesis, they appeared excellent candidates to initiate the structural simplification or “synthetic coarse-gaining” of antimicrobial compounds. We prepared compounds related to the core structure 1, which contains alternating 1,3 phenylene diamine units connected by an isophthalic acid.
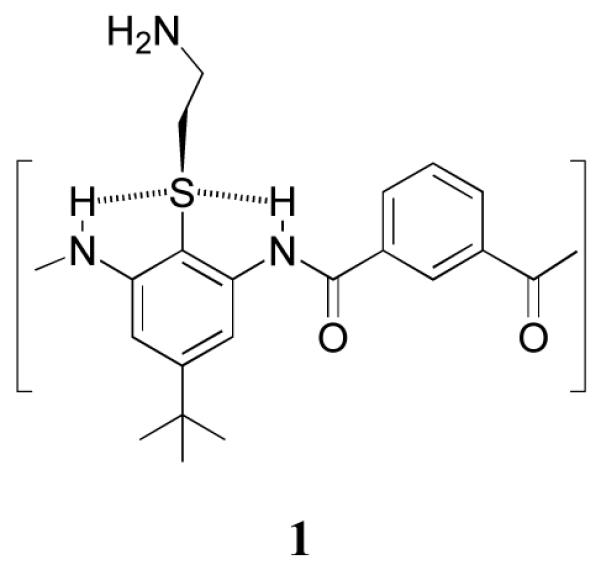
A thioether provided a convenient point for attachment of basic groups while also rigidifying the molecule through the formation of intramolecular hydrogen bonds. The segregation between the polar aminoethyl-thioether from the hydrophobic t-butyl group provided facial amphiphilicity to the overall structure. Quantum mechanical calculations,58 molecular dynamics simulations,58 and crystallographic analysis of model compounds (Figure 3) confirmed that the thioether indeed hydrogen bonded with the neighboring amide. In both amides and ureas, the planar arrangement between the central ring and the carbonyl is enforced by NH-thioether hydrogen bonding, and possibly a non-conventional CH-hydrogen bond59.
Fig. 3.

Crystal structures of model thioether-containing arylamide (a), arylurea (b), and a dialkoxy-substituted arylamide (c and d)
Initially we synthesized oligomers of repeating structure 160. Compounds with two to three repeats had antibacterial activity with MICs as low as ~15 μg/mL, although they lysed human red blood cells (RBCs) at similar or lower concentrations. Longer polymers showed lower antibacterial activity. Interestingly, the simple three-ring structure, 2 (Table 1, Fig 4), showed reasonable activity, and served as a core fragment for elaboration of more active compounds61. Addition of amino acid substituents to this core structure resulted in compounds with very good potency and high specificity for bacteria over mammalian cells. Positively charged residues, particularly Arg (3 in Table 1), increased the potency and the safety of the compounds61.
Table 1.
Conformational tuning of properties of antimicrobial oligomersa.
| Compound | Structure | MIC (μM) S. aureusb |
E. Coli D31 |
HC50 (μM)c |
|---|---|---|---|---|
| 2 | 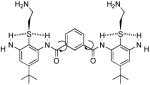 |
50 | 12.5 | 14 |
| 3 | 7.8 | 3.9 | 445 | |
| 4 | 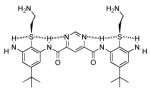 |
0.80 | 0.80 | 18 |
| 5 | 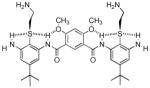 |
0.87 | 0.87 | 145 |
| 6 | 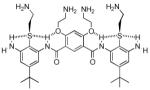 |
0.66 | 5.3 | 593 |
| 7 | 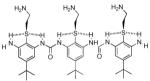 |
0.70c | 0.70 | 5 |
| 8 | 0.2 | 2.8 | 63-90 | |
| 9 | 0.20 | 0.14 | 40 | |
| 10 | 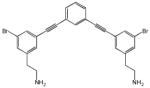 |
0.20 | 0.10 | 440 |
Data were collected using the Hancock method; numbers quoted here may differ somewhat from those in the original publications if a different method was originally used to determine MIC’s.
Tetracycline and streptomycin-resistant S. aureus.
The concentration required to cause 50% lysis of a suspension of red blood cells.
Fig. 4.

Optimization of the activity of arylamide oligomers, including the introduction of hydrogen-bonding substitutents and optimization of pendent functional groups.
Within this initial design, containing a central isophthalic acid group, the two backbone ring—carbonyl single bonds remained rotatable58,62. The addition of internal hydrogen bonds within arylamides can effectively limit these torsional degrees of freedom63-65, which we reasoned might decrease conformational flexibility and increase potency.
Introduction of a pyrimidine in compound 466 or dialkoxygroups in 558,67 allowed the desired hydrogen bonding (Fig. 3c). Another strategy involved the design of an arylurea scaffold68 (7), which can form a planar, fully hydrogen-bonded array. All three of these modifications led to marked improvements in antimicrobial activity.
Optimization of both the total charge as well as the hydrophobic content proved to be particularly important to the design of compounds that are highly active and non-toxic in animals. The introduction of flourous amino acids into antimicrobial peptides can lead to increases in potency69. Similarly, we have found that the replacement of t-butyl groups with trifluoromethyl groups in these arylamides leads to increases in both potency and safety in compounds such as 8 and 9. These compounds, like most of those in Table 1, are significantly more potent than the magainin derivative, MSI-78, which has an MIC, against these two strains, of 12.5 μg/mL.
