

Abstract
The activation of STAT3 has been linked with carcinogenesis through survival, proliferation, and angiogenesis of tumor cells. Agents that can suppress STAT3 activation have potential not only for prevention but also for treatment of cancer. In the present report, we investigated whether plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), an analogue of Vitamin K and isolated from chitrak (Plumbago zeylanica), an Ayurvedic medicinal plant, can modulate the STAT3 pathway. We found that plumbagin inhibited both constitutive and IL-6-inducible STAT3 phosphorylation in multiple myeloma (MM) cells and this correlated with the inhibition of c-Src, JAK1, and JAK2 activation. Vanadate, however, reversed the plumbagin-induced downregulation of STAT3 activation, suggesting the involvement of a protein tyrosine phosphatase. Indeed, we found that plumbagin induced the expression of the protein tyrosine phosphatase, SHP-1; and silencing of the SHP-1 abolished the effect of plumbagin. This agent also downregulated the expression of STAT3-regulated cyclin D1, Bcl-xL, and VEGF, activated caspase-3, induced PARP cleavage, and increased the sub-G1 population of MM cells. Consistent with these results, overexpression of constitutive active STAT3 significantly reduced the plumbagin-induced apoptosis. When compared with AG490, a rationally designed STAT3/JAK2 inhibitor, plumbagin was found more potent in suppressing proliferation of cells. Plumbagin also significantly potentiated the apoptotic effects of thalidomide and bortezomib in MM cells. Overall, these results suggest that the plumbagin inhibits STAT3 activation pathway through induction of SHP-1 and this may mediate sensitization of STAT3 overexpressing cancers to chemotherapeutic agents.
Keywords: Plumbagin, STAT3, JAK1, JAK2, SHP-1, Apoptosis
Introduction
The modern era of cancer therapy began in 1941 with the introduction of nitrogen mustard, a chemical warfare agent, as an effective treatment for cancer, and then chemotherapeutic agents in 1971. Search for specificity and safety, led to the discovery of targeted cancer therapies, first introduced in 1991, are also toxic, lack efficacy and are highly expensive. The use of anti-cancer agents derived from traditional therapies (sometimes referred to as alternate or complementary therapies) provides a novel opportunity to improve the existing standard of care for cancer and other diseases. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone, an analogue of vitamin K3) is one such agent. Plumbagin is a naturally occurring yellow pigment found in the plants of the Plumbaginaceae, Droseraceae, Ancestrocladaceae, and Dioncophyllaceae families. The root of chitrak (Plumbago zeylanica), a major source of plumbagin, has been used in Indian medicine since the period of Charaka as an anti-atherogenic, cardiotonic, hepatoprotective, and neuroprotective agent (1). The active principle, plumbagin, is also present along with a series of other structurally related naphthoquinones in the roots, leaves, bark, and wood of Juglans regia (variously known as the English walnut, Persian walnut, and California walnut), Juglans cinerea (butternut and white walnut), and Juglans nigra (black walnut) (2, 3).
Plumbagin has been shown to exert anti-cancer activities against a wide variety of tumor cells, including breast cancer (4), lung cancer (5, 6), ovarian cancer (7), acute promyelocytic leukemia (8), melanoma (9), and prostate cancer (10, 11). How plumbagin mediates these anti cellular effects is not fully understood. It has been shown to induce G2/M cell cycle arrest through downregulation of cyclin B1, cyclin A, CDC2, and CDC25C (4, 6, 8, 9, 12). Plumbagin regarded as redox recycling quinone and induces superoxide radicals (13); inhibits AKT (4, 6), NF-κB (14), and topoisomerase II (13); downregulates the expression of survivin and EGFR (6); and induce p21 (4, 12), p53 (5), and JNK (5, 9). Plumbagin has been shown to bind NADPH oxidase (15), an estrogen-receptor-α (7) and multidrug resistance linked ATP-binding cassette drug transporter [ABCG2] (16) and inhibit their activity. In animals, plumbagin has shown to exhibit anti-cancer (5, 10, 17, 18), radiosensitizer of tumor cells (19), anti-bacterial (20), and anti-arthritic potential (21). The latter was mediated through the inhibition of neutrophil activation, collagenase activation, and angiogenesis (21). Plumbagin can also radio sensitize melanoma and cervical cancer cells (22). Because several of these effects require the activation of the transcription factor signal transducer and activator of transcription (STAT)-3, we postulated that plumbagin mediates its effects through modulation of this pathway.
STAT proteins are known to play an essential role in tumorigenesis (23). STAT3, one member of the STAT family, is often constitutively active in many human cancer cells, including multiple myeloma (MM), lymphomas, leukemia, breast cancer, prostate cancer, head and neck squamous cell carcinoma, brain tumor, colon cancer, Ewing's sarcoma, gastric cancer, esophageal cancer, ovarian cancer, nasopharyngeal cancer, and pancreatic cancer (24, 25).
Because of the critical role of STAT3 in tumor cell survival, proliferation, and angiogenesis, we hypothesized that plumbagin mediates its effects in part through modulation of the STAT3 pathway. We tested this hypothesis in MM cells. In our experiments, plumbagin indeed suppressed both constitutive and inducible STAT3 activation. This inhibition decreased gene products linked to cell survival, proliferation and angiogenesis. This correlated with suppression of proliferation, induction of apoptosis, and enhancement of the response to the cytotoxic effects of thalidomide (an inhibitor of TNF expression) and bortezomib (a proteasome inhibitor) in MM cells.
Results
The present study was undertaken to determine the effect of plumbagin on the STAT3 signaling pathway. We investigated the effect of plumbagin on both constitutive and IL-6-inducible STAT3 activation in MM cells. We also evaluated the effect of plumbagin on various mediators of cellular proliferation, cell survival, and apoptosis. The structure of plumbagin is shown in Fig. 1A. The dose and duration of plumbagin used to modulate STAT3 activity did not affect cell viability, indicating that downregulation of STAT3 was not due to cell killing (data not shown).
Figure 1. Plumbagin inhibits constitutively active STAT3 in U266 cells.

