

Abstract
The present study is designed to investigate the effects of interleukin-4 (IL-4) on expression of interleukin-6 (IL-6), as well as to examine the role of distinct sources of reactive oxygen species (ROS) in this process. Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) and enzyme-linked immunosorbent assay (ELISA) showed that IL-4 significantly up-regulated the mRNA and protein expression of IL-6 in human aortic endothelial cells (HAEC) and C57BL/6 mice. Dihydroethidium (DHE) and dichlorofluorescein (DCF) fluorescence staining demonstrated that IL-4 significantly increased ROS generation in HAEC. A significant and dose-dependent inhibition of IL-4-induced IL-6 expression was observed in HAEC pre-treated with antioxidants, such as pyrrolidine dithiocarbamate (PDTC) and epigallocatechin gallate (EGCG), indicating that IL-4-induced IL-6 expression is mediated via an ROS-dependent mechanism. Additionally, pharmacological inhibitor of NADPH oxidase (NOX) significantly attenuated IL-4-induced ROS generation and IL-6 expression in HAEC. Furthermore, the disruption of NOX gene dramatically and significantly reduced IL-4-induced IL-6 expression in NOX knockout mice (B6.129S6-Cybbtm1Din/J). In contrast, overexpression of IL-6 in IL-4-activated HAEC was not affected by inhibiting other ROS generating pathways, such as xanthine oxidase, arachidonic acid metabolism, and the mitochondrial electron transport chain. These results demonstrate that IL-4 up-regulates IL-6 expression in vascular endothelium through NOX-mediated ROS generation.
Keywords: IL-4, IL-6, Vascular endothelium, Reactive oxygen species, NADPH oxidase
1. Introduction
Although the contribution of T-helper 1 (Th1) and T-helper 2 (Th2) cell responses to the development of atherosclerosis remains unclear, pro-inflammatory cytokines secreted by Th1 cells (“Th1 cytokines”) have been implicated in the initiation and progression of atherosclerosis. For example, tumor necrosis factor-α (TNF-α) is well known pro-atherogenic cytokine, and pathophysiological role of TNF-α in atherosclerosis has been extensively investigated both in vitro and in vivo [1]. Recent evidence, however, has shown that the development of atherosclerosis was not completely abolished even though disruption of TNF-α gene significantly diminished the severity of this disease in ApoE-deficient mice [2]. These findings suggest that other pathways may contribute to the disease progression. Indeed, there is evidence that Th2 cells might play a role while little is known concerning the possible contribution of Th2 cytokines to the vascular inflammation and atherosclerosis [3–20].
IL-4 is a pleiotropic immunomodulatory cytokine secreted by Th2 cells (“Th2 cytokine”) and was traditionally considered as an anti-inflammatory cytokine [16,18]. However, growing body of evidence has suggested that IL-4 is pro-atherogenic and may play a critical role in the progression of atherosclerosis. For example, recent studies from our laboratory and others have demonstrated that IL-4 induces pro-inflammatory environments by overexpressing a number of pro-inflammatory mediators, such as vascular cell adhesion molecule-1 (VCAM-1), E-selectin, monocyte chemoattractant protein-1 (MCP-1), and IL-6 in human vascular endothelial cells [6–8,10–14,19,20]. It was also found that IL-4 synergistically increases IL-1β-, TNF-α-, or lipopolysaccharide (LPS)-induced VCAM-1 expression in vascular endothelium [3,4,15]. In addition to in vitro cell culture studies, the pro-atherogenic effects of IL-4 have been investigated in animal models of atherosclerosis. King et al. [9] have shown that transplantation of bone marrow stem cells isolated from IL-4-deficient (IL-4−/−) mice led to decreased atherosclerotic lesion formation in LDL receptor-deficient (LDLR−/−) mice. Furthermore, a significant reduction in atherosclerotic plaque area was observed in IL-4−/−/ApoE−/− mice compared to ApoE−/− mice [5]. These findings suggest that there may be potential, novel pathways by which IL-4 exerts its pro-atherogenic effects. The molecular signaling mechanisms responsible for IL-4-induced pro-inflammatory pathways in vascular endothelium, however, remain largely unknown.
Evidence indicates the pivotal role of a pro-inflammatory cytokine IL-6 in the pathogenesis of cardiovascular disease including atherosclerosis [21,22]. For example, the mRNA and protein expression of IL-6 have been detected in human atherosclerotic lesions [23,24]. IL-6 has also been found in the atherosclerotic plaques of ApoE knockout mice aorta, and administration of exogenous IL-6 in ApoE knockout mice exacerbated atherosclerotic lesion formation [25,26]. These studies clearly demonstrate that the IL-6-mediated pro-inflammatory environment in vascular endothelium is crucial for the initiation and development of atherosclerosis.
The present study was designed to investigate the effects of IL-4 on expression of IL-6 in vitro and in vivo. We also examined the potential role of distinct sources of ROS in this process. We have demonstrated that IL-4 significantly up-regulates mRNA and protein expression of IL-6 in vascular endothelium. Additionally, the present study has provided the first novel evidence indicating that NADPH oxidase plays a pivotal role in IL-4-induced ROS generation and IL-6 expression in vitro and in vivo.
2. Materials and methods
2.1. Cell culture
Primary human aortic endothelial cells (HAEC) were purchased from Cascade Biologics™ (Portland, OR) and cultured in Medium 200 supplemented with Low Serum Growth Supplement (LSGS) in a 37°C, 5% CO2/95% air, humidified cell culture incubator.
2.2. Animals
Male C57BL/6 mice (6 weeks old) were purchased from Harlan (Indianapolis, IN) and maintained under environmentally controlled conditions, and subject to a 12 h light/dark cycle with food and water ad libitum. Additionally, NOX knockout mice (NOX gp91phox−/− or B6.129S6-Cybbtm1Din/J, male, 6 weeks old) and matched wild-type controls with the same genetic background (C57BL/6J, male, 6 weeks old) were obtained from The Jackson Laboratory (Bar Harbor, ME). Animals (n=4 to 5) received a single intraperitoneal injection of either phosphate buffered saline (PBS) or 100 μg/kg of IL-4, and were humanely sacrificed by CO2 inhalation. The aortas from mice in each group were isolated and dissected gently free of adhering tissues. Isolated aortic samples were frozen and stored at −80°C until analysis. Additionally, blood was obtained by cardiac puncture, and blood plasmas were prepared, aliquoted, frozen, and stored at −80°C. Freshly thawed blood plasmas were analyzed immediately. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and this study was approved by Virginia Tech Institutional Animal Care and Use Committee (IACUC).