Highly active short antimicrobial phenylene ethynylene
Tew and coworkers have dispensed with the amide bond altogether in their design of a phenylene ethynylene scaffold70 with outstanding antibacterial activity and selectivity71. Both polymeric materials as well as short oligomers were investigated. The most active compound (structure 10 shown in Table 1) contains three aromatic rings. Antibacterial screening showed clearly that the short three-ring oligomer, 10, was highly potent against a large panel of pathogenic and potential biowarfare bacteria.
Biological mode of action
AMPs generally act directly on the cytoplasmic membrane, although this might not be the primary target of some peptides. Indeed, the phenylene ethynylene derivative, 1071,72, is able to cause leakage of dyes from phospholipid vesicles, at concentrations similar to those required to kill bacteria. A similar time course is also seen for the loss of the membrane potential in bacteria. The selectivity of AMPs for bacterial membranes is believed to result from differences between the phospholipid compositions of mammalian and bacterial membranes. Bacterial membranes tend to be more negatively charged and lack cholesterol. In many cases1, more subtle effects relating to phospholipid composition are involved; primary phospholipids in mammalian membranes include zwitterionic phosphatidylcholine (PC) and negatively charged phosphatidylserine (PS) headgroups, while bacterial membranes are rich in zwitterionic phosphatidylethanolamine (PE) and negatively charged phosphatidylglycerol (PG) lipids including cardiolipin. The differences in the physical properties of these various lipids can be as important as the overall charge of the membrane51,52,54,73. In particular, the area covered by the headgroup of PE is less than that of its hydrophobic tail, causing this phospholipid to be less stable in planar bilayer phases than PC. This feature appears to be particularly important for the high selectivity of compound 1071,72, which is much more effective at inducing leakage of PG-PE than PS-PC vesicles. X-ray scattering suggests that this compound induces structural changes favoring hexagonal phases in PE-rich membranes74. Epand and coworkers have observed similar results for antimicrobial foldamers and polymers, which also require PE for activity51,52,54,73. Although the phenylene ethynylene compounds showed a good relationship between membrane-perturbing effects and activity, some of the most potent arylamides cause complete membrane depolarization of S. aureus at concentrations somewhat higher than those required to cause rapid cell death. This finding might reflect a mechanism of action that is distinct from membrane perturbation, or it might simply indicate that only a small degree of perturbation is required to ultimately trigger cell death.
The mechanism of action of these foldamers are of interest with respect to the mechanisms of host defense peptides. Like many AMPs, they are too short to span the hydrophobic length of the bilayer, so they presumably work by a mechanism resembling the “carpet” mechanism of AMP.75 What is critical for this mechanism is that the compounds bind with a sufficiently favorable free energy of association and penetrate the outer leaflet of the bacterial membrane during the initial steps of the process. Furthermore, the compounds must have sufficient polar character to provide water solubility with which to reach the host membrane. Additionally, if the compounds are too hydrophobic, they will bind indiscriminately to mammalian membranes leading to high toxicity. Our data clearly shows that there is no unique combination of molecular features that is absolutely required to satisfy these provisions. Flexible peptides frequently adopt more ordered conformations upon binding; in these structures appropriately placed hydrophobic or charged groups can easily mitigate any loss in conformational entropy required for binding. Thus, for each scaffold their exists a delicate balance between charge, hydrophobicity and rigidity that must be fine-tuned to optimize both potency and selectivity.
Antimicrobial polymers
Studies with antimicrobial foldamers have also informed the design of a variety of arylamides, phenylene ethynylenes, polyacrylate (Fig. 5), and hydrocarbon-based polymers and materials. While an exhaustive review of other antimicrobial polymers would be beyond the scope of this Account, it is important to note that Gelman and coworkers have also developed polymeric antimicrobial agents, whose mechanisms of action have been extensively investigated and compared with more peptidic AMPs73,76. Also, cationic polymers with antibacterial and biocidal activity are well known.77-82, in some cases incorporated into materials or immobilized on surfaces76-78.
Fig. 5.

Structure of antimicrobial polyacrylamide and polynorbornenyl polymers.
Our own work in this area has focused on defining features required for selective antimicrobial activity. The phenylene ethynylene scaffold was the first polymeric series to demonstrate antibacterial activity and selectivity 71; interestingly, increasing the MW decreased the selectivity factor. This trend of decreasing selectivity with increasing MW was seen with several types of polymers 60,83-85. The decrease in selectivity reflects both increased toxicity to mammalian cells as well as decreased antibacterial activity, possibly due to poor penetration of the large molecules to the cytoplasmic membrane. This is, however, not universally true as amphiphilic polynorbornenes with MW > 3,000 Da have good antibacterial activity and minimal toxicity to mammalian cells 86-88.
Coarse-grained computational models
Antimicrobial oligomers represent minimal, coarse-grained models that embody the molecular features required for activity. In a similar spirit, it has been possible to create computational coarse-grained models of the arylamide class of antimicrobial foldamers to allow longer simulations of their membrane-perturbing activities. At the time of our studies, all-atom molecular dynamics (MD) computer simulations had been used to explore the interactions of peptide based antimicrobials with membranes, but these studies had been limited to relatively short-times89 and only one or a few antimicrobials90. Modern coarse-grain approaches91 have resulted in the development of several models for membrane simulations in the past years92-94. In this method, the molecule is considered to be composed of a series of carefully parameterized interconnected units, each encompassing several atoms. By reducing the number of atoms, fewer distances need to be calculated at each MD step, making a significant increase in computational speed possible.