(A) The structure of plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone). (B) Plumbagin suppresses phospho-STAT3 levels in a dose and time-dependent manner. U266 cells (2 × 106) were first treated with the indicated concentrations of plumbagin for 4 h after incubation; whole-cell extracts were prepared and analyzed for phospho-STAT3 through western blot. Plumbagin suppresses phospho-STAT3 levels in a time-dependent manner. U266 cells (2 × 106) were treated with 5.0 μM plumbagin for the indicated times and analyzed for phospho-STAT3 levels. (C) Plumbagin suppresses STAT3 DNA-binding in a dose and time-dependent manner. U266 cells (2 × 106) were treated with the indicated concentrations of plumbagin for 4 h and nuclear extracts were analyzed for STAT3 DNA-binding by EMSA. For time-dependent assessment cells were treated with 10 μM plumbagin for the indicated times and nuclear extracts were examined for STAT3-DNA binding by EMSA. For super shift analysis, nuclear extracts (NE) from U266 cells were incubated with phospho- and total-STAT3 antibody and preimmune serum (PIS). They were then assayed for STAT3 DNA-binding by EMSA. (D) Plumbagin causes inhibition of translocation of STAT3 to the nucleus. U266 cells (1 × 105) were incubated with or without 5 μM plumbagin for 4 h and then analyzed for the intracellular distribution of STAT3 by immunocytochemistry. The same slides were counterstained for nuclei with Hoechst (50 ng/mL) for 5 min. The results shown are representative of two independent experiments.
Plumbagin inhibits constitutive STAT3 phosphorylation in MM cells
Whether plumbagin can modulate the constitutive STAT3 activation in multiple myeloma cells was investigated. U266 cells were first incubated with different concentrations of plumbagin for 4 h, after incubation, whole-cell extracts were prepared and examined for phosphorylated STAT3 by Western blot analysis using an antibody that recognizes STAT3 phosphorylated at tyrosine 705. As shown in Fig. 1B (left panel), plumbagin inhibited the constitutive phosphorylation of STAT3 in U266 cells, with maximum inhibition occurring at 5 μM. We also determined the incubation time required for plumbagin to suppress STAT3 activation in U266 cells. The inhibition was time-dependent, with maximum inhibition occurring at 4 h (Fig. 1B, right panel).
Whether plumbagin modulates the phosphorylation of STAT3 at serine 727 residue, was also examined. We found that plumbagin inhibited the serine phosphorylation of STAT3 in a dose-dependent manner (Fig. 1B; 3rd panel). This quinone had no effect on the expression of STAT3 protein under these conditions.
Plumbagin inhibits binding of STAT3 to DNA in MM cells
Tyrosine phosphorylation causes dimerization of STAT3, leading to its translocation to the nucleus, where it binds to DNA and regulates gene transcription (26), we therefore determined whether plumbagin suppresses DNA-binding activity of STAT3. EMSA analysis of nuclear extracts prepared from plumbagin-treated U266 cells showed that it caused a decrease in STAT3 DNA-binding activity in a dose-dependent (Fig. 1C, left panel) and time-dependent manner (Fig. 1C, middle panel). These results show that plumbagin abrogates the DNA-binding ability of STAT3. Supershift analysis indicated that the binding of STAT3 to the DNA was blocked by anti-phospho-STAT3 antibody, thus confirming that the protein/DNA complex observed is indeed STAT3 (Fig. 1C, right panel).
Plumbagin depletes nuclear pool of STAT3 in MM cells
Because nuclear translocation is central to the function of transcription factors and because it is not certain whether phosphorylation is mandatory for nuclear transport of STAT3 and its oncogenic functions (27), we investigated whether plumbagin suppresses nuclear retention of STAT3. Fig. 1D clearly demonstrates that plumbagin inhibited the translocation of STAT3 to the nucleus in U266 cells.
Plumbagin inhibits IL-6-induced STAT3 phosphorylation in human MM cells
Because IL-6 is a growth factor for MM cells and this effect is mediated through the activation of STAT3 (28), we determined whether plumbagin could inhibit IL-6-induced STAT3 phosphorylation. MM.1S cells, which lack constitutively active STAT3, were treated with IL-6 at different concentrations and then examined for phosphorylated STAT3. IL-6 induced phosphorylation of STAT3 at a concentration of 10 ng/mL within 5 min (Fig. 2A). Pretreatment of cells with plumbagin suppressed this IL-6-induced STAT3 phosphorylation. Exposure of cells to plumbagin for 1 h was sufficient to completely suppress IL-6-induced STAT3 phosphorylation (Fig. 2B).
Figure 2. Plumbagin downregulates IL-6-induced phospho-STAT3.
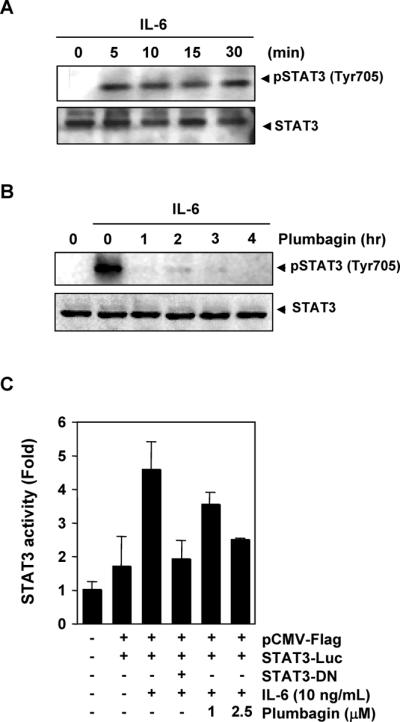
(A) MM1.S cells (2 × 106) were treated with IL-6 (10 ng/mL) for the indicated times. Whole-cell extracts were prepared, and phospho-STAT3 level was detected by western blot. The same blot was stripped and reprobed with STAT3 antibody to verify equal protein loading. (B) MM1.S cells (2 × 106) were treated with 5 μM plumbagin for the indicated times and then stimulated with IL-6 (10 ng/mL) for 30 min. Whole-cell extracts were then prepared and analyzed for phospho-STAT3 by Western blotting. The same blot was stripped and reprobed with STAT3 antibody to verify equal protein loading. (C) A293 cells (5 × 105) were transfected with STAT3-luciferase (STAT3-Luc) plasmid, incubated for 24 h, and treated with 1 and 2.5 μM plumbagin for 4 h and then stimulated with IL-6 (10 ng/mL) for 24 h. Whole-cell extracts were then prepared and analyzed for luciferase activity. Cells were cotransfected with β-gal and the data were normalized with β-galactosidase assay (data not shown). The results shown are representative of two independent experiments.
Plumbagin suppresses IL-6-induced STAT3-dependent reporter gene expression
Our results showed that plumbagin inhibited the phosphorylation, nuclear translocation, and DNA-binding activity of STAT3. We next determined whether plumbagin affects STAT3-dependent gene transcription. We employed A293 cells for this study because these cells do not constitutively express STAT3 and are easy to transfect. When cells transiently transfected with the STAT3-Luc construct were stimulated with IL-6, STAT3-mediated luciferase gene expression significantly increased. Dominant-negative STAT3 blocked this increase, indicating specificity. When the cells were pretreated with plumbagin, IL-6-induced STAT3 activity was inhibited in a dose-dependent manner (Fig. 2C).