2.3. Cell viability assay
Cell viability assay was performed with the standard 3-[(4,5-dimethythiazol-2-yl]2,5-diphenyltetrazolium bromide (MTT) conversion assay. The cell viability of HAEC was not affected by treatment with apocynin at 1 mM, which was used in the present study (Data not shown).
2.4. Real-time RT-PCR
The aortas of mice were homogenized with 1.6 mm stainless steel beads and 1 ml of TRI Reagent (Sigma-Aldrich, St. Louis, MO) using a setting of 8 for 15 min in a tissue homogenizer (The Bullet Blender™ Next Advance Inc., Averill Park, NY), and total RNA was isolated from tissue homogenates as described previously [27]. In cell culture studies, total RNA was isolated from HAEC using RNeasy Mini Kit (Qiagen, Valencia, CA) according to the protocol of the manufacturer. 1 μg of total RNA was reverse-transcribed at 25°C for 15 min, 42°C for 45 min, and 99°C for 5 min in 20 μl of 5 mM MgCl2, 10 mM Tris-HCl, pH 9.0, 50 mM KCl, 0.1% Triton X-100, 1 mM dNTP, 1 unit/μl of recombinant RNasin ribonuclease inhibitor, 15 units/μg of AMV reverse transcriptase, and 0.5 μg of random hexamers. Amplifications of individual genes were performed on ABI 7300 Sequence Detection System (Applied Biosystems, Foster City, CA) using TaqMan ® Universal PCR Master Mix, gene-specific TaqMan PCR probes and primers, and a standard thermal cycler protocol (50°C for 2 min before the 1st cycle, 95°C for 15 s, and 60°C for 1 min, repeated 45 times). For specific probes and primers of PCR amplifications, TaqMan® Gene Expression Assay Reagents for human IL-6, human glyceraldehydes-3-phosphate dehydrogenase (GAPDH), mouse IL-6, mouse GAPDH, and mouse β-actin were obtained from Applied Biosystems. The threshold cycle (CT), which indicates the fractional cycle number at which the amount of amplified target gene reaches a fixed threshold, from each well was determined by the Applied Biosystems Sequence Detection Software v1.2.3. Relative quantification, which represents the change in gene expression from real-time quantitative PCR experiments between treated group and untreated control group, was calculated by the comparative CT method as previously described previously [28–30]. The data was analyzed using equation 2−ΔΔCT, where ΔΔCT = [CT of target gene - CT of housekeeping gene]treated group − [CT of target gene - CT of housekeeping gene]untreated control group. For the treated samples, evaluation of 2−ΔΔCT indicates the fold change in gene expression, normalized to a housekeeping gene (GAPDH or β-actin), and relative to the untreated control.
2.5. ELISA
IL-6 concentrations in cell culture supernatants and mouse blood plasmas were measured using a Human or Mouse Quantikine ® ELISA Kit (R&D Systems Inc., Minneapolis, MN).
2.6. Detection of ROS
The intracellular levels of ROS such as superoxide anion and hydrogen peroxide were measured by DHE and DCF fluorescence staining using a Zeiss AXIO Imager A1m fluorescence microscope equipped with AxioCam MRc5 Digital Imaging System (Carl Zeiss MicroImaging, Inc., Thornwood, NY). Briefly, HAEC were grown on the glass slide in the Lab-Tek® II Chamber Slide™ System (Nalge Nunc International Corp., Naperville, IL). After treatment with IL-4, the cells were loaded with either DHE or carboxy-H2DCF-DA (Invitrogen Corp., Carlsbad, CA) at concentration of 5 μM in PBS for 30 min at 37°C in 5% CO2/95% air, humidified cell culture incubator. HAEC monolayers were washed with PBS and examined on a Zeiss AXIO Imager A1m fluorescence microscope. Cell images were acquired with 20 × objective by AxioCam MRc5 Digital Imaging System. The DHE or DCF fluorescence intensity of acquired digital images was quantified by MATLAB® Imaging Processing Software (The Mathworks™ Natick, MA) as described previously [31]. Data were expressed in fluorescence arbitrary units (×109).
2.7. Statistical analysis
Statistical analysis of data was completed using SigmaStat 3.5 (Systat Software, Inc., Point Richmond, CA). One-way ANOVA was used to compare mean responses among the treatments. For each endpoint, the treatment means was compared using Bonferroni least significant difference procedure. Differences among the means were considered significant at P < 0.05.
3. Results
3.1. Effects of IL-4 on IL-6 expression in HAEC and mice
Since IL-6 has been shown to play an important role in inflammatory responses in vascular endothelium [21–26], we examined the effects of IL-4 on the mRNA and protein expression of IL-6 in vitro and in vivo. Quantitative real-time RT-PCR showed that treatment of HAEC with the increasing concentrations of IL-4 (0.1, 1.0, and 10 ng/ml) significantly and dose-dependently up-regulated the mRNA expression of IL-6 (Fig. 1A). Consistent with the data on gene expression, exposure of HAEC to IL-4 resulted in a significant and dose-dependent up-regulation of IL-6 protein expression (Fig. 1B). To determine whether IL-4 induces IL-6 mRNA expression at the transcriptional level, HAEC were pre-treated for 1 h with actinomycin D, an inhibitor of RNA transcription, and then incubated with 10 ng/ml of IL-4- for 4 h (mRNA expression) or 16 h (protein expression). As illustrated in new Fig. 2, IL-4-mediated increases in mRNA and protein levels of IL-6 were completely abolished by actinomycin D. These data clearly demonstrated that IL-4 induces IL-6 expression at the transcriptional level.
Fig. 1.

Effects of IL-4 on IL-6 expression in vitro and in vivo. HAEC were either untreated (Control) or treated with the indicated concentrations of IL-4 for 4 h (A) or 16 h (B). The mRNA and protein expression levels of IL-6 were determined by real-time RT-PCR (A) and ELISA (B). Mice were administered a single intraperitoneal injection of either PBS (Control) or 100 μg/kg of IL-4, and exposed for 4, 8, and 24 h. The mRNA and protein expression levels of IL-6 in mouse aortas and blood plasma were determined by real-time RT-PCR (C) and ELISA (D). Values represent the mean ± SEM (n=4). * p < 0.05 vs. Control.
