

Abstract
Many cancers are characterized by changes in protein phosphorylation as a result of kinase dysregulation. Disruption of Abl kinase signaling through the Philadelphia chromosome (causing the Bcr-Abl mutation) in chronic myeloid leukemia (CML) has provided a paradigm for development of kinase inhibitor drugs such as the specific inhibitor imatinib (also known as STI571 or Gleevec). However, since patients are treated indefinitely with this drug to maintain remission, resistance is increasingly becoming an issue. While there are many ways to detect kinase activity, most lack the ability to ‘multiplex’ the analysis (to detect more than one substrate simultaneously). Here we report a novel biosensor for detecting Abl kinase activity and sensitivity to inhibitor in live, intact cells overexpressing a CML model Abl kinase construct. This straightforward methodology could eventually provide a new tool for detecting kinase activity and inhibitor drug response in cancer cells that overexpress oncogenic kinases.
Keywords: biosensor, kinase, peptide, assay, chronic myelogenous leukemia, Gleevec, biotechnology
Introduction
Protein phosphorylation plays a major role in the mechanisms underlying control of cell cycle, differentiation and development. Consequently, disruption of these processes can lead to cell cycle dysregulation and uncontrolled cellular proliferation, resulting in malignancy. c-Abl is a non-receptor tyrosine kinase that is critical to normal function of cells in many tissues, but is particularly important to growth and differentiation of hematopoietic cells. Even though it is relatively ubiquitously expressed, in normal cells Abl is very tightly regulated, particularly via autoinhibition through intramolecular contacts between a) its protein-protein interaction domains (SH3 and SH2) and ligand sequences elsewhere in the protein, and b) interaction of an N-terminal myristoyl group with a binding pocket in the protein.(1) This helps maintain low activity of the kinase at normal endogenous levels until its role in specific mechanisms is activated at the right time and place by other events in the cell. As a result, changes in its structure or expression level can cause abnormal Abl kinase activity and lead to blood cancers such as chronic myelogenous leukemia (CML).(2, 3) CML is characterized by the expression of a Bcr-Abl fusion gene which results from reciprocal translocation of chromosomes 9 and 22, giving rise to the Philadelphia chromosome.(4) The Bcr-Abl fusion protein features replacement of some of the autoinhibitory portions of c-Abl with the amino-terminal domain of Bcr (which promotes oligomerization and thus autophosphorylation), resulting in disruption of its regulation and constitutive Abl kinase activity.(5, 6)
The direct involvement of Bcr-Abl in the pathology of CML created a paradigm for small molecule inhibitors of kinases as potential anti-cancer therapeutics. Imatinib mesylate (Gleevec or STI571) inhibits Bcr-Abl kinase activity(7) and induces remission in patients in the early stages of CML, providing a blockbuster “magic bullet” for CML and changing global directions in cancer research. Unfortunately, many patients develop imatinib resistance, usually because they acquire mutations in the kinase domain of Bcr-Abl that abrogate imatinib binding.(8, 9) However, ‘off-target’ resistance is also seen, where even though Bcr-Abl itself still responds to other signaling pathways are upregulated to compensate for Bcr-Abl inhibition and promote cell survival.(10, 11) The resistance to imatinib is normally not clinically detected until the recurring disease has progressed into blast crisis, particularly when ‘off-target’ mechanisms are present that cannot be observed using the simple rt-PCR test for mutations in the kinase domain. Therefore there is a major need to monitor kinase activity in the contexts of these resistance mechanisms to guide the development of next-generation inhibitors or alternative chemotherapies.
Most types of kinase assays either do not detect the activity of the kinases in intact cells or cannot be multiplexed to detect more than one activity at a time in the same cell. Previous use of a TAT-labeled peptide substrate demonstrated high specificity for detecting activation of Akt in single, live cells using a capillary electrophoresis detection technique.(12) This technique was very specific and extremely sensitive, and provided novel access to kinase activation information inside the cell. Other modern kinase biosensors can provide exquisite specificity and sensitivity in real-time assays, but are either inappropriate for use in whole, live cells (e.g. homogeneous fluorescence-based assays(13, 14)) or require transfection and expression of biosensor proteins (e.g. FRET biosensors(15-17)). Here we begin to address some of these issues by delivering an artificial peptide-based substrate selective for Abl kinase, purifying the peptide from lysed cells using an affinity tag and detecting the kinase activity using MALDI-TOF mass spectrometry to monitor phosphorylation. Ultimately, our goal is to develop a modular biosensor methodology that will provide access to quantifiable cellular signaling information about inhibitor drug response for many kinases at the same time in living cells. Mass spectrometry provides an ideal detection method for this, since peaks for multiple peptides can be detected within the same sample in the same analysis. Using the assay described here, we can detect intracellular c-Abl kinase activation and sensitivity to imatinib in a workflow that is amenable to multiplexed adaptation.