Plumbagin suppresses constitutive activation of c-Src
How plumbagin inhibits STAT3 activation in MM cells was investigated. STAT3 has also been reported to be activated by soluble tyrosine kinases of the Src kinase families (29). Hence, we determined the effect of plumbagin on constitutive activation of Src kinase in U266 cells. We found that plumbagin suppressed the constitutive phosphorylation of c-Src kinase at tyrosine 416 (Fig. 3A). The levels of total non-phosphorylated c-Src kinase protein remained unchanged under the same conditions.
Figure 3.

(A) Plumbagin suppresses phospho-Src levels. U266 cells (2 × 106) were treated with the indicated concentrations of plumbagin for 4 h, and whole-cell extracts were prepared. Aliquots (30 μg) of those extracts were resolved on 10% SDS-PAGE, electro transferred onto nitrocellulose membranes, and probed for phospho-Src antibody. The same blot was stripped and reprobed with Src antibody to verify equal protein loading. (B) Plumbagin suppresses phospho-JAK1 and JAK2 expression. U266 cells (2 × 106) were treated with the indicated concentrations of plumbagin for 4 h, and whole-cell extracts were prepared. Aliquots (30 μg) of those extracts were resolved on 10% SDS-PAGE, electro transferred onto nitrocellulose membranes, and probed for phospho-JAK1 and phospho-JAK2 antibody. The same blots were stripped and reprobed with JAK1 and JAK2 antibody to verify equal protein loading. (C) To compare the effects of plumbagin and AG490 on phospho-STAT3, U266 cells were treated with 5μM concentration of plumbagin for 4 h and 100 μM AG490 for 4 h, whole-cell extracts were prepared, and 30 μg portions of those extracts were resolved on 10% SDS-PAGE and probed against phospho-STAT3 and STAT3. (D) Plumbagin suppresses JAK2 activity. U266 cells (2 × 106) were treated with the indicated concentrations of plumbagin for 4 h, whole-cell extracts were prepared and the kinase assay was performed. The results shown are representative of two independent experiments.
Plumbagin suppresses constitutive activation of JAK1 and JAK2
STAT3 has been reported to be activated by soluble tyrosine kinases of the Janus family (JAK) (30), so we determined whether plumbagin affects constitutive activation of JAK1 and JAK2 in U266 cells. We found that plumbagin suppressed the constitutive phosphorylation of JAK1 at tyrosine 1022/1023 and JAK2 at tyrosine 1007/1008 (Fig. 3B). The levels of non-phosphorylated JAK1 and JAK2 protein remained unchanged under the same conditions (Fig. 3B, bottom panel).
Plumbagin is more effective than AG490 in inhibiting the activation of STAT3
AG490 is the best-known rationally designed inhibitor of JAK2 kinase linked to STAT3 activation (30). How does the relative activity of plumbagin compares with that of AG490 for the suppression of phosphorylation of STAT3, was examined. As shown in Fig. 3C, plumbagin at 5 μM was found to be more effective than AG490 at 100 μM.
Plumbagin suppresses constitutive JAK2 activity
JAK2 has also been linked with STAT3 phosphorylation. We therefore investigated whether plumbagin affects JAK2 activity in U266 cells, by using the immune complex kinase assays. We found that plumbagin also suppressed the activity of JAK2 (Fig. 3D).
Tyrosine phosphatases are involved in plumbagin-induced inhibition of STAT3 activation
Because protein tyrosine phosphatases have been implicated in inactivation of STAT3 (31), we determined whether plumbagin-induced inhibition of STAT3 tyrosine phosphorylation could be due to activation of a protein tyrosine phosphatase (PTPase). Treatment of U266 cells with the broad-acting PTPase inhibitor sodium pervanadate prevented the plumbagin-induced inhibition of STAT3 activation (Fig. 4A). This suggests that PTPase are involved in plumbagin-induced inhibition of STAT3 activation.
Figure 4.

(A) Pervanadate reverses the phospho-STAT3 inhibitory effect of plumbagin. U266 cells (2 × 106) were treated with the indicated concentrations of pervanadate and 5 μM plumbagin for 4 h, and whole-cell extracts were prepared. 30 μg of protein extracts were resolved on 7.5% SDS-PAGE gel, electro transferred onto nitrocellulose membranes, and probed for phospho-STAT3 and STAT3. (B) Plumbagin induces the expression of SHP-1 protein. U266 cells (2 × 106) were treated with the indicated concentrations of plumbagin for 4 h. After incubation, whole-cell extracts were prepared, and 30 μg of protein was resolved on 10% SDS-PAGE, electro transferred onto nitrocellulose membranes, and probed for SHP-1 antibody. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. (C) Effect of SHP-1 knockdown on plumbagin-induced expression of SHP-1. SCC4 cells (1 × 105) were transfected with either SHP-1-specific or scrambled siRNA (50 nM). After 48 h, cells were treated with 5 μM plumbagin for 4 h, and whole-cell extracts were subjected to Western blot analysis for SHP-1. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. Transfection with SHP-1 siRNA reverses plumbagin-induced suppression of STAT3 activation. The same whole-cell extracts were subjected to phospho-STAT3 and STAT3. The results shown are representative of two independent experiments.
Plumbagin induces the expression of SHP-1 in MM cells
SHP-1 is a non-transmembrane PTPase expressed most abundantly in hematopoietic cells (32). This PTPase is an important negative regulator of JAK/STAT signaling in leukemias and lymphomas (33). We therefore examined whether plumbagin can modulate expression of SHP-1 in U266 cells. Cells were incubated with different concentrations of plumbagin for 4 h; whole-cell extracts were prepared and examined for SHP-1 protein by Western blot analysis. As shown in Fig. 4B, plumbagin induced the expression of SHP-1 protein in U266 cells.
SHP-1 siRNA down-regulate the expression of SHP-1 and reverses the inhibition of STAT3 activation by plumbagin
We determined whether the suppression of SHP-1 expression by siRNA would abrogate the inhibitory effect of plumbagin on STAT3 activation. Western blotting showed that plumbagin-induced SHP-1 expression was effectively abolished in the cells treated with SHP-1 siRNA; treatment with scrambled siRNA had no effect (Fig. 4C). We also found that plumbagin failed to suppress STAT-3 activation in cells treated with SHP-1 siRNA (Fig. 4C, lower panel). These siRNA results corroborate our earlier evidence of the critical role of SHP-1 in suppression of STAT-3 phosphorylation by plumbagin.
Plumbagin inhibits the proliferation of MM cells and is more effective than AG490
Because plumbagin downregulated the expression of cyclin D1, the gene critical for cell proliferation, we investigated whether plumbagin inhibits the proliferation of MM cells by using the MTT method. Plumbagin inhibited proliferation of cells in a dose-dependent manner (Fig. 5A).