Fig. 2.
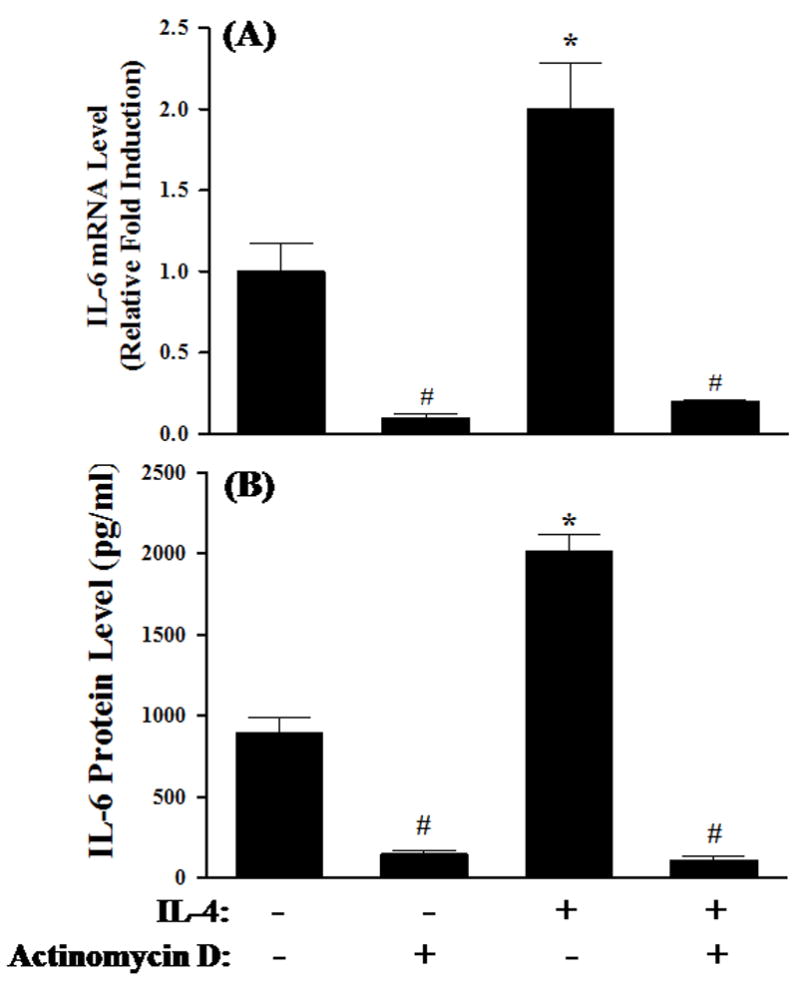
Effects of actinomycin D on IL-4-induced IL-6 expression. HAEC were pre-treated for 1 h with 5 μg/ml of actinomycin D, an inhibitor of RNA transcription, before exposure to 10 ng/ml of IL-4 for 4 h (A) or 16 h (B). The mRNA and protein expression levels of IL-6 were determined by real-time RT-PCR (A) and ELISA (B). Values represent the mean ± SEM (n=4). * p < 0.05 vs. Control. # Values in the cultures pre-treated with actinomycin D are statistically different from those in respective controls (p < 0.05).
We also performed a series of animal experiments to examine the ability of IL-4 to induce IL-6 expression in vivo. As depicted in Fig. 1C, real-time RT-PCR analysis demonstrated a significant up-regulation of IL-6 mRNA expression in mouse aortas isolated at 4 h after intraperitoneal administration of 100 μg/kg of IL-4. In addition, a significantly enhanced expression of IL-6 protein was observed in blood plasmas collected from IL-4-injected mice (Fig. 1D). These results provide the first direct evidence demonstrating that IL-4 induces IL-6 expression in both HAEC and mice.
3.2. Effects of IL-4 on the intracellular level of ROS
It is generally accepted that ROS play a critical role in atherogenesis by regulating the expression of pro-inflammatory mediators such as cytokines, chemokines, and adhesion molecules in vascular endothelium [11,13,20,32,33]. In the present study, the levels of the intracellular ROS were measured by DHE and DCF fluorescence staining to examine whether IL-4-induced IL-6 expression in HAEC is mediated through ROS generation. When DHE is oxidized to ethidium by superoxide anion, ethidium intercalates within the cell’s DNA and stain its nucleus a bright fluorescent red. Dichlorofluorescin diacetate (H2DCF-DA) is a stable, non-polar compound that readily diffuses into the cells and is converted to a non-fluorescent polar derivative dichlorofluorescin (DCF-H) by intracellular esterases. DCF-H can be oxidized to the highly fluorescent compound DCF by hydrogen peroxide or other peroxides produced by the cells. As illustrated in Fig. 3, the levels of both DHE and DCF fluorescence were significantly and time-dependently increased after exposure of HAEC to IL-4, indicating that IL-4 treatment induces the generation of ROS such as superoxide anion and hydrogen peroxide. To determine whether IL-4-mediated overexpression of IL-6 is mediated via an ROS-dependent mechanism, we examined the effects of antioxidants on the mRNA and protein expression of IL-6 in IL-4-treated HAEC. As shown in Fig. 4, pre-treatment of HAEC with PDTC markedly attenuated IL-4-mediated up-regulation of IL-6 mRNA and protein expression in a dose-dependent manner (Fig. 4A and 4B). A similar effect was also observed in IL-4-activated HAEC pre-treated with EGCG (Fig. 4C and 4D). Both PDTC and EGCG have been widely used as antioxidant compounds to investigate redox regulation of the intracellular signaling pathways and of cell function [11,13,34,35]. These results suggest that IL-4-induced IL-6 expression in HAEC is associated with an increase in intracellular ROS generation.
Fig. 3.