Coarse-grain MD allowed μs simulations with a relevant number of arylamides per bilayer95 at coarse-grained oligomer/phospholipid (CGO/PL) from 1:256 to 1:14, spanning surface-concentrations that experimentally give rise to from none to very rapid vesicle lysis. The antimicrobial was initially placed in the aqueous phase just above one side of the membrane to eliminate the requirement for diffusion to the membrane surface, which can be rate-limiting in experimental studies. Different results were observed at the two extreme CGO/PL ratios. At low CGO/PL ratios the antimicrobial inserts into the bilayer with its long axis parallel to the membrane surface and its apolar groups penetrating into the membrane (Fig. 6). By contrast, at the high CGO/phospholipid ratios required for lysis of bilayers in experimental systems95, the drugs initially inserts with an ensemble of different angles. The very large imbalance in the concentration of the oligomers between the proximal versus the distal leaflets of the bilayer leads to a metastable state with pronounced buckling of the proximal leaflet of the bilayer. This imbalance provides a strong driving force for translocation of the drug molecules to the opposite side of the bilayer; the buckling and disordering of the bilayer structure might additionally lower the kinetic barrier for translocation. The CGOs frequently diffuse in pairs across the bilayer, often with accompanying ions and water molecules. Following the translocation step the oligomers are oriented predominantly parallel to the bilayer, although with less extensive ordering than in the simulations at low antimicrobial/phospholipid ratios. In the final configuration the ammonium end of the molecule locates near the glycerol backbone, significantly below the highly polar zwitterionic headgroups. Thus, the polarity gradient of the membrane is altered, with the introduction of charged groups that have a high preference to be hydrated, at otherwise relatively non-polar regions of the bilayer. The result is an increased permeability to water in the simulations.
Fig. 6.

Snap-shot from a coarse-grained molecular dynamics simulation of the action of an arylamide on the bilayer at an oligomer/lipid ratio of 1:256 (left) and 1:14 (right)95. The backbone of the foldamer is shown in blue and yellow, while the headgroups of the membrane and polar groups of the foldamer are shown in purple and red. The lipid chains are shown as sticks. The right panel represents an intermediate time point at which foldamers have inserted deeply, and are beginning translocate across the bilayer.
These simulations provide pictorial detail to prior models inferred from experimental studies. They explain the dependence of lysis on a high order of the peptide concentration — sufficient concentrations of the oligomers must accumulate in the cis leaflet of the bilayer to induce membrane disruption and to provide a driving force for oligomer translocation. Secondly, the simulations are in agreement with the experimental observation that membrane disruption is a biphasic process. In the initial step the oligomers bind to and strongly buckle and otherwise asymmetrically perturb the bilayer. The subsequent translocation step appears to occur with concomitant formation of pathways for solvent and solute diffusion. Finally, following equilibration of the compounds on both sides of the bilayer, the membrane reaches a state that is intrinsically more permeable than the initial membrane. There are also differences between the coarse-grained and experimental observations. The first is in the time scale of the events, because experimental measurements require an initial diffusion to the membrane surface, which can be rate-determining. The second difference is that the size of our system precludes the observation of large long-lived pores or other openings in the membrane. Thus, the increase in permeability to water and ions observed in our simulations may be a prelude to more deep-seated disruption of the membrane, induced by osmotic imbalances in cellular systems.
Potential of synthetic foldamers as antimicrobials for fundamental and applied research
In summary, beginning with peptides of molecular weights in the range of 2,000 to 5,000 Da, it has been possible first to prepare β-peptides with significantly lower molecular weights, followed by arylamides and small molecules with molecular weights in the range of 600 — 1000 Da67,96. The simplicity of these compounds makes them outstanding candidates for fundamental computational and experimental studies of the mechanism of membrane disruption. Furthermore, proteomic and genomic studies will illuminate the mechanisms by which bacteria respond to these agents as compared to natural AMPs.
From a more practical perspective the lower MW and non-peptidic structures of arylamides have allowed us to address previous problems — including problems with tissue distribution, toxicity, and cost of materials — that plagued the development of peptidic analogues of AMP’s as intravenously administered antibiotics1. Experimental studies in vitro have shown that bacteria cannot easily develop resistance to this class of compounds 67, suggesting they might not suffer from some of the same problems of resistance encountered in other classes of antibiotics with more specific targets. Indeed, a compound related to 8 and 9 successfully completed the first phase of human clinical trials as an i.v. antibiotic for treatment of multi-drug resistant Staphylococcal aureus96.
Fig. 2.

Structure of β3-amino acid (left) and an antimicrobial β-peptide, (β3HAla-β3HLys β3HVal)4-CONH2 (middle), and the natural α-helical peptide magainins (right). Hydrophboic residues are in green, and basic sidechains in blue.
ACKNOWLEDGMENT
We thank our many coworkers for their contributions to these studies and grants GM54616 and AI74866 from NIH for support of this work.
