

CONSPECTUS
For decades, triflic acid, methyl triflate and trialkylsilyl triflate reagents have served synthetic chemistry well as clean, strong electrophilic sources of H+, CH3+ and R3Si+ respectively. However, a number of weakly basic substrates are unreactive towards these reagents. In addition, triflate anion can express undesired nucleophilicity towards electrophilically activated substrates.
In this Account, we describe methods that replace triflate-based electrophilic reagents with carborane reagents. Using carborane anions of type CHB11R5X6− (R = H, Me, X; X = Br, Cl) – members of a class of notably inert, weakly nucleophilic anions – instead of triflate significantly increases the electrophilicity of these reagents and shuts down subsequent nucleophilic chemistry of the anion. Thus, H(carborane) acids cleanly protonate benzene, phosphabenzene, C60 etc. while triflic acid does not. Similarly, CH3(carborane) reagents can methylate substrates that are inert to boiling neat methyl triflate, including benzene, phosphabenzenes, phosphazenes, and the pentamethylhydrazinium ion which forms the dipositive ethane analogue, Me6N22+. Methyl carboranes are also surprisingly effective in abstracting hydride from simple alkanes to give isolable carbocation salts e.g. t-butyl cation.
Trialkylsilyl carborane reagents, R3Si(carborane), abstract halides from substrates to produce cations of unprecedented reactivity. For example, fluoride is extracted from freons to form carbocations, chloride is extracted from IrCl(CO)(PPh3)2 to form a coordinatively-unsaturated iridium cation that undergoes oxidative addition with chlorobenzene at room temperature, and silylation of cyclo-N3P3Cl6 produces a catalyst for the polymerization of phosphazenes that functions at room temperature. Although currently too expensive for widespread use, carborane reagents are nevertheless of considerable interest as specialty reagents for making reactive cations and catalysts.
1. Introduction
Chemists often reach for triflic acid, methyl triflate (or a Me3O+ Meerwein oxonium salt) and trimethylsilyl triflate when strongly electrophilic sources of H+, Me+ and Me3Si+ are required. Over the last half century, these triflate reagents have replaced their corresponding halides (e.g. HCl, MeI and Me3SiI) in a wide variety of applications.
The success of triflate reagents is due to the lower basicity and chemical inertness of the triflate anion (CF3SO3−) relative to halides. Triflate is a better leaving group in the reagent and is less likely to interfere as a nucleophile in subsequent chemistry of the electrophilically activated substrate. The triflate anion also lends useful solubility to compounds, particularly in coordination compounds of both main group and transition metal elements, thereby promoting cation-like reactivity.1 Finally, triflic acid is available on a tank car scale, making triflate reagents affordable.
Triflate reagents now find themselves in a position related to halide reagents 50 years ago. Many weakly basic molecules (e.g. benzene, C60, phosphabenzenes, phosphazenes, Xe, etc.) are not protonated, alkylated or silylated by triflate reagents, or if they are, their cationic products react in unwanted ways with the triflate nucleophile. Traditionally, electrophilic reagents have been rendered more potent by the addition of a Lewis acid such as AlCl3 in Friedel-Crafts-type chemistry, or SbF5 in superacid chemistry. However, the addition of a Lewis acid can result in messy reaction conditions, the presence of halide can interfere with desired outcomes and excess Lewis acid can suppress the nucleophilicity of the substrate via adduct formation. In addition, SbF5 is a potent oxidant. It decomposes many molecules and glassware is corroded when HF is produced from trace water.
Carborane reagents avoid most of these difficulties. Carborane acids protonate a number of weakly basic molecules that are not protonated by triflic acid. Moreover, the inertness of a carborane anion as the conjugate base means that ensuing nucleophilic or oxidative chemistry is suppressed. Methyl carboranes can react with molecules that are inert to boiling neat methyl triflate. Trialkylsilyl carboranes are able to silylate substrates that are unreactive towards trialkylsilyl triflates. Because they are expensive to produce, however, carborane reagents are not yet available for bulk use. Nevertheless, sufficient new organic and inorganic chemistry has been demonstrated with carborane reagents over the past few years to warrant an account of their potential as new specialty reagents for electrophilic chemistry.