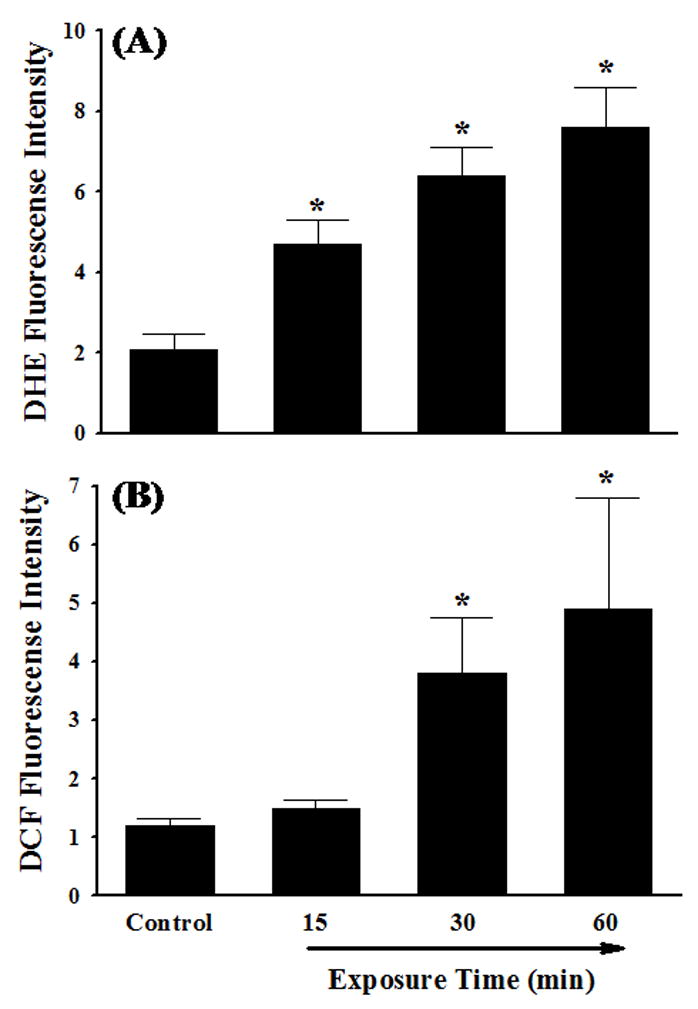
Effects of IL-4 on generation of ROS. HAEC were incubated with either PBS (Control) or 10 ng/ml of IL-4 for 15 min, 30 min, and 60 min. Intracellular levels of superoxide and hydrogen peroxide were determined by DHE (A) and DCF (B) fluorescence staining. ROS values are in arbitrary units of fluorescence intensity (×109). Values represent the mean ± SEM (n=4). * p < 0.05 vs. Control.
Fig. 4.
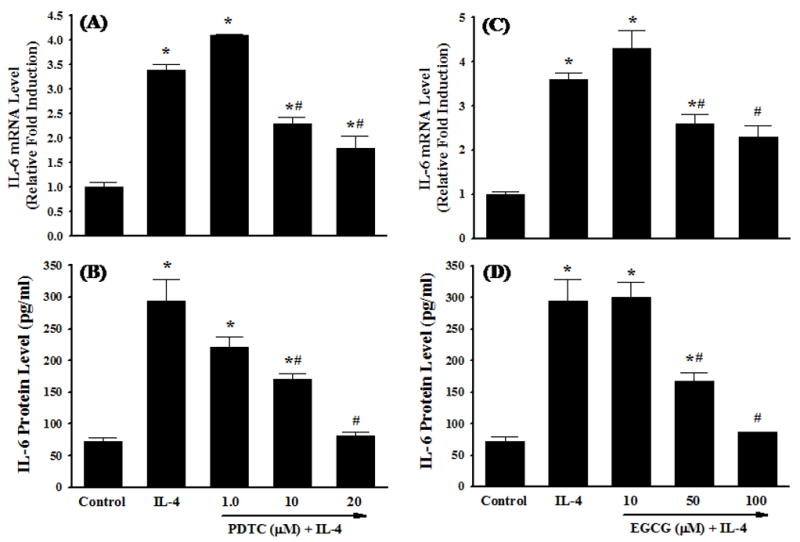
Effects of antioxidants on IL-4-induced IL-6 expression. HAEC were pre-treated with the indicated concentrations of PDTC or EGCG for 1 h and then exposed to either PBS (Control) or 10 ng/ml of IL-4 for 4 h (A and C) or 16 h (B and D). The mRNA and protein expression levels of IL-6 were determined by real-time RT-PCR (A and C) and ELISA (B and D). Values represent the mean ± SEM (n=4). * p < 0.05 vs. Control, # p < 0.05 vs. IL-4.
3.3. Role of NOX in IL-4-induced IL-6 expression
Previous studies have shown that NOX is a key source of enzymatic generation of ROS in a variety of cell types including endothelial cells [36]. To determine whether NOX is involved in IL-4-induced ROS generation and IL-6 expression, HAEC were pre-treated with apocynin, an inhibitor of NOX, for 30 min and then exposed to 10 ng/ml of IL-4. Fig. 5 depicted that inhibition of NOX by apocynin significantly attenuated IL-4-induced overexpression of IL-6 mRNA and protein in HAEC. Additionally, pre-treatment of HAEC with apocynin at 1.0 mM significantly suppressed IL-4-induced ROS generation as assessed by DHE and DCF fluorescence staining (Fig. 6), indicating that IL-4 up-regulates IL-6 expression in HAEC through NOX-mediated ROS generation. Additionally, NOX knockout mice and matched wild-type controls were employed to examine the crucial role of NOX in IL-4-induced IL-6 expression in vivo. As shown in Fig. 7, consistent with the data on IL-4-induced up-regulation of IL-6 expression in mice (Fig. 1C), IL-4 administration resulted in a significant increase in the mRNA expression of IL-6 in the wild-type mice. On the other hand, the disruption of NOX gp91phox gene dramatically and significantly attenuated IL-4-induced IL-6 expression in the NOX knockout mice. These results strongly suggest that NOX plays a pivotal role in IL-4-induced ROS generation and IL-6 expression in vitro and in vivo.
Fig. 5.