Figure 5.

(A) Effects of AG490 and plumbagin on the proliferation of cells. U266 cells were plated in triplicate, treated with the indicated concentrations of plumbagin and AG490 for 24 h and then subjected to the MTT assay. (B) Plumbagin causes accumulation of cells in Sub-G1 phase. U266 cells (2 × 106) were synchronized by incubation overnight in the absence of serum and then treated with 2.5 μM plumbagin for the indicated times, after which the cells were washed, fixed, stained with PI, and analyzed for DNA content by flow cytometry. (C) Plumbagin downregulates the expression of cell proliferative, anti-apoptotic and angiogenic proteins. U266 cells (2 × 106) were treated with 2.5 μM plumbagin for the indicated time, after which whole-cell extracts were prepared and 30 μg portions of those extracts were resolved on 10% SDS-PAGE and probed against Bcl-xL, cyclin D1, and VEGF antibodies. The same blots were stripped and reprobed with β-actin antibody to verify equal protein loading. (D) Plumbagin induces caspase-3 dependent PARP cleavage. U266 cells were treated with 2.5 μM plumbagin for the indicated times, and whole-cell extracts were prepared, separated on SDS-PAGE, and subjected to Western blot against caspase-3 and PARP antibodies. The results shown are representative of two independent experiments.
To determine how the potency of plumbagin compares with that of well-established and rationally designed inhibitors of the STAT3 pathway, we examined the anti-proliferative activity of AG490 and plumbagin side by side. As shown in Fig. 5A, plumbagin was more potent than AG490 in suppressing the proliferation of MM cells. These results coincide with their relative effects on STAT3 phosphorylation
Plumbagin causes the accumulation of the cells in the sub-G1 phase of the cell cycle
Because D-type cyclin are required for the progression of cells from the G1 phase of the cell cycle to S phase (34), and because a rapid decline in levels of cyclin D1 was observed in plumbagin-treated cells, we determined the effect of plumbagin on cell cycle phase distribution. We found that plumbagin caused a significant accumulation of the cell population in the sub-G1 phase. After 48 h, 22% of the cell population had accumulated in sub-G1 phase, which is indicative of apoptosis (Fig. 5B).
Plumbagin downregulates the expression of cyclin D1, Bcl-xL, and VEGF
STAT3 activation has been shown to regulate the expression of various gene products involved in cell survival, proliferation, and angiogenesis. We investigated whether the expressions of the anti-apoptotic proteins Bcl-xL, cell cycle regulator protein, cyclin D1, and VEGF which are all reported to be regulated by STAT3 (24, 25, 35) were modulated by plumbagin. Plumbagin treatment downregulated expression of these proteins in a time-dependent manner, with maximum suppression observed at 36 h followed by 48 h of treatment (Fig. 5C).
Plumbagin activates caspase-3 and causes PARP cleavage
We examined whether suppression of constitutively active STAT3 in U266 cells by plumbagin leads to PARP cleavage. Treatment of U266 cells with plumbagin induced caspase-3–dependent cleavage of a 118-kDa PARP protein into an 87-kDa fragment (Fig. 5D).
Overexpression of constitutively active STAT3 rescues plumbagin-induced apoptosis
We assessed whether the overexpression of constitutive active STAT3 can rescue plumbagin-induced apoptosis. We used A293 cells to furnish this experiment because these cells are easily tranfectable as compared to U266 and do not express STAT3 constitutively. A293 cells were transfected with constitutively active STAT3 plasmid for 24 h, and cells were incubated with plumbagin for the next 24 h and examined for apoptosis by esterase staining assay. The results show that the forced expression of STAT3 significantly reduces the plumbagin-induced apoptosis (Fig. 6A).
Figure 6.

(A) Overexpression of constitutive STAT3 rescues A293 cells from plumbagin-induced cytotoxicity. First, A293 cells were transfected with constitutive STAT3 plasmid. After 24 h of transfection, the cells were treated with 2.5 μM plumbagin for 24 h, and then the cytotoxicity was determined by Live/Dead assay and 20 random fields were counted. (B) Plumbagin potentiates the apoptotic effect of thalidomide and bortezomib. U266 cells (1 × 106) were treated with 2.5 μM plumbagin and 10 μg/mL thalidomide or 20 nM bortezomib alone or in combination for 24 h at 37°C. Cells were stained with a Live/Dead assay reagent for 30 min and then analyzed under a fluorescence microscope. The results shown are representative of two independent experiments.
Plumbagin potentiates the apoptotic effect of bortezomib and thalidomide in MM cells
Bortezomib, an inhibitor of proteasome, and thalidomide, an inhibitor of TNF expression, have been approved for the treatment of MM patients (36). We investigated whether plumbagin can potentiate the effect of these drugs. For this, U266 cells were treated with plumbagin together with either thalidomide or bortezomib. The cells were then examined for apoptosis by using the Live-Dead assay, which determines plasma membrane stability using esterase staining. As shown in Fig. 6B, plumbagin significantly enhanced the apoptotic effects of thalidomide from 12% to 36% and of bortezomib from 12% to 40%.
Discussion
The goal of this study was to determine whether plumbagin exerts its anti-cancer effects through the modulation of the STAT3 signaling pathway in MM cells. We found that this quinone suppressed both constitutive and IL-6-inducible STAT3 activation in parallel with the inhibition of c-Src and JAK1 and JAK2 activation. Plumbagin stimulated the expression of the non-transmembrane PTPase SHP-1. Plumbagin also downregulated the expression of STAT3-regulated gene products, including Bcl-xL, cyclin D1, and VEGF. It induced inhibition of proliferation, induced apoptosis, and significantly potentiated the apoptotic effects of bortezomib and thalidomide in MM cells.
We found that plumbagin could suppress both constitutive and inducible STAT3 activation in MM cells. Exposure of cells to 5 μM plumbagin for 4 h was needed to fully abolish STAT3 activation. In comparison, exposure of cells to 100 μM AG490 (a rationally designed inhibitor of JAK2) for 8 h was required to suppress STAT3 activation (37). Because we used an antibody that recognizes the phosphorylation of serine and tyrosine located at position 727 and 705 respectively, in the STAT3 protein, plumbagin seems to inhibit this phosphorylation. Constitutive activation of STAT3 has been reported in a large variety of tumors, including breast cancer, prostate cancer, head and neck squamous cell carcinoma, lymphomas and leukemia, brain tumor, colon cancer, Ewing's sarcoma, gastric cancer, esophageal cancer, ovarian cancer, nasopharyngeal cancer, and pancreatic cancer (24). Hence, the suppression of constitutively active STAT3 in MM cells raises the possibility that this novel STAT3 inhibitor might also inhibit constitutively activated STAT3 in other types of cancer cells. It is possible that the anti-proliferative effects of plumbagin reported previously against breast cancer, lung cancer, ovarian cancer, acute promyelocytic leukemia, melanoma, and prostate cancer cells (4–11) could be due to downregulation of the STAT3 pathway. Furthermore, we observed that overexpression of STAT3 also rescues the apoptotic effects of plumbagin, strengthening our hypothesis that anti-proliferative effects of plumbagin are mediated through the abrogation of the STAT3 signaling pathway.