Materials and Methods
Cell Culture
HEK293 FKBP-Abl cells (provided by the laboratory of Prof. Jean Y. Wang, UCSD-Moores Cancer Center)(18) were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% (v/v) heat-inactivated fetal bovine serum, 1% (v/v) penicillin/streptomycin and 2 mM L-glutamine in a CO2-containing (5%) humidified environment at 37 °C.
Peptide synthesis and purification
Amino acids and other peptide synthesis reagents were obtained from Peptides International (Louisville, KY), except for the Fmoc-Biocytin (Kbiotin) (obtained from Akaal Organics, Long Beach, CA) and the photocleavable residue (3-(2-nitrobenzyl)-3-aminopropionic acid), which was purchased from Lancaster Synthesis and Fmoc-protected by Dr. J. Thomas Ippoliti's lab at the University of St. Thomas (St. Paul, MN). Peptide was synthesized at 50 μmol scale on CLEAR-Amide resin (mixed with glass beads to avoid clumping) using solid-phase Fmoc chemistry (Fmoc-protected amino acid monomers at 100 mM final concentration) with a Prelude Parallel Peptide Synthesizer (Protein Technologies, Tucson, AZ, USA), using ‘fast’ protocols (coupling time: 10 min, deprotection time: 2 × 2 min) with HCTU/NMM (95 mM/200 mM final concentrations) as coupling reagent. Peptides were analyzed with HPLC/MS (Accela/LTQ, Thermo-Finnegan) and MALDI TOF/TOF (Voyager 4800, Applied Biosystems Foster City, CA, USA) and purified using a C18 reverse-phase column on Agilent Technologies 1200 Series Preparative HPLC system (Santa Clara, CA, USA). See supporting information for LC/MS analysis of the purified peptide.
A portion of the peptide was labeled with Alexafluor-555-maleimide (Invitrogen) in 6 M guanidinium-HCl, 100 mM phosphate buffer at pH 6.5. Reaction progress was checked by MALDI-TOF MS and found to be complete after 30 min. The labeled peptide was purified using a C18 Maxi-Clean cartridge (500 mg, Alltech Associates) and lyophilized. See supporting information for LC/MS analysis of labeled peptide.
Peptide uptake experiment
Uninduced HEK293 cells were plated in 24-well plates at a density of 0.5 × 106 cells/ml and allowed to recover for 24 h. Wells were treated with Alexafluor-555-labeled biosensor peptide for 15 min or 4 h. Cells were treated with Hoechst 33342 (1 ng/ml) either before peptide treatment (for 15 min timepoint) or during the final 30 min of incubation (for 4 h timepoint). Wells were washed gently with PBS (3 × 1 ml), covered in PBS (500 μl) and imaged using a Zeiss Axiovert fluorescence microscope (20× objective, using phase contrast, DAPI and rhodamine filters).
Biosensor assay
FKBP-Abl cells were plated at 2.5 × 106 cells/mL (1 ml/well), allowed to recover for 48h and FKBP-Abl expression was induced (doxycycline, 2 μM, 18h). As a control, some wells were left uninduced. The biosensor peptide (1-100 μM) and activating dimerizer AP20187 (10 nM) were added in fresh medium with or without imatinib (10 μM). Additional controls (FKBP-Abl expression and activation as described above, no peptide treatment) were performed for determining fold change as a result of peptide treatment. Cells were then incubated for 4 h at 37°C, collected, washed once with phosphate-buffered saline (PBS) and flash frozen in PhosphoSafe Extraction Reagent (Novagen, San Diego, CA) containing freshly dissolved complete protease inhibitor (Roche, NJ).