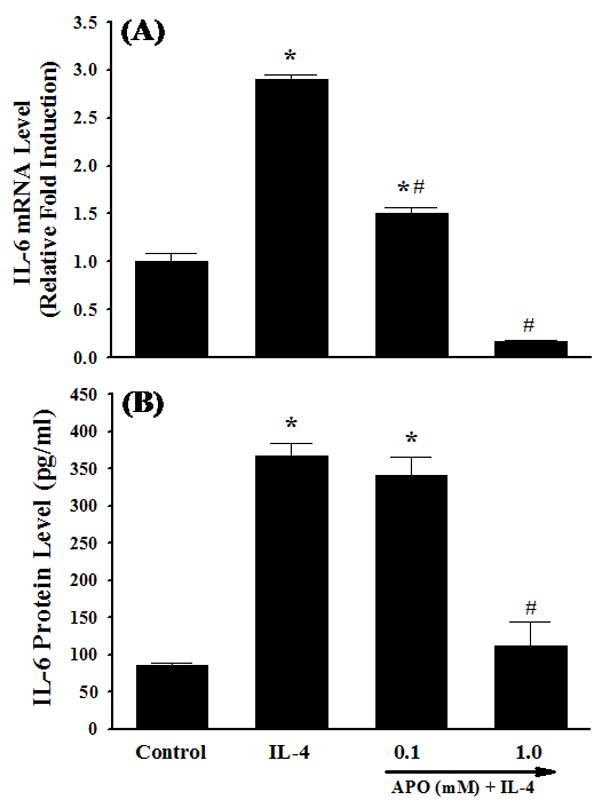
Effects of NOX inhibitor on IL-4-induced IL-6 expression. HAEC were pre-treated with the indicated concentrations of apocynin (APO) for 30 min, and then incubated with either PBS (Control) or 10 ng/ml of IL-4 for 4 h (A) or 16 h (B). The mRNA and protein expression levels of IL-6 were determined by real-time RT-PCR (A) and ELISA (B). Values represent the mean ± SEM (n=4). * p < 0.05 vs. Control, # p < 0.05 vs. IL-4.
Fig. 6.
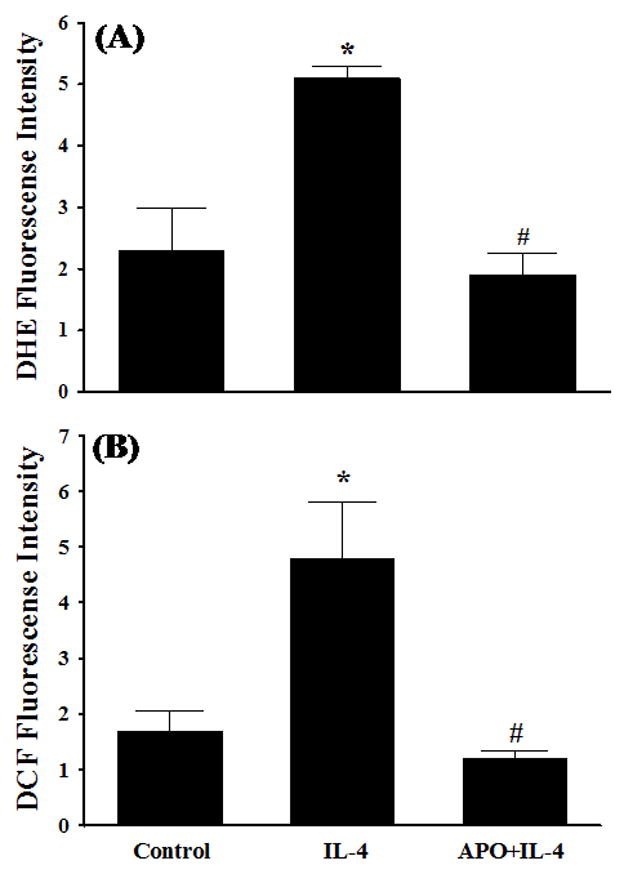
Effects of NOX inhibitor on IL-4-induced ROS generation. HAEC were pre-treated with 1.0 mM apocynin (APO) for 30 min, and then incubated with either PBS (Control) or 10 ng/ml of IL-4 for 60 min. Intracellular levels of ROS generation were determined by DHE (A) and DCF (B) fluorescence staining. ROS values are in arbitrary units of fluorescence intensity (×109). Values represent the mean ± SEM (n=4). * p < 0.05 vs. Control, # p < 0.05 vs. IL-4.
Fig. 7.
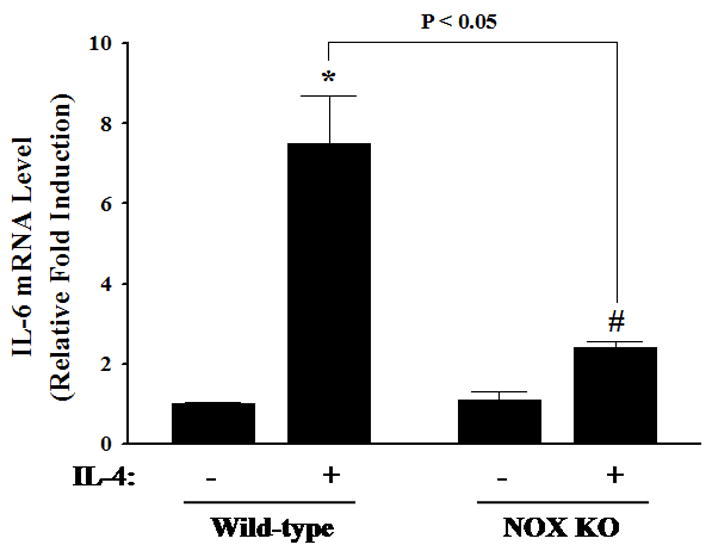
Effects of IL-4 on IL-6 expression in wild-type and NOX knockout mice. Mice were administered a single intraperitoneal injection of either PBS or 100 μg/kg of IL-4, and exposed for 4 h. The mRNA expression levels of IL-6 in mouse aortas were determined by real-time RT-PCR. Values represent the mean ± SEM (n=5). * p < 0.05 vs. Wild-type control, # p < 0.05 vs. NOX KO control.
3.4. Role of xanthine oxidase, arachidonic acid metabolism, and mitochondrial electron transport chain in IL-4-induced IL-6 expression
Another enzymatic source of ROS generation is xanthine oxidase [36]. To investigate the potential contribution of xanthine oxidase-mediated ROS generation to IL-6 expression in IL-4-activated HAEC, the cells were pre-treated with allopurinol (ALLO), an inhibitor of xanthine oxidase, for 30 min and then incubated with 10 ng/ml of IL-4 for 16 h. Pre-treatment of HAEC with allopurinol at 1.0 and 10 μM did not affect IL-4-mediated up-regulation of IL-6 expression (Fig. 8), indicating that xanthine oxidase-mediated ROS generation is not associated with IL-4-induced IL-6 expression in HAEC.
Fig. 8.