Biography
Gregory N. Tew received a B.S. in Chemistry from North Carolina State University in 1995 and performed undergraduate research with Prof. D. A. Shultz. In 2000, he earned his Ph.D. from the University of Illinois-Urbana under Samuel Stupp after which he joined the Faculty at the Polymer Science and Engineering Department at the University of Massachusetts, Amherst. Before starting there, he spent one year in William DeGrado’s laboratory at the University of Pennsylvania Medical School. Current research interests include bioinspired and biomimetic macromolecules, supramolecular polymer science, molecular self organization, and novel hydrogels for regenerative medicine. This work has lead to awards from the National Science Foundation (CAREER), the Office of Naval Research, the Army Research Office, 3M, DuPont and the Presidential Early Career Award for Scientists and Engineers.
Richard W. Scott received his Ph.D. at the University of Pennsylvania in the Department of Microbiology. His post-doctoral training was in molecular biology at the Institute for Cancer Research, Fox Chase, PA. He joined E. I. DuPont as a Principal Investigator in the Central Research and Development Department and then joined Cephalon, Inc. to build and lead the molecular biology department. Over an eleven year career at Cephalon, Dr. Scott became Vice-President of Biology where he led discovery teams targeting neurodegenerative disorders as well as the development of genetically-based animal models for Alzheimers disease and other neurodegenerative disorders. Presently, Dr. Scott is Vice President of Research at PolyMedix, Inc. and has led research teams responsible for the identification and selection of two lead compounds presently in Phase 1 clinical trials; one of which is a small foldamer mimic of the antimicrobial peptides that is being developed as an anti-Staphylococcal IV agent. Dr. Scott has over 50 peer-reviewed journal publications and book chapters, and is an inventor on 6 US patents
Michael L. Klein received his BSc and PhD in Chemistry from the University of Bristol (UK). He was a postdoctoral fellow in Italy, UK and USA before joining the Chemistry Division of the NRCC in Ottawa, Canada, where he rose through the ranks from Associate to Principal Research Officer. In 1987 he returned to the United States as Professor of Chemistry at the University of Pennsylvania, where from 1993 he was the Hepburn Professor of Physical Science and Director of the Laboratory for Research on the Structure of Matter. In 2009 he moved to Temple University as the Carnell Professor of Science and Director of the Institute for Computational Molecular Science. His research interests involve probing the structure and dynamics of molecular systems using computer simulation techniques; research topics range from soft matter to chemical biology.
Bill DeGrado received his B.A. in chemistry from Kalamazoo College (1978) and his Ph.D. in organic chemistry from the University of Chicago (1981). He spent the subsequent 15 years engaged in research at DuPont and DuPont-Merck Pharmaceutical Company. He joined the University of Pennsylvania in 1996, where he is the Raiziss Professor in the Department of Biochemistry and Biophysics, and an adjunct member of the Chemistry Department. The DeGrado laboratory works on experimental and computational studies aimed at understanding molecular interactions and dynamics, with particular emphasis on the design of proteins, peptides, and peptide mimics. Specific areas of investigation include pharmaceutical chemistry, structural biology, and biochemistry.
REFERENCES
- (1).Zasloff M. antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- (2).Tossi A, Sandri L, Giangaspero A. Amphipathic, α-Helical Antimicrobial Peptides. Biopolymers. 2000;55:4–30. doi: 10.1002/1097-0282(2000)55:1<4::AID-BIP30>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- (3).Boman HG. Innate immunity and the normal microflora. Immunol Rev. 2000;173:5–16. doi: 10.1034/j.1600-065x.2000.917301.x. [DOI] [PubMed] [Google Scholar]
- (4).Dervan PB. Design of Sequence-Specific DNA-Binding Molecules. Science. 1986;232:464–471. doi: 10.1126/science.2421408. [DOI] [PubMed] [Google Scholar]
- (5).Smith AB, 3rd, Knight SD, Sprengeler PA, Hirschmann R. The design and synthesis of 2,5-linked pyrrolinones. A potential non- peptide peptidomimetic scaffold. Bioorg Med Chem. 1996;4:1021–34. doi: 10.1016/0968-0896(96)00094-6. [DOI] [PubMed] [Google Scholar]
- (6).Hamuro Y, Geib SJ, Hamilton AD. Oligoanthranilamides; Non-peptide Subunits that Show Intramolecular Hydrogen Bonding Control of Secondary Structure. J. Am. Chem. Soc. 1996;118:7529–7541. [Google Scholar]
- (7).Stadler AM, Kyritsakas N, Graff R, Lehn JM. Formation of RACK- and grid-type metallosupramolecular architectures and generation of molecular motion by reversible uncoiling of helical ligand strands. Chemistry. 2006;12:4503–22. doi: 10.1002/chem.200501202. [DOI] [PubMed] [Google Scholar]