We also observed that plumbagin suppressed the nuclear translocation and DNA-binding activity of STAT3. However, the dose of plumbagin required to suppress DNA binding is higher than the dose required suppressing phosphosrylation. We speculate that phosphorylation of STAT3 is an early event and nuclear translocation is a late event. These results suggested that plumbagin may be affecting the early event at 5 μM but not the late event. This has been recently recognized that non-phosphorylated forms of STATs also shuttle between nucleus and cytoplasm at all times in a constitutive manner (38) suggesting that inhibition of phosphorylation of STAT3 is not sufficient to inhibit its nuclear translocation. STAT3 phosphorylation plays a critical role in transformation and proliferation of tumor cells (35). All Src-transformed cell lines have persistently activated STAT3, and dominant-negative STAT3 blocks transformation (27). The effects of plumbagin on STAT3 phosphorylation correlated with the suppression of upstream protein tyrosine kinases JAK1, JAK2, and c-Src. Dominant-negative STAT3 has also been shown to induce apoptosis in cells with constitutively active STAT3 (39). Our results are in agreement with a recent report that plumbagin can inhibit phosphorylation of JAK2 and STAT3 in prostate cancer cells (10), and suggest the potential contribution of JAK1 and c-Src in downregulation of STAT3 activation by plumbagin.
We also found evidence that the plumbagin-induced inhibition of STAT3 activation involves a PTPase. Numerous PTPases have been implicated in STAT3 signaling, including SHP-1, SHP-2, TC-PTP, PTEN, PTP-1D, CD45, and PTP-episilon (40–42). The type of PTPase involved in the downregulation of STAT3 phosphorylation is not clear. We found that plumbagin induced both the protein and mRNA for SHP-1(data not shown). Loss of SHP-1 has been shown to enhance JAK3/STAT3 signaling in ALK-positive anaplastic large-cell lymphoma (31).
Previously we have shown that plumbagin can also suppress NF-κB activation (14). Whether suppression of STAT3 activation by plumbagin is linked to inhibition of NF-κB activation is not clear. The p65 subunit of NF-κB has been shown to interact with STAT3 (43). STAT3 and NF-κB, however, are activated in response to different cytokines: whereas IL-6 is a major activator of STAT3, TNF is a potent activator of NF-κB. Interestingly, erythropoietin has been shown to activate both NF-κB through the activation of JAK2 kinase (44). Thus it is possible that suppression of JAK activation is the critical target for inhibition of both NF-κB and STAT3 activation by plumbagin.
We also found that plumbagin suppressed the expression of STAT3-regulated genes, including cyclin D1, and the anti-apoptotic gene products. Constitutively active STAT3 can contribute to oncogenesis by protecting cancer cells from apoptosis; this implies that suppression of STAT3 activation by agents such as plumbagin could facilitate apoptosis. Expression of Bcl-xL is regulated by STAT3 (45) and is overexpressed in MM cells (46). Bcl-xL can also block cell death induced by a variety of chemotherapeutic agents, in parallel with an increase in chemo resistance (47). The downregulation of the expression of Bcl-xL is likely linked with the ability of plumbagin to induce apoptosis in MM cells. VEGF is a critical growth factor for angiogenesis of the tumor and is also regulated by STAT3. Plumbagin also downregulated the expression of this growth factor.
Recently, a proteasome inhibitor (bortezomib) and a TNF inhibitor (thalidomide) were approved for the treatment of multiple myeloma (36, 48). We found that plumbagin potentiates the apoptotic effect of bortezomib and thalidomide in MM cells. We argue that the pharmacologic safety of plumbagin and its ability to downregulate the expression of several genes involved in cell survival and chemo resistance provide sufficient rationale for further testing in patients with MM.
In addition to plumbagin, previously we have shown that juglone and 1,4-naphthoquinone but not menadione (vitamin K3) can modulated NF-κB activation (14). Whether these analogues can also modulate STAT3 activation is not clear. Unlike plumbagin, shikonin (49) and compound 5 (50), that are analogues of vitamin K, are know to inhibit protein tyrosine phosphates. Thus it is possible that not all analogues of vitamin K exhibit similar effects on STAT3 activation.
The downregulation of EGFR signaling, inhibition of expression of survivin (6), suppression of angiogenesis (21), and radio sensitization of tumors (22) attributed to plumbagin could be due to modulation of the STAT3 pathway as described here. Various animal studies have indicated that plumbagin is well tolerated (5, 17–21). Also, doses of plumbagin used in the present study are achievable in vivo as supported by the studies of Hsieh et al that maximal plasma concentration of plumbagin in rat following treatment with 3 mg/kg i.v. or 100 mg/kg p.o. was shown to be about 1.0 and 1.85 μM with corresponding t1/2 of about 108 and 1028 min, respectively (51).
MM that has relapsed after conventional-dose therapy or stem-cell transplantation is typically treated with high-dose corticosteroids, thalidomide, or bortezomib. However, significant numbers of patients does not respond to these agents. Moreover, prolonged exposure leads to the development of resistance and toxicity, and progression-free and overall survival times are short. Collectively, the lack of toxicity of plumbagin and its ability to suppress STAT3 activation, inhibit IL-6-induced STAT3 and JAK1 phosphorylation, downregulate the expression of cyclin D1, Bcl-xL and VEGF, inhibit cell proliferation, induce apoptosis, and potentiate the effect of bortezomib and thalidomide indicate the need for further preclinical studies preceding human trials.