Effects of selective inhibitors of other ROS generating pathways on IL-4-induced IL-6 expression. HAEC were pre-treated with xanthine oxidase inhibitor, such as allopurinol (ALLO; 1.0 and 10 μM), arachidonic acid metabolism inhibitors, such as eicosatetraynoic acid (ETYA; 10 and 50 μM), nordihydroguaiaretic acid (NDGA; 1.0 and 10 μM), and N-2-(cyclohexyloxy)-4-nitrophenyl-methanesulfonamide (NS-398; 0.1 and 1.0 μM), or mitochondrial electron transport chain inhibitors, such as rotenone (ROTN; 0.1 and 1.0μM), thenoyltrifluoroacetone (TTFA; 0.1 and 1.0 μM), and antimycin A (ANTI; 10 and 100 μM), for 30 min and then incubated with either PBS (Control) or 10 ng/ml of IL-4 for 16 h. The protein expression levels of IL-6 were determined by ELISA. Values represent the mean ± SEM (n=4). * p < 0.05 vs. Control.
It has been proposed that metabolic pathways of arachidonic acid by several enzymes, including cyclooxygenase (COX) and lipoxygenase (LOX), are associated with the generation of ROS [37]. To evaluate whether arachidonic acid metabolism is involved in IL-4-induced IL-6 expression in HAEC, the cells were pre-treated with several inhibitors, such as 5,8,11,14-eicosatetraynoic acid (ETYA), nordihydroguaiaretic acid (NDGA), and N-2-(cyclohexyloxy)-4-nitrophenyl-methanesulfonamide (NS-398), for 30 min and then incubated with 10 ng/ml of IL-4 for 16 h. As shown in Fig. 8, none of these inhibitors had significant effects on IL-4-induced IL-6 expression. These results suggest that arachidonic acid metabolism-mediated ROS generation is not involved in IL-4-induced IL-6 expression in HAEC.
Mitochondrial electron transport chain is another major source of intracellular ROS production [36]. To determine the involvement of mitochondria in ROS-mediated IL-6 overexpression in response to IL-4, HAEC were pre-treated with the mitochondrial electron transport chain inhibitors, such as rotenone (ROTN), thenoyltrifluoroacetone (TTFA), and antimycin A (ANTI), for 30 min to selectively block the electron flows between mitochondrial respiratory chain complexes, and then incubated with 10 ng/ml of IL-4 for 16 h. As shown in Fig. 8, inhibitions of mitochondrial respiratory chain complexes did not exert any significant effects on IL-6 expression by IL-4-activated HAEC. These results suggest that mitochondrial electron transport chain-mediated ROS generation is not associated with IL-4-induced IL-6 expression in HAEC.
4. Discussion
Cardiovascular disease (CVD) is the leading cause of illness and death in the United States. Indeed, an estimated 80,000,000 American adults (approximately 1 in 3) have one or more types of CVD, and the estimated direct and indirect cost of CVD for 2009 is $475.3 billion [38]. Although the exact cause of this disease remains unsolved, compelling evidence indicates that cardiovascular risk factors (hypercholesterolemia, hypertension, obesity, etc.) increase vascular ROS generation that play an important role in the development of CVD [32,39]. For example, increased production of superoxide was observed in atherosclerotic human coronary arteries [40,41]. It has become apparent that endothelial dysfunction is closely associated with an increased risk of atherosclerosis and vascular endothelial cells are particularly sensitive to oxidative stress [42,43]. Heitzer et al. [44] demonstrated that elevated production of vascular ROS is linked to impaired endothelial function and progression of atherosclerosis in patients with coronary artery disease. In addition, oxidative stress up-regulates the expression of pro-inflammatory mediators such as cytokines, chemokines, and adhesion molecules in vascular endothelium [32,45], which is one of the earliest steps in the development of atherosclerotic lesion formation [46].
The present study demonstrated that IL-4 induces IL-6 expression in endothelial cells and mouse aortas. However, we cannot exclude possible involvement of other cell types, such as smooth muscle cells and macrophages, in IL-4-induced IL-6 expression in aortas. Therefore, we are currently conducting a series of immunofluorescence staining with dual-labeling procedures to further investigate the potential contribution of specific type of cells, such as endothelial cells, smooth muscle cells, and macrophages, to the induction of IL-6 expression in mouse aortas after treatment with IL-4.
Recent studies by our group and others highlight that IL-4 may be considered a pro-oxidative cytokine which increases the oxidizing potential of target cells [11,13,14,47]. For example, treatment with IL-4 resulted in a dose-dependent increase in intracellular ROS and subsequent overexpression of redox-responsive genes such as VCAM-1 and MCP-1 in human umbilical vein endothelial cells (HUVEC) [10,11,13]. The intracellular sources of IL-4-induced ROS generation in vascular endothelium, however, remain unknown. In the present study, we demonstrate that ROS generation in HAEC by selective intracellular source is associated with the IL-4-mediated signaling pathways leading to the expression of IL-6. Several distinct pathways generating ROS appear responsible for the signal transduction cascade of vascular inflammation and progression of atherosclerosis [36]. Although our previous studies [11,13] have demonstrated that intracellular ROS generation may be associated with IL-4-mediated overexpression of pro-inflammatory mediators in human vascular endothelial cells, distinct sources of ROS involved in the IL-4-initiated signal transduction pathways have not been reported. Therefore, we examined the effects of various pharmacological inhibitors of ROS generating pathways, such as NOX, xanthine oxidase, arachidonic acid metabolism, and the mitochondrial electron transport chain, on IL-4-induced IL-6 expression in HAEC. We used apocynin and allopurinol to selectively inhibit NOX and xanthine oxidase. As inhibitors of arachidonic acid metabolism, we used ETYA as a combined COX and LOX inhibitor, NDGA as a specific inhibitor of 5-LOX, and NS-398 as a selective inhibitor of COX-2. Additionally, we used rotenone as an inhibitor of complex I which blocks the electron flow from NADH dehydrogenase (complex I) to ubiquinone. TTFA was used as a complex II inhibitor which interferes with the electron transport from succinate dehydrogenase (complex II) to ubiquinone. We also used antimycin A to selectively inhibit the electron flow at complex III. Among these ROS generating pathways, the present study demonstrated that the inhibition of NOX by apocynin significantly attenuated IL-4-induced ROS generation and IL-6 expression in HAEC while it was not affected by inhibiting other ROS generating pathways, such as xanthine oxidase, arachidonic acid metabolism, and the mitochondrial electron transport chain. The potential role of NOX in IL-4-induced IL-6 expression was also confirmed by pre-treatment of HAEC with DPI, a structurally unrelated NOX inhibitor. Even though pharmacological inhibitors of NOX have been widely used in a number of previous in vitro and in vivo studies [17,36,48], it is also known that they are not highly selective or specific. In the present study, in addition to the pharmacological approaches using NOX inhibitors such as apocynin and DPI, genetic approach using the mice with targeted disruption of NOX gp91phox (B6.129S6-Cybbtm1Din/J) further provided robust evidence in support of a central role for NOX in IL-4-induced IL-6 expression in vascular endothelium. These novel findings suggest that at least one distinct ROS generating pathway, such as NOX, mediates IL-6 induction in human aortic endothelial cells and mouse aortas by IL-4.