- (8).Cuccia LA, Lehn JM, Homo JC, Schmutz M. Encoded Helical Self-Organization and Self-Assembly into Helical Fibers of an Oligoheterocyclic Pyridine - Pyridazine Molecular Strand. Angew Chem Int Ed Engl. 2000;39:233–237. doi: 10.1002/(sici)1521-3773(20000103)39:1<233::aid-anie233>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- (9).Gardinier KM, Khoury RG, Lehn JM. Enforced helicity: efficient access to self-organized helical molecular strands by the imine route. Chemistry. 2000;6:4124–31. doi: 10.1002/1521-3765(20001117)6:22<4124::aid-chem4124>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- (10).Nakano T, Okamoto Y. Synthetic helical polymers: conformation and function. Chem Rev. 2001;101:4013–38. doi: 10.1021/cr0000978. [DOI] [PubMed] [Google Scholar]
- (11).Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, et al. Peptoids: a modular approach to drug discovery. Proc Natl Acad Sci U S A. 1992;89:9367–71. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cho CY, Moran EJ, Cherry SR, Stephans JC, Fodor SPA, Adams CL, Sundaram A, Jacobs JW, Schultz PG. An unnatural biopolymer. Science. 1993;261:1303–1305. doi: 10.1126/science.7689747. [DOI] [PubMed] [Google Scholar]
- (13).Prince RB, Saven JG, Wolynes PG, Moore JS. Cooperative conformational transitions in phenylene ethynylene oligomers: Chain-length dependence. J. Am. Chem. Soc. 1999;121:3114–3121. [Google Scholar]
- (14).Nelson JC, Saven JG, Moore JS, Wolynes PG. Solvophobically driven folding of nonbiological oligomers. Science. 1997;277:1793–1796. doi: 10.1126/science.277.5333.1793. [DOI] [PubMed] [Google Scholar]
- (15).Gellman SH. Foldamers: A Manifesto. Acc. Chem. Res. 1998;31:173–180. [Google Scholar]
- (16).Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. A field guide to foldamers. Chem Rev. 2001;101:3893–4012. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- (17).Seebach D, Beck AK, Bierbaum DJ. The world of beta- and gamma-peptides comprised of homologated proteinogenic amino acids and other components. Chem Biodivers. 2004;1:1111–239. doi: 10.1002/cbdv.200490087. [DOI] [PubMed] [Google Scholar]
- (18).Cheng RP, Gellman SH, DeGrado WF. beta-Peptides: from structure to function. Chem Rev. 2001;101:3219–32. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- (19).Horne WS, Gellman SH. Foldamers with Heterogeneous Backbones. Acc Chem Res. 2008 doi: 10.1021/ar800009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Seebach D, Hook DF, Glattli A. Helices and other secondary structures of beta- and gamma-peptides. Biopolymers. 2006;84:23–37. doi: 10.1002/bip.20391. [DOI] [PubMed] [Google Scholar]
- (21).Kritzer JA, Stephens OM, Guarracino DA, Reznik SK, Schepartz A. beta-Peptides as inhibitors of protein-protein interactions. Bioorg Med Chem. 2005;13:11–6. doi: 10.1016/j.bmc.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Patch JA, Barron AE. Helical peptoid mimics of magainin-2 amide. J Am Chem Soc. 2003;125:12092–3. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- (23).Goodman CM, Choi S, Shandler S, DeGrado WF. Foldamers as versatile frameworks for the design and evolution of function. Nat Chem Biol. 2007;3:252–62. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Weinstein RA. Controlling antimicrobial resistance in hospitals: infection control and use of antibiotics. Emerg Infect Dis. 2001;7:188–92. doi: 10.3201/eid0702.010206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Opar A. Bad bugs need more drugs. Nature Reviews Drug Discovery. 2007;6:943–944. [Google Scholar]
- (26).Klevens R. E., J. R. Monina, Richards CL, Horan TC, Gaynes RP, Pollock DA, Cardo DM. Public Health Reports. 2007;122:6. doi: 10.1177/003335490712200205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bishop JL, Finlay BB. Friend or foe? Antimicrobial peptides trigger pathogen virulence. Trends Mol Med. 2006;12:3–6. doi: 10.1016/j.molmed.2005.11.001. [DOI] [PubMed] [Google Scholar]
- (28).Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–50. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- (29).Shai Y. From innate immunity to de-novo designed antimicrobial peptides. Curr Pharm Des. 2002;8:715–25. doi: 10.2174/1381612023395367. [DOI] [PubMed] [Google Scholar]
- (30).Van Bambeke F, Mingeot-Leclercq MP, Struelens MJ, Tulkens PM. The bacterial envelope as a target for novel anti-MRSA antibiotics. Trends Pharmacol Sci. 2008;29:124–34. doi: 10.1016/j.tips.2007.12.004. [DOI] [PubMed] [Google Scholar]
- (31).Finlay BB, Hancock RE. Can innate immunity be enhanced to treat microbial infections? Nat Rev Microbiol. 2004;2:497–504. doi: 10.1038/nrmicro908. [DOI] [PubMed] [Google Scholar]
- (32).Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun. 1998;244:253–7. doi: 10.1006/bbrc.1998.8159. [DOI] [PubMed] [Google Scholar]