The chemistry described in this account is possible because carborane anions of the type CHB11R5X6− (R = H, Me, X; X = halogen) (Figure 1) are amongst the least basic and most chemically inert anions presently known.2,3 Their exceptional stability derives from σ aromaticity in the icosahedral CB11 core. In addition, the delocalized negative charge is screened by a layer of halogen substituents. By all measures, they are weaker bases,4 weaker nucleophiles and more weakly coordinating anions than triflate.5 In main group and transition metal coordination chemistry, anions such as the perfluorinated tetraphenylborates (F20-BPh4−) are often competitive with carborane anions as the most weakly coordinating anions,6,7 and they are cheaper, but with potent hard electrophiles such as H+, CH3+ and R3Si+, the corresponding reagents do not exist or are problematic to use. The acid H(F20-BPh4−), although predicted to be extremely strong,8 cannot be prepared because the anion is unstable with respect to B-phenyl bond cleavage.9 Only the much weaker dietherate acid, [H(Et2O)2+][F20-BPh4−] is known.10 There are no reports of CH3(F20-BPh4). R3Si(F20-BPh4) reagents11 do not crystallize well for easy handling and may be unwittingly contaminated with silane in the form of the [R3Si-H-SiR3]+ cation.12
Fig. 1.
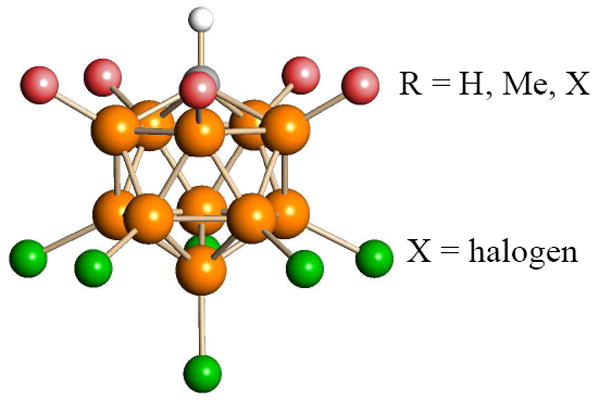
Carborane anions of the type CHB11R5X6−.
The choice of a specific carborane anion for a particular reagent is made on the basis of inertness, basicity, solubility and ease of preparation. Typically, the parent carborane, CHB11H11−, has to be at least hexa-halogenated at boron (in the 7-12 positions, antipodal to C at 1) to achieve the required level of inertness. Inertness increases with the extent of halogenation, presumably because halide substituents screen the negative charge and create an impervious layer that protects the CB11 core from chemical attack. Anion basicity decreases in the order I > Br > Cl > F which can be ascribed to decreasing polarizability of the halide. Thus, the undecachloro reagent R3Si(CHB11Cl11) is a stronger silylating reagent than the corresponding hexachloro reagent, which is stronger than the corresponding hexabromo reagent. The fluorinated carborane13 and borane14 anions developed in the Strauss lab should give even stronger electrophilic reagents15 but these anions are only available to those willing to work with F2. The easiest carboranes to prepare are the hexa- and undeca-halo anions of the more conveniently used halogens. For the convenience of readers, the most commonly used synthetic procedures are gathered together in the Supporting Information along with typical NMR and IR characterization data. Salts of the undecachloro anion, CHB11Cl11−,16 have better solubility in chlorocarbon solvents than most other carborane anions which, together with its extreme inertness and good crystallizing ability, has made it a favorite in our laboratories. With the hexane-soluble permethylated carborane, unique electrophilic Li+ chemistry has been developed in the Michl labs.17,18
2. Protonation
Anhydrous carborane acids, H(carborane), are prepared by treatment of solid trialkylsilyl carboranes with condensed HCl (Eq. 1).
| (1) |
They are solids having bridged-proton linear chain structures (Fig. 2).19 By a number of measures, they are the strongest Brønsted acids presently known, yet they are also the gentlest.4 This seeming paradox can be understood by recognizing that carborane acids separate the protonating capability of an acid from the nucleophilicity and oxidizing capacity of its conjugate base in a manner not previously achieved. Nitric acid decomposes molecules because protonation activates them towards attack by the nitrate ion as a nucleophile and oxidant. This occurs to a much lesser extent with triflic acid, and even less so with carborane acids because of the exceptional inertness of their conjugate base anions.
Fig. 2.