Materials and Methods
Reagents
Plumbagin (with purity greater than 97%) was purchased from Sigma-Aldrich (St. Louis, MO). A 100 mM solution of plumbagin was prepared in dimethyl sulfoxide, stored as small aliquots at −70°C, and then diluted as needed in cell culture medium. Hoechst 33342, MTT, Tris, glycine, NaCl, SDS, and BSA were purchased from Sigma-Aldrich. RPMI 1640, fetal bovine serum (FBS), 0.4% trypan blue vital stain, and antibiotic-antimycotic mixture were obtained from Invitrogen (Grand Island, NY). Rabbit polyclonal antibodies to STAT3 and mouse monoclonal antibodies against phospho-STAT3 (Tyr705) and Ser 727, Bcl-xL, SHP-1, cyclin D1, procaspase-3, JAK2 and PARP were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Goat anti-rabbit horseradish peroxidase (HRP) conjugate was purchased from Bio-Rad (Hercules, CA). Antibodies to phospho-Src (Tyr416), Src, phospho-JAK1 (Tyr1022/1023), phospho-JAK2 (Tyr1007/1008) and JAK1 were purchased from Cell Signaling Technology (Beverly, MA). Goat anti-mouse HRP was purchased from Transduction Laboratories (Lexington, KY) and goat anti-rabbit Alexa 594 from Invitrogen. AG490 was obtained from Calbiochem (San Diego, CA). Bacteria-derived recombinant human IL-6 was kindly provided by Novartis Pharmaceuticals (East Hanover, NJ). The siRNA for SHP-1 and the scrambled control were obtained from Ambion (Austin, TX). Bortezomib (Velcade, PS-341) was obtained from Millennium (Cambridge, MA) and thalidomide from Tocris Cookson (St. Louis, MO). GST-JAK2 substrate was kindly provided by Dr. Zhizhuang Joe Zhao (Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, OK). The constitutively active STAT3 construct was kindly provided by Dr. John DiGiovanni from The University of Texas M.D. Anderson Cancer Center (Smithville, TX).
Cell lines
Human MM cell lines U266 and MM.1S (dexamethasone-sensitive) were obtained from the American Type Culture Collection (Manassas, VA). Cell line U266 (ATCC TIB-196) is a plasmacytoma of B cell origin and is known to produce monoclonal antibodies and IL-6. The MM.1S cell line, established from the peripheral blood cells of a patient with IgA myeloma, secretes λL chain, is negative for the presence of the EBV genome, and expresses leukocyte antigen DR, plasma cell Ag-1, and T9 and T10 antigens. U266, MM.1S cells were cultured in RPMI 1640 medium containing 1X antibiotic-antimycotic solution with 10% FBS. Human embryonic kidney (A293) and human head and neck (SCC4) cells were maintained in DMEM medium containing 1X antibiotic-antimycotic solution with 10% FBS.
Electrophoretic mobility shift assay for STAT3-DNA binding
STAT3-DNA binding was analyzed by EMSA using a 32P-labeled high-affinity sis-inducible element (hSIE) probe (5'-CTTCATTTCCCGT AAATCCCT AAA GCT-3' and 5'-AGCTTTAGGGATTTACGGGAAATGA-3') as previously described (26). Briefly, nuclear extracts were prepared from plumbagin-treated cells and incubated with the 32P labeled hSIE probe. The resultant DNA-protein complex was separated from free oligonucleotide on 5% native polyacrylamide gels. For super shift assays, nuclear extracts prepared from U266 cells were incubated with antibodies against either phospho-STAT3 or STAT3 for 30 min at 37 °C before the complex was analyzed by EMSA. Preimmune serum was used as control. The dried gels were visualized, and the radioactive bands were quantified with the Storm 820 imaging system and Image quant software (GE Healthcare, Piscataway, NJ).
Western blotting
To detect various proteins, U266 cells (2 × 106) were treated with plumbagin. The cells were then washed and extracted by incubation for 30 min on ice in 0.05 ml of buffer containing 20 mM HEPES, pH 7.4, 2 mM EDTA, 250 mM NaCl, 0.1% NP-40, 2 μg/mL leupeptin, 2 μg/mL aprotinin, 1 mM PMSF, 0.5 μg/mL benzamidine, 1 mM DTT, and 1 mM sodium vanadate. The lysate was centrifuged and the supernatant was collected. Whole-cell extract protein (30 μg) was resolved on 7.5–12% SDS-PAGE, electro transferred onto a nitrocellulose membrane, blotted with antibodies, and then detected by chemiluminescence (ECL; GE Healthcare).
Immunocytochemistry for STAT3 localization
Plumbagin-treated MM cells were plated on a glass slide by centrifugation using a Cytospin 4 (Thermoshendon, Pittsburg, PA), air-dried for 1 h at room temperature, and fixed in 4% formaldehyde. After a brief washing in PBS, slides were blocked with 5% normal goat serum for 1 h and then incubated with rabbit polyclonal anti-human STAT3 antibody (dilution, 1:100). After overnight incubation, the slides were washed and then incubated with goat anti-rabbit IgG-Alexa 594 (1:100) for 1 h and counterstained for nuclei with Hoechst (50 ng/mL) for 5 min. Stained slides were mounted with mounting medium (Sigma-Aldrich) and analyzed under an epifluorescence microscope (Labophot-2; Nikon, Tokyo, Japan). Pictures were captured using a Photometrics Coolsnap CF color camera (Nikon) and MetaMorph version 4.6.5 software (Universal Imaging, Downingtown, PA).
STAT3 luciferase reporter assay
A293 cells were plated in six-well plates with 5 × 105 per well in DMEM containing 10% FBS. The STAT3-responsive elements linked to a luciferase reporter gene was transfected with wild-type or dominant-negative STAT3-Y705F (STAT3F). Transfections were done according to the manufacturer's protocols using Fugene-6 (Roche). At 24 h post transfection, cells were pretreated with plumbagin for 4 h and then induced by IL-6 for additional 24 h before being washed and lysed in luciferase lysis buffer (Promega). Luciferase activity was measured with a luminometer by using a luciferase assay kit (Promega) and was normalized to β-galactosidase activity. All luciferase experiments were done in triplicate and repeated three or more times. The data show the mean and the SD of the mean of the experiments.
Transfection with SHP-1 siRNA
SCC4 cells were plated in each well of six-well plates and allowed to adhere for 24 h. On the day of transfection, 12 μL Hiperfect transfection reagent (Qiagen) were added to 50 nmol/L SHP-1 siRNA in a final volume of 100 μL culture medium. After 48 h of transfection, cells were treated with plumbagin for 4 h and whole-cell extracts were prepared for SHP-1, STAT3, and phospho-STAT3 analysis by Western blot.
MTT assay
The anti proliferative effect of plumbagin and AG490 against the MM cell lines was determined by the MTT dye uptake method.
Live/Dead assay
Viability of cells was also determined by using the Live/Dead assay (Invitrogen), which measures intracellular esterase activity and plasma membrane integrity.
Flow cytometric analysis
To determine the effect of plumbagin on the cell cycle, U266 cells were first synchronized by serum starvation and then exposed to plumbagin. The cells were then washed, fixed with 70% ethanol, and incubated for 30 min at 37°C with 0.1% RNase A in PBS. Cells were then washed again, resuspended, and stained in PBS containing 25 μg/mL propidium iodide (PI) for 30 min at room temperature. Cell distribution across the cell cycle was analyzed with a FACS Calibur flow cytometer (Becton Dickinson, Bedford, MA).