- (33).Kragol G, Lovas S, Varadi G, Condie BA, Hoffmann R, Otvos L., Jr. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry. 2001;40:3016–26. doi: 10.1021/bi002656a. [DOI] [PubMed] [Google Scholar]
- (34).del Castillo FJ, del Castillo I, Moreno F. Construction and characterization of mutations at codon 751 of the Escherichia coli gyrB gene that confer resistance to the antimicrobial peptide microcin B17 and alter the activity of DNA gyrase. J Bacteriol. 2001;183:2137–40. doi: 10.1128/JB.183.6.2137-2140.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell. 2005;122:461–72. doi: 10.1016/j.cell.2005.05.030. [DOI] [PubMed] [Google Scholar]
- (36).McPhee JB, Bains M, Winsor G, Lewenza S, Kwasnicka A, Brazas MD, Brinkman FS, Hancock RE. Contribution of the PhoP-PhoQ and PmrA-PmrB two-component regulatory systems to Mg2+-induced gene regulation in Pseudomonas aeruginosa. J Bacteriol. 2006;188:3995–4006. doi: 10.1128/JB.00053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Perron GG, Zasloff M, Bell G. Experimental evolution of resistance to an antimicrobial peptide. Proc Biol Sci. 2006;273:251–6. doi: 10.1098/rspb.2005.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Buckling A, Brockhurst M. Microbiology: RAMP resistance. Nature. 2005;438:170–1. doi: 10.1038/438170a. [DOI] [PubMed] [Google Scholar]
- (39).Shai Y, Oren Z. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides. 2001;22:1629–41. doi: 10.1016/s0196-9781(01)00498-3. [DOI] [PubMed] [Google Scholar]
- (40).DeGrado WF. Design of peptides and proteins. Adv. Prot. Chem. 1988;39:51–124. doi: 10.1016/s0065-3233(08)60375-7. [DOI] [PubMed] [Google Scholar]
- (41).Chen Y, Mant CT, Farmer SW, Hancock RE, Vasil ML, Hodges RS. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J Biol Chem. 2005;280:12316–29. doi: 10.1074/jbc.M413406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Prenner EJ, Kiricsi M, Jelokhani-Niaraki M, Lewis RN, Hodges RS, McElhaney RN. Structure-activity relationships of diastereomeric lysine ring size analogs of the antimicrobial peptide gramicidin S: mechanism of action and discrimination between bacterial and animal cell membranes. J Biol Chem. 2005;280:2002–11. doi: 10.1074/jbc.M406509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Braunstein A, Papo N, Shai Y. In vitro activity and potency of an intravenously injected antimicrobial peptide and its DL amino acid analog in mice infected with bacteria. Antimicrob Agents Chemother. 2004;48:3127–9. doi: 10.1128/AAC.48.8.3127-3129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shai Y, Makovitzky A, Avrahami D. Host defense peptides and lipopeptides: modes of action and potential candidates for the treatment of bacterial and fungal infections. Curr Protein Pept Sci. 2006;7:479–86. doi: 10.2174/138920306779025620. [DOI] [PubMed] [Google Scholar]
- (45).Makovitzki A, Baram J, Shai Y. Antimicrobial lipopolypeptides composed of palmitoyl Di- and tricationic peptides: in vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry. 2008;47:10630–6. doi: 10.1021/bi8011675. [DOI] [PubMed] [Google Scholar]
- (46).Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinber DA, wilcoxen KM, Ghadiri MR. Antibacterial agents based on the cyclic D,L-alpha-peptide architecture. Nature. 2001;412:452–455. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- (47).Hamuro Y, Schneider JP, DeGrado WF. De novo design of antibacterial beta-peptides. J. Amer. Chem. Soc. 1999;121:12200–12201. [Google Scholar]
- (48).Liu D, DeGrado W. De Novo Design, Synthesis, and Characterization of Antimicrobial Beta-peptides. J Am Chem Soc. 2001;123:7553–7559. doi: 10.1021/ja0107475. [DOI] [PubMed] [Google Scholar]
- (49).Porter EA, Wang X, Lee HS, Weisblum B, Gellman SH. Non-haemolytic beta-amino-acid oligomers. Nature. 2000;404:565. doi: 10.1038/35007145. [DOI] [PubMed] [Google Scholar]
- (50).Arvidsson PI, Ryder NS, Weiss HM, Hook DF, Escalante J, Seebach D. Exploring the antibacterial and hemolytic activity of shorter- and longer-chain beta-, alpha,beta-, and gamma-peptides, and of beta-peptides from beta2-3-aza- and beta3-2-methylidene-amino acids bearing proteinogenic side chains--a survey. Chem Biodivers. 2005;2:401–20. doi: 10.1002/cbdv.200590020. [DOI] [PubMed] [Google Scholar]
- (51).Epand RF, Schmitt MA, Gellman SH, Epand RM. Role of membrane lipids in the mechanism of bacterial species selective toxicity by two alpha/beta-antimicrobial peptides. Biochim Biophys Acta. 2006 doi: 10.1016/j.bbamem.2006.01.018. [DOI] [PubMed] [Google Scholar]
- (52).Epand RF, Schmitt MA, Gellman SH, Sen A, Auger M, Hughes DW, Epand RM. Bacterial species selective toxicity of two isomeric alpha/beta-peptides: role of membrane lipids. Mol Membr Biol. 2005;22:457–69. doi: 10.1080/09687860500370562. [DOI] [PubMed] [Google Scholar]
- (53).Porter EA, Weisblum B, Gellman SH. Use of parallel synthesis to probe structure-activity relationships among 12-helical beta-peptides: evidence of a limit on antimicrobial activity. J Am Chem Soc. 2005;127:11516–29. doi: 10.1021/ja0519785. [DOI] [PubMed] [Google Scholar]