In conclusion, we have demonstrated that IL-4 significantly up-regulates mRNA and protein expression of IL-6 in vascular endothelium. Additionally, the present study has provided the first novel evidence indicating that NADPH oxidase plays a pivotal role in IL-4-induced ROS generation and IL-6 expression in vitro and in vivo. The present study will contribute to a better understanding of the molecular signaling mechanisms by which IL-4 mediates endothelial dysfunction and development of atherosclerosis. It will also provide insights to novel therapeutic approaches for atherosclerosis specifically targeted against pro-oxidative and pro-inflammatory pathways in vascular endothelium.
Acknowledgments
This study was supported in part by grants from National Institutes of Health/National Heart, Lung, and Blood Institute (HL085229) and National Science Foundation Macromolecular Interfaces with Life Sciences-Integrative Graduate Education and Research Traineeship (MILES-IGERT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–81. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 2.Ohta H, Wada H, Niwa T, Kirii H, Iwamoto N, Fujii H, et al. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180:11–7. doi: 10.1016/j.atherosclerosis.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 3.Barks JL, McQuillan JJ, Iademarco F. TNF-α and IL-4 synergistically increase vascular cell adhesion molecule-1 expression in cultured vascular smooth muscle cells. J Immunol. 1997;159:4532–87. [PubMed] [Google Scholar]
- 4.Blease K, Seybold J, Adcock IM, Hellewell PG, Burke-Gaffney A. Interleukin-4 and lipopolysaccharide synergize to induce vascular cell adhesion molecule-1 expression in human lung microvascular endothelial cells. Am J Respir Cell Mol Biol. 1998;18:620–30. doi: 10.1165/ajrcmb.18.5.3052. [DOI] [PubMed] [Google Scholar]
- 5.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galéa P, Thibault G, Lacord M, Bardos P, Lebranchu Y. IL-4, but not tumor necrosis factor-alpha, increases endothelial cell adhesiveness for lymphocytes by activating a cAMP-dependent pathway. J Immunol. 1993;151:588–96. [PubMed] [Google Scholar]
- 7.Hong HY, Lee HY, Kwak W, Yoo J, Na MH, So IS, et al. Phage display selection of peptides that home to atherosclerotic plaques: IL-4 receptor as a candidate target in atherosclerosis. J Cell Moll Med. 2008;12:2003–14. doi: 10.1111/j.1582-4934.2008.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang H, Lavoie-Lamoureux A, Moran K, Lavoie JP. IL-4 stimulates the expression of CXCL-8, E-selectin, VEGF, and inducible nitric oxide synthase mRNA by equine pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1147–54. doi: 10.1152/ajplung.00294.2006. [DOI] [PubMed] [Google Scholar]
- 9.King VL, Szilvassy SJ, Daugherty A. Interleukin-4 deficiency decreases atherosclerotic lesion formation in a site-specific manner in female LDL receptor −/− mice. Arterioscler Thromb Vasc Biol. 2002;22:456–61. doi: 10.1161/hq0302.104905. [DOI] [PubMed] [Google Scholar]
- 10.Lee YW, Eum SY, Chen KC, Hennig B, Toborek M. Gene expression profile in interleukin-4-stimulated human vascular endothelial cells. Mol Med. 2004;10:19–27. doi: 10.2119/2004-00024.lee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee YW, Hennig B, Toborek M. Redox-regulated mechanisms of interleukin-4-induced MCP-1 expression in human vascular endothelial cells. Am J Physiol Heart Circ Physiol. 2003;284:H185–92. doi: 10.1152/ajpheart.00524.2002. [DOI] [PubMed] [Google Scholar]
- 12.Lee YW, Hirani AA. Role of interleukin-4 in atherosclerosis. Arch Pharm Res. 2006;29:1–15. doi: 10.1007/BF02977462. [DOI] [PubMed] [Google Scholar]
- 13.Lee YW, Kühn H, Hennig B, Neish AS, Toborek M. IL-4-induced oxidative stress upregulates VCAM-1 gene expression in human endothelial cells. J Mol Cell Cardiol. 2001;33:83–94. doi: 10.1006/jmcc.2000.1278. [DOI] [PubMed] [Google Scholar]
- 14.Lee YW, Kühn H, Kaiser S, Hennig B, Daughterty A, Toborek M. Interleukin 4 induces transcription of the 15-lipoxygenase I gene in human endothelial cells. J Lipid Res. 2001;42:783–91. [PubMed] [Google Scholar]
- 15.Masinovsky B, Urdal D, Gallatin WM. IL-4 acts synergistically with IL-1β to promote lymphocyte adhesion to microvacular endothelium by induction of vascular cell adhesion molecule-1. J Immunol. 1990;145:2886–95. [PubMed] [Google Scholar]
- 16.Paul WE. Interleukin-4: a prototypic immunoregulatory lymphokine. Blood. 1991;77:1859–70. [PubMed] [Google Scholar]
- 17.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2− and systolic blood pressure in mice. Circ Res. 2001;89:408–11. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 18.Rocken M, Racke M, Shevach EM. IL-4-induced immune deviation as antigen-specific therapy for inflammatory autoimmune disease. Immunol Today. 1996;17:225–31. doi: 10.1016/0167-5699(96)80556-1. [DOI] [PubMed] [Google Scholar]
- 19.Rollins BJ, Pober JS. Interleukin-4 induces the synthesis and secretion of MCP-1/JE by human endothelial cells. Am J Pathol. 1991;138:1315–9. [PMC free article] [PubMed] [Google Scholar]
- 20.Walch L, Massade L, Dufilho M, Brunet A, Rendu F. Pro-atherogenic effect of interleukin-4 in endothelial cells: modulation of oxidative stress, nitric oxide and monocyte chemoattractant protein-1 expression. Atherosclerosis. 2006;187:285–91. doi: 10.1016/j.atherosclerosis.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 21.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–72. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 22.Ross R. Atherosclerosis is an inflammatory disease. Am Heart J. 1999;138:S419–20. doi: 10.1016/s0002-8703(99)70266-8. [DOI] [PubMed] [Google Scholar]