Approximately 100 μg total protein was treated with UV light (302 nm) for 5 min to cleave the biotinylated substrate portion from the rest of the molecule, followed by capture of the segment of interest via the biotin group using Streptavidin-coated MagneSpheres (20 μL, Promega, WI) in a MagnaBot 96-well magnetic capture device (Promega). Beads were pre-washed in 0.1% octylgucoside/PBS (3 × 150 μl), incubated with lysate for 15-60 min on a short-radius plate shaker (400 rpm), washed again with 0.1% octylgucoside/PBS (3 × 150 μl) and rinsed with ddH2O (6 × 150 μl) to remove salts. The peptide was eluted from the beads using MALDI-TOF sample preparation buffer (20 μL) containing acetonitrile/water/trifluoroacetic acid (ACN/H2O/TFA) (75/25/0.1%), collected by magnetic separation and co-spotted on a MALDI target with the matrix solution (alpha-cyano-4-hydroxycinnamic acid, CHCA), in ACN/H2O/TFA (50/50/0.1%). Samples were analyzed using a Voyager 4800 MALDI-TOF/TOF (Applied Biosystems) in linear positive and negative modes and the relative levels of phosphopeptide signal quantified as previously described.(19) Data plotted are from a combination of biological (n≥3) and technical (three spots) replicate analyses. One-sample t-tests were used to determine statistical significance.
Western blot analysis
A separate portion of the lysates (200 μg protein per sample) was denatured with NuPAGE Laemmli protein gel loading buffer (Invitrogen, Carlsbad, CA, USA) and run on a 4-12% Bis-Tris NuPAGE gel (Invitrogen). Proteins were transferred to nitrocellulose membranes (BioRad) for 18h at 15 V at 4 °C. Membranes were blocked in 5% milk/TBS-T for 1h at r.t. and cut into two segments: >15 kD, containing endogenous proteins, and <15 kD, containing biosensor peptide.
The >15 kD segment was analyzed with immunoblotting using a cocktail of antibodies for phosphorylation of c-Abl (Y245) (1:1000, rabbit) (Cell Signaling Technologies), STAT5 (Y695) (1:5000, rabbit) and Crkl (Y207) (1:1000, rabbit) (both Abcam). The antibody cocktail included either anti-tubulin (1:100,000, rat) (Millipore) or anti-elF4 (1:1000, rabbit) (Cell Signaling Technologies) as loading controls. Membranes were incubated with the cocktail for 24 h at 4 °C. The <15 kD segment was analyzed with immunoblotting using a cocktail of antiphosphotyrosine antibody 4G10 (1:1000, mouse) (Millipore) and streptavidin labeled with DyLight-649 (1:1000) (Pierce, Rockford, IL), incubated for 24 h at 4 °C.
Membranes were washed (3 × 5 min/ TBS-T) and visualized by incubating for 1h at room temperature with goat-derived secondary antibodies (1:10,000 for anti-mouse and anti-rabbit and 1:20,000 for anti-rat in 3% milk/TBS-T) tagged with IRDye-680 or 800 (LiCor) and scanned using a LiCor Odyssey infrared scanner. Band densities from scan images were determined using QuantityOne software (BioRad). pAbl(Y245), pStat5(Y695) and pCrkL(Y207) band densities were analyzed from the same blot, divided by loading control and normalized to the ‘no peptide’ control lane to obtain the fold change. Bands from the ‘no induction’ blots were normalized to the ‘no peptide’ control lane in the ‘induced’ experiment (15 min peptide treatment) to show the lower levels of protein phosphorylation seen when FKBP-Abl is not induced and activated. Data were plotted in GraphPad Prism from biological replicate analyses (n≥3). One-sample t-tests were used to determine statistical significance.
Results and discussion
The biosensor peptide substrate was synthesized using solid-phase peptide chemistry to contain the following functional modules (Fig. 1): a highly specific kinase substrate for c-Abl,(20) a photocleavable linker to release the substrate sequence for analysis (21), a Src-homology 3 (SH3)-binding ligand,(22) to enhance interaction with the kinase(23, 24), a TAT peptide to aid intracellular delivery of cargo across the plasma membrane, (12, 25) and a biotin tag to facilitate isolation of the peptide from the complex biological mixture of cell lysate. See supporting information for analytical data. A portion of the peptide was also functionalized with Alexafluor-555-maleimide for confirmation of intracellular uptake and distribution using fluorescence microscopy.
Figure 1. A cartoon representation of the peptide-based biosensor for the c-Abl kinase.