- (54).Epand RF, Raguse TL, Gellman SH, Epand RM. Antimicrobial 14-helical beta-peptides: potent bilayer disrupting agents. Biochemistry. 2004;43:9527–35. doi: 10.1021/bi049414l. [DOI] [PubMed] [Google Scholar]
- (55).Schmitt MA, Weisblum B, Gellman SH. Unexpected relationships between structure and function in alpha,beta-peptides: antimicrobial foldamers with heterogeneous backbones. J Am Chem Soc. 2004;126:6848–9. doi: 10.1021/ja048546z. [DOI] [PubMed] [Google Scholar]
- (56).Violette A, Fournel S, Lamour K, Chaloin O, Frisch B, Briand JP, Monteil H, Guichard G. Mimicking helical antibacterial peptides with nonpeptidic folding oligomers. Chem Biol. 2006;13:531–8. doi: 10.1016/j.chembiol.2006.03.009. [DOI] [PubMed] [Google Scholar]
- (57).Schmitt MA, Weisblum B, Gellman SH. Interplay among folding, sequence, and lipophilicity in the antibacterial and hemolytic activities of alpha/beta-peptides. J Am Chem Soc. 2007;129:417–28. doi: 10.1021/ja0666553. [DOI] [PubMed] [Google Scholar]
- (58).Doerksen RJ, Chen B, Liu D, Tew GN, Degrado WF, Klein ML. Controlling the Conformation of Arylamides: Computational Studies of Intramolecular Hydrogen Bonds between Amides and Ethers or Thioethers. Chemistry, A European Journal. 2004;10:5008–5016. doi: 10.1002/chem.200400176. [DOI] [PubMed] [Google Scholar]
- (59).Steiner T. CH••O hydrogen bonding in crystals. Cryst. Rev. 2003;9:177–238. [Google Scholar]
- (60).Tew GN, Liu D, Chen B, Doerksen RJ, Kaplan J, Carroll PJ, Klein ML, DeGrado WF. De novo design of biomimetic antimicrobial polymers. Proc Natl Acad Sci U S A. 2002;99:5110–4. doi: 10.1073/pnas.082046199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Liu D, Choi S, Chen B, Doerksen RJ, Clements DJ, Winkler JD, Klein ML, DeGrado WF. Nontoxic membrane-active antimicrobial arylamide oligomers. Angew Chem Int Ed Engl. 2004;43:1158–62. doi: 10.1002/anie.200352791. [DOI] [PubMed] [Google Scholar]
- (62).Pophristic V, Vemparala S, Ivanov I, Liu Z, Klein ML, DeGrado WF. Controlling the shape and flexibility of arylamides: a combined ab initio, ab initio molecular dynamics, and classical molecular dynamics study. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:3517–26. doi: 10.1021/jp054306+. [DOI] [PubMed] [Google Scholar]
- (63).Huc I. Aromatic oligoamide foldamers. European Journal of Organic Chemistry. 2004:17–29. [Google Scholar]
- (64).Kolomiets E, Berl V, Odriozola I, Stadler AM, Kyritsakas N, Lehn JM. Contraction/extension molecular motion by protonation/deprotonation induced structural switching of pyridine derived oligoamides. Chemical Communications. 2003:2868–2869. doi: 10.1039/b311578j. [DOI] [PubMed] [Google Scholar]
- (65).Nowick JS. Exploring beta-sheet structure and interactions with chemical model systems. Acc Chem Res. 2008;41:1319–30. doi: 10.1021/ar800064f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Tang H, Doerksen RJ, Jones TV, Klein ML, Tew GN. Biomimetic facially amphiphilic antibacterial oligomers with conformationally stiff backbones. Chem Biol. 2006;13:427–35. doi: 10.1016/j.chembiol.2006.02.007. [DOI] [PubMed] [Google Scholar]
- (67).Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc Natl Acad Sci U S A. 2009;106:6968–73. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Tang H, Doerksen RJ, Tew GN. Synthesis of urea oligomers and their antibacterial activity. ChemComm. 2005;12:1537–1539. doi: 10.1039/b413679a. [DOI] [PubMed] [Google Scholar]
- (69).Gottler LM, Lee HY, Shelburne CE, Ramamoorthy A, Marsh EN. Using fluorous amino acids to modulate the biological activity of an antimicrobial peptide. Chembiochem. 2008;9:370–3. doi: 10.1002/cbic.200700643. [DOI] [PubMed] [Google Scholar]
- (70).Rennie J, Arnt L, Tang H, Nusslein K, Tew GN. Simple oligomers as antimicrobial peptide mimics. J Ind Microbiol Biotechnol. 2005;32:296–300. doi: 10.1007/s10295-005-0219-0. [DOI] [PubMed] [Google Scholar]
- (71).Nusslein K, Arnt L, Rennie J, Owens C, Tew GN. Broad-spectrum antibacterial activity by a novel abiogenic peptide mimic. Microbiology. 2006;152:1913–8. doi: 10.1099/mic.0.28812-0. [DOI] [PubMed] [Google Scholar]
- (72).Arnt L, Rennie JR, Linser S, Willumeit R, Tew GN. Membrane activity of biomimetic facially amphiphilic antibiotics. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:3527–32. doi: 10.1021/jp054339p. [DOI] [PubMed] [Google Scholar]
- (73).Epand RF, Mowery BP, Lee SE, Stahl SS, Lehrer RI, Gellman SH, Epand RM. Dual mechanism of bacterial lethality for a cationic sequence-random copolymer that mimics host-defense antimicrobial peptides. J Mol Biol. 2008;379:38–50. doi: 10.1016/j.jmb.2008.03.047. [DOI] [PubMed] [Google Scholar]
- (74).Yang L, Gordon VD, Mishra A, Som A, Purdy KR, Davis MA, Tew GN, Wong GCL. Synthetic Antimicrobial Oligomers Induce a Composition-Dependent Topological Transition in Membranes. J. Am. Chem. Soc. 2007;129:12141–12147. doi: 10.1021/ja072310o. [DOI] [PubMed] [Google Scholar]