- 23.Kishikawa H, Shimokama T, Watanabe T. Localization of T lymphocytes and macrophages expressing IL-1, IL-2 receptor, IL-6 and TNF in human aortic intima: role of cell mediated immunity in human atherogenesis. Virchows Arch A Pathol Anat Histopathol. 1993;423:433–42. doi: 10.1007/BF01606532. [DOI] [PubMed] [Google Scholar]
- 24.Seino Y, Ikeda U, Ikeda M, Yamamoto K, Misawa Y, Hasegawa T, et al. Interleukin 6 gene transcripts are expressed in human atherosclerotic lesions. Cytokine. 1994;6:87–91. doi: 10.1016/1043-4666(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 25.Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19:2364–7. doi: 10.1161/01.atv.19.10.2364. [DOI] [PubMed] [Google Scholar]
- 26.Sukovich DA, Kauser K, Shirley FD, DelVecchio V, Halks-Miller M, Rubanyi GM. Expression of interleukin-6 in atherosclerotic lesions of male apoE-knockout mice. Arterioscler Thromb Vasc Biol. 1998;18:1498–505. doi: 10.1161/01.atv.18.9.1498. [DOI] [PubMed] [Google Scholar]
- 27.Toborek M, Lee YW, Kaiser S, Hennig B. Measurement of inflammatory properties of fatty acids in human endothelial cells. Methods Enzymol. 2002;352:198–219. doi: 10.1016/s0076-6879(02)52020-6. [DOI] [PubMed] [Google Scholar]
- 28.Deng X, Li H, Tang YW. Cytokine expression in respiratory syncytial virus-infected mice as measured by quantitative reverse-transcriptase PCR. J Virol Methods. 2003;107:141–6. doi: 10.1016/s0166-0934(02)00211-2. [DOI] [PubMed] [Google Scholar]
- 29.Lee YW, Lee WH. Protective effects of genistein on pro-inflammatory pathways in human brain microvascular endothelial cells. J Nutr Biochem. 2008;19:819–25. doi: 10.1016/j.jnutbio.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Kreft M, Milisav I, Potokar M, Zorec R. Automated high throughput colocalization analysis of multichannel confocal images. Comput Methods Programs Biomed. 2004;74:63–7. doi: 10.1016/S0169-2607(03)00071-3. [DOI] [PubMed] [Google Scholar]
- 32.Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999;13:1231–8. [PubMed] [Google Scholar]
- 33.Gimbrone MA, Bevilacqua MP, Cybulsky MI. Endothelial-dependent mechanisms of leukocyte adhesion in inflammation and atherosclerosis. Ann NY Acad Sci. 1990;598:77–85. doi: 10.1111/j.1749-6632.1990.tb42279.x. [DOI] [PubMed] [Google Scholar]
- 34.Iseki A, Kambe F, Okumura K, Niwata S, Yamamoto R, Hayakawa T, et al. Pyrrolidine dithiocarbamate inhibits TNF-α-dependent activation of NF-κB by increasing intracellular copper level in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2000;276:88–92. doi: 10.1006/bbrc.2000.3452. [DOI] [PubMed] [Google Scholar]
- 35.Tipoe GL, Leung TM, Hung MW, Fung ML. Green tea polyphenols as an anti-oxidant and anti-inflammatory agent for cardiovascular protection. Cardiovasc Hematol Disord Drug Targets. 2007;7:135–44. doi: 10.2174/187152907780830905. [DOI] [PubMed] [Google Scholar]
- 36.Basta G, Lazzerini G, Del Turco S, Ratto GM, Schmidt AM, De Caterina R. At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. Arterioscler Thromb Vasc Biol. 2005;25:1401–7. doi: 10.1161/01.ATV.0000167522.48370.5e. [DOI] [PubMed] [Google Scholar]
- 37.Oltman CL, Kane NL, Miller FJ, Jr, Spector AA, Weintraub NL, Dellsperger KC. Reactive oxygen species mediate arachidonic acid-induced dilation in porcine coronary microvessels. Am J Physiol Heart Circ Physiol. 2003;285:H2309–15. doi: 10.1152/ajpheart.00456.2003. [DOI] [PubMed] [Google Scholar]
- 38.The American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2009 update. Circulation. 2009;119:e1–161. doi: 10.1161/CIRCULATIONAHA.108.191259. [DOI] [PubMed] [Google Scholar]
- 39.Hennig B, Toborek M, McClain CJ, Diana JN. Nutritional implications in vascular endothelial cell metabolism. J Am Coll Nutr. 1996;15:345–58. doi: 10.1080/07315724.1996.10718609. [DOI] [PubMed] [Google Scholar]
- 40.Guzik TJ, Sadowski J, Guzik B, Jopek A, Kapelak B, Przybylowski P, et al. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler Thromb Vasc Biol. 2006;26:333–9. doi: 10.1161/01.ATV.0000196651.64776.51. [DOI] [PubMed] [Google Scholar]
- 41.Sorescu D, Weiss D, Lassegue B, Clempus RE, Szocs K, Sorescu GP, et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–35. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- 42.Hennig B, Chow CK. Lipid peroxidation and endothelial cells injury: implication in atherosclerosis. Free Radical Biol Med. 1988;4:99–106. doi: 10.1016/0891-5849(88)90070-6. [DOI] [PubMed] [Google Scholar]
- 43.Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2008;10:1713–65. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- 44.Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673–8. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- 45.Yla-Herttuala S. Gene expression in atherosclerotic lesions. Hertz. 1992;17:270–6. [PubMed] [Google Scholar]
- 46.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 47.Brinckmann R, Topp MS, Zalan I, Heydeck D, Ludwig P, Kühn H, et al. Regulation of 15-lipoxygenase expression in lung epithelial cells by interleukin-4. Biochem J. 1996;318:305–312. doi: 10.1042/bj3180305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bedard K, Krause K. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]