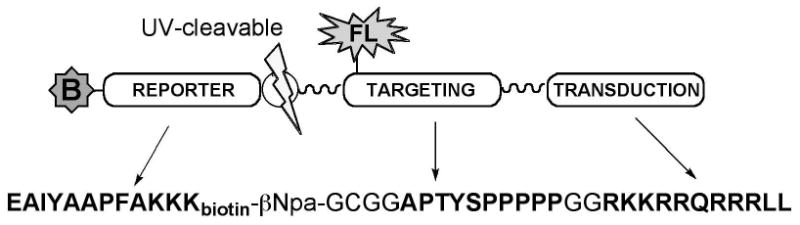
The substrate contains a biotinylated “reporter” module that is phosphorylated by c-Abl; a “targeting” module binds the Abl SH3 domain; and a “transduction” module (the TAT peptide), to aid the peptide in crossing the membrane barrier. The reporter and targeting sequences are connected via a photocleavable linker to allow release of the reporter from the backbone of the biosensor.
To develop this method for detecting Abl activity in intact cells we used an engineered CML model cell line: FKBP-Abl, a TET-On inducible, selectively activatable construct of Abl kinase overexpressed in HEK293 cells.(18) In these cells, the Abl kinase is fused to an FKBP-like domain, which is dimerized by an FKBP-binding molecule, AP20187, resulting in trans-autophosphorylation and kinase activation that mimics the oligomerization-based constitutive activation of Bcr-Abl. This overexpression system serves two purposes: it functions as a model for Bcr-Abl overexpression in leukemia, and also provides correlative evidence that the Abl kinase is responsible for phosphorylating the peptide biosensor in a cellular context. Engineered overexpression systems are commonly used perform experiments to demonstrate signaling specificity towards endogenous substrates, and thus enable us to make reasonable conclusions about the specificity of the biosensor developed here before moving on to apply this biosensor to non-engineered systems such as cancer cell lines. Phosphorylation of the biosensor peptide was tested with and without overexpression of the construct, in the presence and absence of imatinib in order to correlate observed phosphorylation of the biosensor peptide with activation of the engineered kinase.
Uptake and distribution of the biosensor peptide were assessed using fluorescence microscopy. HEK293 cells were treated with the biosensor peptide carrying an Alexafluor-555 label (25 μM). Uptake was rapid (strong signal observed within 30 min), consistent with what is known about transduction and distribution of macromolecules via the TAT peptide.(26) Peptide persisted through at least 4 h of treatment (Fig. 2). In these cells the peptide was mainly localized in endocytic-like compartments outside the nucleus, as expected from recent literature studies of TAT-mediated uptake.(26) Since the peptide remains concentrated in these compartments for up to four hours (which also results in the highest levels of phosphorylation), it appears that kinase/substrate contact is largely occurring at these structures. Given that phosphorylation levels are relatively high even with the peptide concentrated in these compartments, it is possible that either the substrate portion of the peptide is displayed on the cytoplasmic surface of the vesicles (allowing contact with free cytoplasmic Abl kinase construct) or that the overexpressed Abl kinase associates with the trafficking machinery around these vesicles. c-Abl has recently been shown to play a role in B-cell receptor endocytosis,(27) therefore it is possible that the overexpressed Abl construct participates in trafficking in these cells as well. More comprehensive imaging studies are ongoing to examine this observation, including the interaction between the peptide and the kinase, the biological relevance of the localization and the potential role and/or association of Abl kinase in this specific intracellular distribution.
Figure 2. Cellular uptake and distribution of substrate peptide.
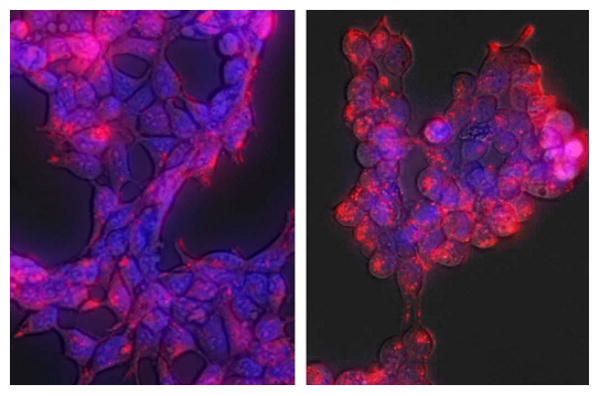
HEK293 cells were treated with Alexafluor-555-labeled Abl biosensor peptide (red, 25 μM) for 15 min (left panel) or 4 h (right panel) at 37 °C in media. Nuclei in the live cells were stained with Hoechst 33342 (blue). Cells were washed with PBS and visualized using a Zeiss Axiovert fluorescence microscope within 5 min. Peptide was rapidly taken up and distributed throughout the cell (15 min), persisting up to 4 h.