Kinase assay
To determine the effect of plumbagin on JAK2 activation, we performed an immunocomplex kinase assay using GST-JAK2 as the substrate. Briefly, the JAKs complex from whole-cell extracts was precipitated with antibody against JAK2 and treated with protein A/G-agarose beads (Pierce, Rockford, IL). After 2 h, the beads were washed with whole-cell extract buffer and then resuspended in a kinase assay mixture containing 50 mM HEPES (pH 7.4), 20 mM MgCl2, 2 mM DTT, 20 μCi of [32P]ATP, 10 μM unlabeled ATP, and 2 μg of substrate GST-JAK2. After incubation at 30°C for 30 min, the reaction was terminated by boiling with SDS sample buffer for 5 min. Finally, the protein was resolved on 10% SDS-PAGE, the gel was dried, and the radioactive bands were visualized with the Storm 820 imaging system. To determine the total amounts of JAK2 in each sample, 30 μg of whole-cell proteins was resolved on 10% SDS-PAGE, electro transferred to a nitrocellulose membrane, and then blotted with anti-JAK2 antibody.
Transfections with Constitutive STAT3 Construct
A293 cells were plated in chamber slides in DMEM containing 10% FBS. After 24 h, the cells were transfected with constitutive STAT3-plasmid by FuGene 6 according to manufacturer's protocol (Roche, Indianapolis, IN). Cells were treated with plumbagin for 24 h, and viability of the cells was determined by Live/Dead assay.
Statistical analysis
The statistical analysis was done using Student's t-test with Microsoft excel software.
Acknowledgments
We would like to thank Michael Worley for carefully editing the manuscript and providing valuable comments. Dr. Aggarwal is the Ransom Horne, Jr., Professor of Cancer Research. This work was supported by a grant from the Clayton Foundation for Research (to B.B. Aggarwal), a program project grant from National Institutes of Health (NIH CA-124787-01A2), and grant from Center for Targeted Therapy of M. D. Anderson Cancer Center.
References
- 1.Tilak JC, Adhikari S, Devasagayam TP. Antioxidant properties of Plumbago zeylanica, an Indian medicinal plant and its active ingredient, plumbagin. Redox Rep. 2004;9:219–27. doi: 10.1179/135100004225005976. [DOI] [PubMed] [Google Scholar]
- 2.Binder RG, Benson ME, Flath RA. Eight 1,4-Naphthoquinones from Juglone. Phytochemistry. 1989;28:2799–801. [Google Scholar]
- 3.Hedin PA, Collum DH, Langhans VE, Graves CH. Distribution of Juglone and related compounds in pecan and their effects on Fusicladium effusum. J Agric Food Chem. 1980;28:340–2. [Google Scholar]
- 4.Kuo PL, Hsu YL, Cho CY. Plumbagin induces G2-M arrest and autophagy by inhibiting the AKT/mammalian target of rapamycin pathway in breast cancer cells. Mol Cancer Ther. 2006;5:3209–21. doi: 10.1158/1535-7163.MCT-06-0478. [DOI] [PubMed] [Google Scholar]
- 5.Hsu YL, Cho CY, Kuo PL, Huang YT, Lin CC. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) induces apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation at serine 15 in vitro and in vivo. J Pharmacol Exp Ther. 2006;318:484–94. doi: 10.1124/jpet.105.098863. [DOI] [PubMed] [Google Scholar]
- 6.Gomathinayagam R, Sowmyalakshmi S, Mardhatillah F, Kumar R, Akbarsha MA, Damodaran C. Anticancer mechanism of plumbagin, a natural compound, on non-small cell lung cancer cells. Anticancer Res. 2008;28:785–92. [PubMed] [Google Scholar]
- 7.Thasni KA, Rakesh S, Rojini G, Ratheeshkumar T, Srinivas G, Priya S. Estrogen-dependent cell signaling and apoptosis in BRCA1-blocked BG1 ovarian cancer cells in response to plumbagin and other chemotherapeutic agents. Ann Oncol. 2008;19:696–705. doi: 10.1093/annonc/mdm557. [DOI] [PubMed] [Google Scholar]
- 8.Zhao YL, Lu DP. Effects of plumbagin on the human acute promyelocytic leukemia cells in vitro. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2006;14:208–11. [PubMed] [Google Scholar]
- 9.Wang CC, Chiang YM, Sung SC, Hsu YL, Chang JK, Kuo PL. Plumbagin induces cell cycle arrest and apoptosis through reactive oxygen species/c-Jun N-terminal kinase pathways in human melanoma A375.S2 cells. Cancer Lett. 2008;259:82–98. doi: 10.1016/j.canlet.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Aziz MH, Dreckschmidt NE, Verma AK. Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer Res. 2008;68:9024–32. doi: 10.1158/0008-5472.CAN-08-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powolny AA, Singh SV. Plumbagin-induced apoptosis in human prostate cancer cells is associated with modulation of cellular redox status and generation of reactive oxygen species. Pharm Res. 2008;25:2171–80. doi: 10.1007/s11095-008-9533-3. [DOI] [PubMed] [Google Scholar]
- 12.Jaiswal AS, Bloom LB, Narayan S. Long-patch base excision repair of apurinic/apyrimidinic site DNA is decreased in mouse embryonic fibroblast cell lines treated with plumbagin: involvement of cyclin-dependent kinase inhibitor p21Waf-1/Cip-1. Oncogene. 2002;21:5912–22. doi: 10.1038/sj.onc.1205789. [DOI] [PubMed] [Google Scholar]
- 13.Kawiak A, Piosik J, Stasilojc G, et al. Induction of apoptosis by plumbagin through reactive oxygen species-mediated inhibition of topoisomerase II. Toxicol Appl Pharmacol. 2007;223:267–76. doi: 10.1016/j.taap.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 14.Sandur SK, Ichikawa H, Sethi G, Ahn KS, Aggarwal BB. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-kappaB activation and NF-kappaB-regulated gene products through modulation of p65 and IkappaBalpha kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem. 2006;281:17023–33. doi: 10.1074/jbc.M601595200. [DOI] [PubMed] [Google Scholar]
- 15.Rossary A, Arab K, Steghens JP. Polyunsaturated fatty acids modulate NOX 4 anion superoxide production in human fibroblasts. Biochem J. 2007;406:77–83. doi: 10.1042/BJ20061009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shukla S, Wu CP, Nandigama K, Ambudkar SV. The naphthoquinones, vitamin K3 and its structural analogue plumbagin, are substrates of the multidrug resistance linked ATP binding cassette drug transporter ABCG2. Mol Cancer Ther. 2007;6:3279–86. doi: 10.1158/1535-7163.MCT-07-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh UV, Udupa N. Reduced toxicity and enhanced antitumor efficacy of betacyclodextrin plumbagin inclusion complex in mice bearing Ehrlich ascites carcinoma. Indian J Physiol Pharmacol. 1997;41:171–5. [PubMed] [Google Scholar]