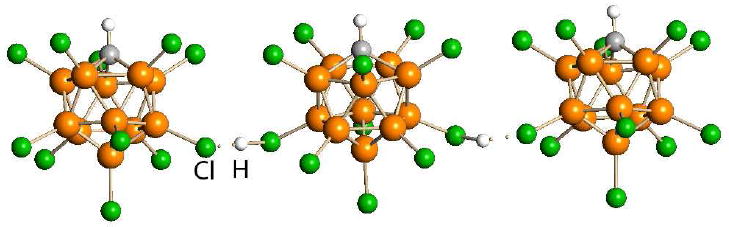
The proton-bridged linear-chain X-ray structure of H(CHB11Cl11).
Except in gas phase calculations,20,8 the strength of 100% carborane acids cannot be quantified in the usual manner because they are solids not liquids. Nevertheless, we know that in dilute solution carborane acids are, at the very least, 100 times stronger than triflic acid because they protonate benzene whereas triflic acid does not.4 Benzenium ion salts, [C6H7+][carborane-], are formed and their remarkable stability allows these Wheland intermediates of electrophilic aromatic substitution to be studied by X-ray crystallography (Fig. 3).9
Fig. 3.
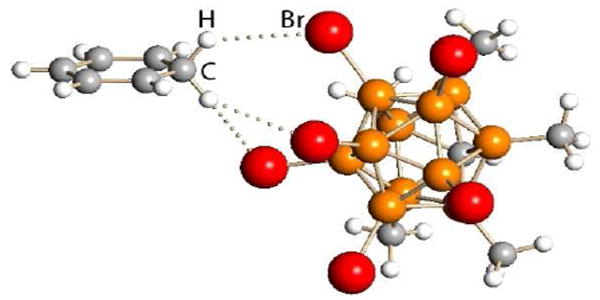
X-ray structure of the benzenium ion salt [C6H7][CHB11Me5Br6] showing H-bonding.
Another illustration of the strong-yet-gentle properties of carborane acids is their use in making the first cations of fullerenes.21 While typical superacids such as HF/SbF5 decompose fullerenes even at dry ice temperatures, carborane superacids protonate C60 at room temperature in o-dichlorobenzene solution to give isolable HC60+ salts. Novel solid state 13C NMR methodology shows that the carbocation center has a 1,2 rather than 1,4 disposition to the site of protonation (Fig. 4).22
Fig. 4.
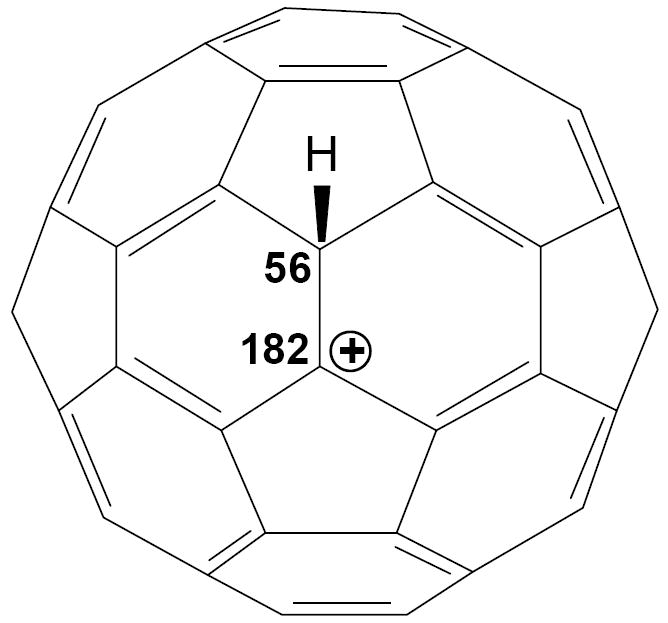
Structure of HC60+ as determined by NMR showing key 13C chemical shifts.
A more recent example is the protonation of the weakly basic P atom in a phosphabenzene to give an isolable salt (Fig. 5).23 Although triflic acid is strong enough to protonate phosphabenzenes, the triflate anion reacts with the protonated product causing a rearrangement reaction, the driving force presumably being that of strong P-O bond formation. The carborane anion is a poorer nucleophile so the reaction is arrested at the protonation stage.
Fig. 5.