To test intracellular phosphorylation of the biosensor peptide, FKBP-Abl cells were treated for 18 h with doxycycline to induce overexpression of the FKBP-Abl protein or left uninduced as a control (which would rely on endogenous levels of Abl or other Abl-related proteins such as Arg to observe phosphorylation of the substrate). Both induced and uninduced cells were then treated with the activating dimerizer AP20187 (10 nM) as well as the biosensor peptide at a range of concentrations (1, 25, 50, 75 and 100 μM) for 15 min and 4 h. After lysing the cells, phosphorylation of the biosensor peptide was analyzed by Western blot and MALDI-TOF MS. For Western blot analysis, peptide was observed between the lowest MW marker and the dye front (as verified by streptavidin binding to the biotinylated peptide, Fig. S4, supporting information). Phosphorylation was detected with antiphosphotyrosine antibody 4G10 and total peptide confirmed and estimated by streptavidin reactivity. For MALDI-TOF MS analysis, peptide was captured from cell lysates (after UV treatment to release the ‘reporter’ segment) in a 96-well plate format using streptavidin conjugated to superparamagnetic nanoparticles and analyzed in both positive and negative linear modes with MALDI-TOF MS. As commonly seen for MALDI-TOF MS and previously observed with similar peptides, the phosphopeptide ionized preferentially in negative mode.(19) Signal from unphosphorylated peptide (EAIYAAPFAKKKbG) was used as an internal standard and relative conversion was estimated by quantifying the peak integrations for the unphosphorylated and phosphorylated species from multiple experimental replicates, as described by us previously.(19)
Overexpression and activation of FKBP-Abl protein resulted in phosphorylation of the peptide after as little as 15 min. Phosphorylation was increased with 4 h of incubation. After the shorter 15 min incubation time, phosphopeptide was best detected using Western blot under our current assay conditions. By 4 h of incubation, however, robust levels of phosphopeptide were observed by MALDI-TOF MS (Fig. 3). The unmodified peptide could be detected by MALDI-TOF following the 15 min incubation time, however the low relative conversion of peptide after that shorter time in this cell model meant that the phosphopeptide was less robustly observable by MS for that timepoint. For MALDI-TOF MS analysis (which can only be quantified in a relative manner), both the unphosphorylated and phosphorylated peptide peaks were detected together (Fig. 3, example spectra) and the sensitivity was determined by the most intense peak in the spectrum. This means that MALDI-TOF MS is not very sensitive for detecting less than ∼1-5% phosphorylation, since the phosphorylated peak is not reliably observed below those levels, and indicates that at 15 min, whether the FKBP-Abl protein expression and activation is induced or not, the conversions are likely below 5%. Western blotting is more sensitive for detecting phosphorylation at low levels of conversion since the 4G10 antibody detects total phosphopeptide regardless of ratio to unphosphorylated peptide. However, Western blotting is lower-throughput than MALDI-TOF MS. Also, the antibody concentrations make it more expensive overall for large numbers of analyses, making it less desirable as a detection method for screening assays. Whereas ELISA-type formats would improve throughput and cost for antibody-based methods, these label-dependent readouts are not as flexible for detection of multiple biosensors at a time. MALDI-TOF MS has nearly unlimited flexibility in this respect. Even though the shorter incubation time did not allow for sensitive, robust detection under these current conditions in this cell system, 4 h of incubation was sufficient to achieve reproducible detection of phosphopeptide by MALDI-TOF MS. These technical limitations will be addressed in future refinements of this assay for other cell systems. Further optimization of peptide affinity capture and enrichment, including separation of the phosphorylated peptide from the unphosphorylated material, will facilitate more robust detection of peptide phosphorylation. Miniaturization of the detection format (to highly concentrate the enriched peptide) for better detection of substrate at lower treatment concentrations (<1 μM) will allow higher relative conversions of the substrate and therefore improve phosphopeptide sensitivity.
Figure 3. Analysis of peptide phosphorylation by Western blot and MALDI-TOF MS.