- (75).Oren Z, Shai Y. model of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers. 1998;6:451–463. doi: 10.1002/(SICI)1097-0282(1998)47:6<451::AID-BIP4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- (76).Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. Mimicry of antimicrobial host-defense peptides by random copolymers. J Am Chem Soc. 2007;129:15474–15476. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]
- (77).Tashiro T. Antibacterial and bacterium adsorbing macromolecules. Macromolecular Materials and Engineering. 2001;286:63–87. [Google Scholar]
- (78).Worley SD, Sun G. Biocidal polymers. Trends in Polymer Science (Cambridge, United Kingdom) 1996;4:364–370. [Google Scholar]
- (79).Stiriba S-E, Frey H, Haag R. Dendritic polymers in biomedical applications: From potential to clinical use in diagnostics and therapy. Angewandte Chemie, International Edition. 2002;41:1329–1334. doi: 10.1002/1521-3773(20020415)41:8<1329::aid-anie1329>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- (80).Thorsteinsson T, Loftsson T, Masson M. Soft antibacterial agents. Current Medicinal Chemistry. 2003;10:1129–1136. doi: 10.2174/0929867033457520. [DOI] [PubMed] [Google Scholar]
- (81).Kenawy E-R, Mahmoud YAG. Biologically active polymers, 6: Synthesis and antimicrobial activity of some linear copolymers with quaternary ammonium and phosphonium groups. Macromolecular Bioscience. 2003;3:107–116. [Google Scholar]
- (82).Pavlikova M, Lacko I, Devinsky F, Mlynarcik D. Quantitative relationships between structure, aggregation properties and antimicrobial activity of quaternary ammonium bolaamphiphiles. Collection of Czechoslovak Chemical Communications. 1995;60:1213–28. [Google Scholar]
- (83).Kuroda K, Degrado WF. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J Am Chem Soc. 2005;127:4128–9. doi: 10.1021/ja044205+. [DOI] [PubMed] [Google Scholar]
- (84).Ivanov I, Vemparala S, Pophristic V, Kuroda K, DeGrado WF, McCammon JA, Klein ML. Characterization of nonbiological antimicrobial polymers in aqueous solution and at water-lipid interfaces from all-atom molecular dynamics. J Am Chem Soc. 2006;128:1778–9. doi: 10.1021/ja0564665. [DOI] [PubMed] [Google Scholar]
- (85).Kuroda K, Caputo GA, DeGrado WF. The role of hydrophobicity in the antimicrobial and hemolytic activities of polymethacrylate derivatives. Chemistry. 2009;15:1123–33. doi: 10.1002/chem.200801523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Ilker MF, Nusslein K, Tew GN, Coughlin EB. Tuning the hemolytic and antibacterial activities of amphiphilic polynorbornene derivatives. J. Am. Chem. Soc. 2004;126:15870–15875. doi: 10.1021/ja045664d. [DOI] [PubMed] [Google Scholar]
- (87).Lienkamp K, Madkour AE, Musante A, Nelson CF, Nusslein K, Tew GN. Antimicrobial polymers prepared by ROMP with unprecedented selectivity: A molecular construction kit approach. J. Am. Chem. Soc. 2008;130:9836–9843. doi: 10.1021/ja801662y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Gabriel GJ, Maegerlein JA, Nelson CF, Dabkowski JM, Eren T, Nüsslein K, Tew GN. Comparison of Facially Amphiphilic versus Segregated Monomers in the Design of Antibacterial Copolymers. Chem. E. J. 2008 doi: 10.1002/chem.200801233. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).La Rocca P, Shai Y, Sansom MS. Peptide-bilayer interactions: simulations of dermaseptin B, an antimicrobial peptide. Biophys Chem. 1999;76:145–59. doi: 10.1016/s0301-4622(98)00232-4. [DOI] [PubMed] [Google Scholar]
- (90).Aliste MP, MacCallum JL, Tieleman DP. Molecular dynamics simulations of pentapeptides at interfaces: salt bridge and cation-pi interactions. Biochemistry. 2003;42:8976–87. doi: 10.1021/bi027001j. [DOI] [PubMed] [Google Scholar]
- (91).Karaborni S, Essellink K, Hilbers P, Smit B, Karthauser J, Vanos N, Zana R. Science. 1994;266:254–256. doi: 10.1126/science.266.5183.254. [DOI] [PubMed] [Google Scholar]
- (92).Nielsen SO, Lopez CF, Ivanov I, Moore PB, Shelley JC, Klein ML. Transmembrane peptide-induced lipid sorting and mechanism of L-alpha-to-inverted phase transition using coarse-grain molecular dynamics. Biophysical Journal. 2004;87:2107–2115. doi: 10.1529/biophysj.104.040311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Izvekov S, Voth GA. J. Phys. Chem. B. 2005;105:4464–4470. [Google Scholar]
- (94).Shelley JC, Shelley MY, Reeder RC, Bandyopadhyay S, Klein ML. J. Phys. Chem. B. 2001;109:2469–2473. [Google Scholar]
- (95).Lopez C, Nielsen S, Srinivas G, WF D, MK K. Probing membrane insertion activity of antimicrobial polymers via coarse-grain molecular dynamics. J Chem Theory Comp. 2006;2:649–655. doi: 10.1021/ct050298p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Scott RW, DeGrado WF, Tew GN. De novo designed synthetic mimics of antimicrobial peptides. Curr Opin Biotechnol. 2008;19:620–7. doi: 10.1016/j.copbio.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]