- 18.Tiwari SB, Pai RM, Udupa N. Temperature sensitive liposomes of plumbagin: characterization and in vivo evaluation in mice bearing melanoma B16F1. J Drug Target. 2002;10:585–91. doi: 10.1080/1061186021000054924. [DOI] [PubMed] [Google Scholar]
- 19.Devi PU, Rao BS, Solomon FE. Effect of plumbagin on the radiation induced cytogenetic and cell cycle changes in mouse Ehrlich ascites carcinoma in vivo. Indian J Exp Biol. 1998;36:891–5. [PubMed] [Google Scholar]
- 20.Abdul KM, Ramchender RP. Modulatory effect of plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) on macrophage functions in BALB/c mice. I. Potentiation of macrophage bactericidal activity. Immunopharmacology. 1995;30:231–6. doi: 10.1016/0162-3109(95)00027-q. [DOI] [PubMed] [Google Scholar]
- 21.Jackson JK, Higo T, Hunter WL, Burt HM. Topoisomerase inhibitors as anti-arthritic agents. Inflamm Res. 2008;57:126–34. doi: 10.1007/s00011-007-7163-6. [DOI] [PubMed] [Google Scholar]
- 22.Nair S, Nair RR, Srinivas P, Srinivas G, Pillai MR. Radiosensitizing effects of plumbagin in cervical cancer cells is through modulation of apoptotic pathway. Mol Carcinog. 2008;47:22–33. doi: 10.1002/mc.20359. [DOI] [PubMed] [Google Scholar]
- 23.Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18:254–67. doi: 10.1038/cr.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aggarwal BB, Sethi G, Ahn KS, et al. Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann N Y Acad Sci. 2006;1091:151–69. doi: 10.1196/annals.1378.063. [DOI] [PubMed] [Google Scholar]
- 25.Aggarwal BB, Kunnumakkara AB, Harikumar KB, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu CL, Meyer DJ, Campbell GS, et al. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science. 1995;269:81–3. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- 27.Brierley MM, Fish EN. Stats: multifaceted regulators of transcription. J Interferon Cytokine Res. 2005;25:733–44. doi: 10.1089/jir.2005.25.733. [DOI] [PubMed] [Google Scholar]
- 28.Kawano M, Hirano T, Matsuda T, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332:83–5. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- 29.Schreiner SJ, Schiavone AP, Smithgall TE. Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J Biol Chem. 2002;277:45680–7. doi: 10.1074/jbc.M204255200. [DOI] [PubMed] [Google Scholar]
- 30.Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84:331–4. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- 31.Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006;108:2796–803. doi: 10.1182/blood-2006-04-017434. [DOI] [PubMed] [Google Scholar]
- 32.Wu C, Sun M, Liu L, Zhou GW. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene. 2003;306:1–12. doi: 10.1016/s0378-1119(03)00400-1. [DOI] [PubMed] [Google Scholar]
- 33.Oka T, Ouchida M, Koyama M, et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62:6390–4. [PubMed] [Google Scholar]
- 34.Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–13. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- 35.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 36.Cavo M. Proteasome inhibitor bortezomib for the treatment of multiple myeloma. Leukemia. 2006;20:1341–52. doi: 10.1038/sj.leu.2404278. [DOI] [PubMed] [Google Scholar]
- 37.Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol. 2003;171:3863–71. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 38.Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol. 2008;19:329–40. doi: 10.1016/j.semcdb.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–15. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 40.Tenev T, Bohmer SA, Kaufmann R, et al. Perinuclear localization of the protein-tyrosine phosphatase SHP-1 and inhibition of epidermal growth factor-stimulated STAT1/3 activation in A431 cells. Eur J Cell Biol. 2000;79:261–71. doi: 10.1078/S0171-9335(04)70029-1. [DOI] [PubMed] [Google Scholar]
- 41.Woetmann A, Nielsen M, Christensen ST, et al. Inhibition of protein phosphatase 2A induces serine/threonine phosphorylation, subcellular redistribution, and functional inhibition of STAT3. Proc Natl Acad Sci U S A. 1999;96:10620–5. doi: 10.1073/pnas.96.19.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H, Baumann H. Dual signaling role of the protein tyrosine phosphatase SHP-2 in regulating expression of acute-phase plasma proteins by interleukin-6 cytokine receptors in hepatic cells. Mol Cell Biol. 1999;19:5326–38. doi: 10.1128/mcb.19.8.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J. 2002;367:97–105. doi: 10.1042/BJ20020588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves crosstalk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–7. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 45.Niu G, Heller R, Catlett-Falcone R, et al. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999;59:5059–63. [PubMed] [Google Scholar]
- 46.Tu Y, Renner S, Xu F, et al. BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer Res. 1998;58:256–62. [PubMed] [Google Scholar]
- 47.Simonian PL, Grillot DA, Nunez G. Bcl-2 and Bcl-XL can differentially block chemotherapy-induced cell death. Blood. 1997;90:1208–16. [PubMed] [Google Scholar]
- 48.Glasmacher A, Hahn C, Hoffmann F, et al. A systematic review of phase-II trials of thalidomide monotherapy in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2006;132:584–93. doi: 10.1111/j.1365-2141.2005.05914.x. [DOI] [PubMed] [Google Scholar]
- 49.Nigorikawa K, Yoshikawa K, Sasaki T, et al. A naphthoquinone derivative, shikonin, has insulin-like actions by inhibiting both phosphatase and tensin homolog deleted on chromosome 10 and tyrosine phosphatases. Mol Pharmacol. 2006;70:1143–9. doi: 10.1124/mol.106.025809. [DOI] [PubMed] [Google Scholar]
- 50.Markovits J, Wang Z, Carr BI, et al. Differential effects of two growth inhibitory K vitamin analogs on cell cycle regulating proteins in human hepatoma cells. Life Sci. 2003;72:2769–84. doi: 10.1016/s0024-3205(03)00188-7. [DOI] [PubMed] [Google Scholar]
- 51.Hsieh YJ, Lin LC, Tsai TH. Measurement and pharmacokinetic study of plumbagin in a conscious freely moving rat using liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;844:1–5. doi: 10.1016/j.jchromb.2006.06.024. [DOI] [PubMed] [Google Scholar]