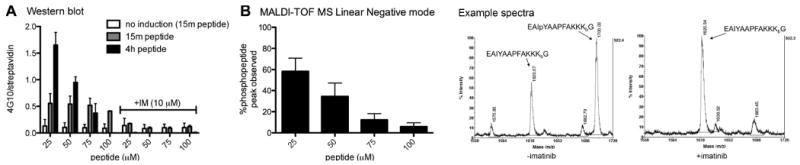
The engineered HEK293 cell line stably expressing a Tet-ON FKBP-Abl construct was plated at 2.5 × 106 cells per well in a six-well plate. Cells were allowed to grow to confluency (48 h) and treated with 2 μM doxycycline for 18h to induce the expression of FKBP-Abl. Abl-activating dimerizer AP20187 (10 nM) and peptide (at the indicated concentrations) were added and cells were incubated at 37 °C for 15 min and 4 hrs (with and without imatinib, 10 μM). A: Western blot analysis: Cell lysates were run on SDS-PAGE (200 μg/lane, 4-12% Bis-Tris gel) and biotinylated peptide (streptavidin-DyLight649) and phosphotyrosine (antiphosphotyrosine antibody 4G10) were detected with immunoblotting. Data are shown as phosphotyrosine signal divided by total peptide (4G10/streptavidin). B: MALDI-TOF analysis: Cell lysates (100 μg) were treated with UV light (302 nm) for 5 min. Peptide was isolated from the lysate via affinity capture with Tetralink Avidin beads using a 96-well filter plate. Peptides were eluted using 50/50/0.1% acetonitrile/H2O/TFA and analyzed using MALDI-TOF MS in linear negative mode. Example spectra: doxycycline-induced and AP20187-treated cells, incubation with 25 μM peptide for 4 h, +/- imatinib (10μM). See supporting information for representative blots and more MALDI spectra for these data.
To demonstrate specificity, when peptide was added to cells that had not been treated with doxycycline to induce overexpression of the FKBP-Abl kinase (‘uninduced’), little to no phosphorylation was seen by either MALDI-TOF MS or Western blot. This means that endogenous levels of Abl and/or Arg kinases were not sufficiently activated to appreciably phosphorylate this substrate under our conditions. Therefore, since phosphorylation of the substrate is so strongly correlated with overexpression and activation of the engineered construct, we conclude that other endogenous kinases are unlikely to contribute to the activity observed. Also, phosphorylation was completely inhibited by imatinib under both conditions, providing further support that the biosensor is relatively specific for the Abl kinase since the cells used in this engineered system do not feature overexpression or abnormally high activation of related imatinib targets. There was a substrate inhibition effect at ≥50 μM peptide, however concentrations below 50 μM did not seem to give rise to substrate inhibition at the times tested here. Therefore lower concentrations are preferable not only because they consume less material and facilitate higher relative conversion levels (and thus sensitivity for detection of phosphopeptide), but also because they avoid disruption of the kinase activity.
Whereas we showed that active Abl kinase in the engineered cells is the most likely protein responsible for phosphorylating the biosensor peptide, it was also important to determine the effects of the peptide on the phosphorylation of native Abl substrates upon activation of the overexpressed construct. The biosensor peptide contains an SH3-binding ligand module. Since SH3 domain interactions are important for regulating the activity of Abl kinase and its association with its substrates,(28) disruption of SH3-domain-related kinase/substrate interactions was a concern. In order to assess artifactual signaling alteration via this domain or other portions of the peptide, we looked for concentration-related effects of the peptide on some endogenous c-Abl substrates: Abl autophosphorylation (at Y245 in the activation loop); Stat5 at Y695 and CrkL at Y207 (both CML-related Bcr-Abl substrates in vivo). Western blot for these endogenous phosphorylation sites (Fig. 4) showed that increasing concentrations (>50 μM) of this biosensor had a significant effect on autophosphorylation of Abl (Y245) (p<0.01) upon extended treatment (4 h), however there was no significant effect on the phosphorylation of the other substrates. Neither Abl autophosphorylation nor substrate phosphorylation were significantly affected by lower amounts of peptide (≤ 25 μM). Therefore this strategy should be useful for detecting biologically relevant Abl kinase activation.
Figure 4. Assessing disruption of endogenous Abl signaling.
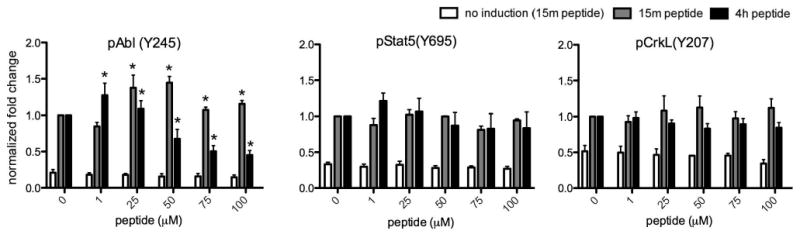
The cells were treated as described in Fig. 3. Lysates were analyzed using SDS PAGE and Western blot (200 μg protein per lane) for pAbl(Y245), pStat5(Y695), pCrkL(Y207) (endogenous Abl sites) and eIF4 or tubulin (loading control). Data shown (n≥3) for each phosphoprotein come from the same blot and are normalized to loading control and represented as fold change from internal control ([peptide] = 0, treated with doxycycline and AP20187 as described in Experimental Methods). *Indicates significant differences from control (p<0.01). See supporting information for representative images of Western blots for these data.
In summary, we have developed a biosensor that can detect Abl kinase overexpression/activation in live cells, and that does not depend on engineering the cells themselves to produce the sensor. We have shown that our biosensor peptide is taken up by live cells and phosphorylated in an Abl kinase-related manner. We are currently characterizing and optimizing the assay conditions, including the arrangement of the peptide modules within the biosensor, and using this sensor to develop an assay for detection of Abl kinase activation and inhibitor drug response in leukemia cell lines. We are also developing analogous biosensor substrates for other kinases related to signaling in leukemia and other cancers, in order to detect more complex profiles of inhibitor drug response in tumor cells. This concept can be extended to build substrate sensors for any enzyme that is capable of modifying a target peptide. By creating sets of biosensor peptides that can each report individual enzyme activities, we aim to develop multiplexed assays where the activation of e.g. many kinases and phosphatases within a pathway can be detected quickly and simultaneously. We believe this format will pave the way to obtaining relatively complex proteomic information about signaling in live cells.(29)
Supplementary Material
Acknowledgments
The authors thank Prof. Jean Y. Wang (UCSD-Moores Cancer Center) for the gift of the FKBP-Abl HEK293 cell line, as well as continued advice and support. We also thank Dr. Julia Kirshner (Purdue University) for microscope use and Drs. H. Charbonneau, R. Geahlen, D. Riese and V. Watts for editing and discussion during preparation of this manuscript. This work was funded by an NIH/NCI Howard Temin Pathway to Independence Award (K99CA127161A) to L.L.P., for which initial mentorship from Profs. Stephen J. Kron and Stephen B. H. Kent at the University of Chicago is very gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, Kuriyan J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–71. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 2.Leibowitz D, Young KS. The molecular biology of CML: a review. Cancer Invest. 1989;7:195–203. doi: 10.3109/07357908909038285. [DOI] [PubMed] [Google Scholar]
- 3.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–30. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 4.Bartram CR, Kleihauer E, de Klein A, Grosveld G, Teyssier JR, Heisterkamp N, Groffen J. C-abl and bcr are rearranged in a Ph1-negative CML patient. Embo J. 1985;4:683–6. doi: 10.1002/j.1460-2075.1985.tb03683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pluk H, Dorey K, Superti-Furga G. Autoinhibition of c-Abl. Cell. 2002;108:247–59. doi: 10.1016/s0092-8674(02)00623-2. [DOI] [PubMed] [Google Scholar]
- 6.Sawyers CL. Disabling Abl-perspectives on Abl kinase regulation and cancer therapeutics. Cancer Cell. 2002;1:13–5. doi: 10.1016/s1535-6108(02)00022-3. [DOI] [PubMed] [Google Scholar]
- 7.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 8.Nardi V, Azam M, Daley GQ. Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Curr Opin Hematol. 2004;11:35–43. doi: 10.1097/00062752-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 10.Wu J, Meng F, Kong LY, Peng Z, Ying Y, Bornmann WG, Darnay BG, Lamothe B, Sun H, Talpaz M, Donato NJ. Association between imatinib-resistant BCR-ABL mutation-negative leukemia and persistent activation of LYN kinase. J Natl Cancer Inst. 2008;100:926–39. doi: 10.1093/jnci/djn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahon FX, Hayette S, Lagarde V, Belloc F, Turcq B, Nicolini F, Belanger C, Manley PW, Leroy C, Etienne G, Roche S, Pasquet JM. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008;68:9809–16. doi: 10.1158/0008-5472.CAN-08-1008. [DOI] [PubMed] [Google Scholar]
- 12.Soughayer JS, Wang Y, Li H, Cheung SH, Rossi FM, Stanbridge EJ, Sims CE, Allbritton NL. Characterization of TAT-mediated transport of detachable kinase substrates. Biochemistry. 2004;43:8528–40. doi: 10.1021/bi036296d. [DOI] [PubMed] [Google Scholar]
- 13.Kristjansdottir K, Rudolph J. A fluorescence polarization assay for native protein substrates of kinases. Anal Biochem. 2003;316:41–9. doi: 10.1016/s0003-2697(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q, Lawrence DS. Phosphorylation-driven protein-protein interactions: a protein kinase sensing system. J Am Chem Soc. 2005;127:7684–5. doi: 10.1021/ja050789j. [DOI] [PubMed] [Google Scholar]
- 15.Fuller BG, Lampson MA, Foley EA, Rosasco-Nitcher S, Le KV, Tobelmann P, Brautigan DL, Stukenberg PT, Kapoor TM. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 2008;453:1132–6. doi: 10.1038/nature06923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seong J, Lu S, Ouyang M, Huang H, Zhang J, Frame MC, Wang Y. Visualization of Src activity at different compartments of the plasma membrane by FRET imaging. Chem Biol. 2009;16:48–57. doi: 10.1016/j.chembiol.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Allen MD. FRET-based biosensors for protein kinases: illuminating the kinome. Mol Biosyst. 2007;3:759–65. doi: 10.1039/b706628g. [DOI] [PubMed] [Google Scholar]
- 18.Jin H, Wang JY. Abl tyrosine kinase promotes dorsal ruffles but restrains lamellipodia extension during cell spreading on fibronectin. Mol Biol Cell. 2007;18:4143–54. doi: 10.1091/mbc.E07-01-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parker L, Engel-Hall A, Drew K, Steinhardt G, Helseth DL, Jr, Jabon D, McMurry T, Angulo DS, Kron SJ. Investigating quantitation of phosphorylation using MALDI-TOF mass spectrometry. J Mass Spectrom. 2008;43:518–27. doi: 10.1002/jms.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Songyang Z, Carraway KL, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, Lorenzo MJ, Ponder BAJ, Mayer BJ, Cantley LC. Catalytic Specificity Of Protein-Tyrosine Kinases Is Critical For Selective Signaling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 21.Parker LL, Brueggemeier SB, Rhee WJ, Wu D, Kent SB, Kron SJ, Palecek SP. Photocleavable peptide hydrogel arrays for MALDI-TOF analysis of kinase activity. Analyst. 2006;131:1097–104. doi: 10.1039/b607180e. [DOI] [PubMed] [Google Scholar]
- 22.Pisabarro MT, Serrano L. Rational design of specific high-affinity peptide ligands for the Abl-SH3 domain. Biochemistry. 1996;35:10634–40. doi: 10.1021/bi960203t. [DOI] [PubMed] [Google Scholar]
- 23.Miller WT. Determinants of substrate recognition in nonreceptor tyrosine kinases. Accounts Of Chemical Research. 2003;36:393–400. doi: 10.1021/ar020116v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu D, Nair-Gill E, Sher DA, Parker LL, Campbell JM, Siddiqui M, Stock W, Kron SJ. Assaying Bcr-Abl kinase activity and inhibition in whole cell extracts by phosphorylation of substrates immobilized on agarose beads. Analytical Biochemistry. 2005;347:67–76. doi: 10.1016/j.ab.2005.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wadia JS, Dowdy SF. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Advanced Drug Delivery Reviews. 2005;57:579–596. doi: 10.1016/j.addr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Ter-Avetisyan G, Tunnemann G, Nowak D, Nitschke M, Herrmann A, Drab M, Cardoso MC. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284:3370–8. doi: 10.1074/jbc.M805550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacob M, Todd LA, Majumdar RS, Li Y, Yamamoto KI, Pure E. Endogenous cAbl regulates receptor endocytosis. Cell Signal. 2009 doi: 10.1016/j.cellsig.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 28.Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 29.Parker LL, Kron SJ. Kinase activation in circulating cells: opportunities for biomarkers for diagnosis and therapeutic monitoring. Expert Opin Med Diagn. 2008;2:33–46. doi: 10.1517/17530059.2.1.33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.