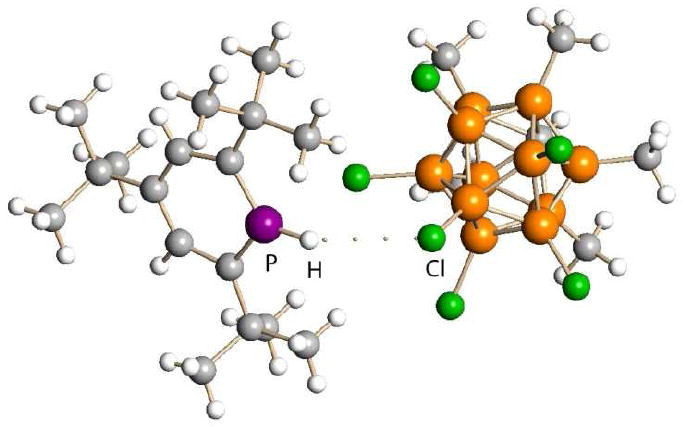
X-ray structure of a protonated phosphabenzene showing ion-pairing to the CHB11Me5Cl6- anion via weak H-bonding.
Carborane acids are so strong that limitations on their use are imposed by the properties of solvents. Dissolution appears to occur only in solvents that are protonated. Not only does the acidity become leveled down to that of H(solvent)n+ (typically n = 1 for arenes and n = 2 for O-atom donor solvents) but the stability of the acid depends on the stability of the protonated solvent. In the case of benzene, solubility is low but the acids have long term stability as C6H7+ salts. H(CHB11Cl11) protonates o-dichlorobenzene but is stable only for ca. 2 hours at room temperature. Dichloromethane eliminates HCl and forms dialkylchloronium ions, even at subambient temperatures.24 Carborane acids are stable in anhydrous SO2 in the form of a proton disolvate, H(SO2)2+, but with a boiling point of 10°C, liquid SO2 is not so easy to handle. Carborane acids irreversibly leach water from glassware to form H3O+ or higher hydrated H(H2O)n+ salts25,26 so vigilance in drying solvents is important. Maintaining the effective concentration of water below ca. 5 × 10-4 M is difficult on a routine basis.
Carborane acids can be sublimed,19 offering a way to overcome the problems of using solvents. Solventless reactions may allow exploitation of the full acidity of carborane acids and lead to protonation of bases hitherto unprotonated. Xenon is such a target.
3. Alkylation
Alkyl carboranes are synthesized by treatment of a triethylsilylium carborane with an alkyl triflate (Eq. 2).27,28
| (2) |
An X-ray structure for R = i-Pr (Fig. 6) reveals alkylation of the carborane at the “lower” pentagonal belt (7-11 positions) but 1H NMR for R = CH3 shows that the structure is fluxional in solution with the R group also occupying the 12-position.28 The ∠C-C-C in the i-propyl group is 116.5° indicating developing carbocationic character. Because of their high reactivity, the preparative reaction of alkyl carboranes is frequently carried out in situ with its intended substrate.
Fig. 6.
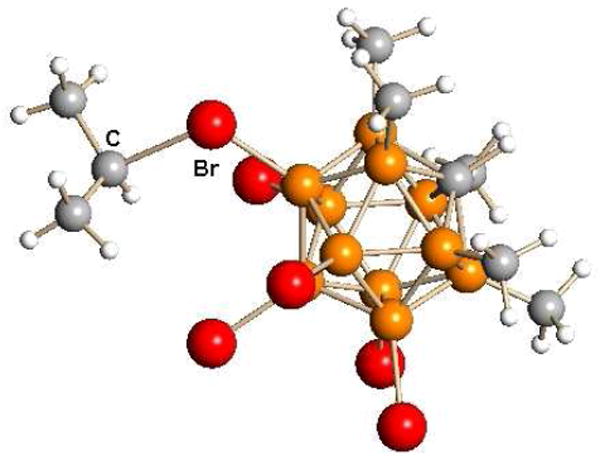
X-ray structure of i-Pr(CHB11Me5Br6) showing alkylation at the 7 position.
The electrophilicity of “methyl+” carborane reagents was revealed to us in a rather startling manner. The most reactive reagent, CH3(CHB11Cl11), was observed to react with hexane solvent while merely attempting to wash its crystals at sub-ambient temperatures. The product was the methyl-cyclo-pentyl carbocation. Indeed, tertiary carbocations are formed readily by the reaction of methyl carborane reagents with C4 or higher hydrocarbons (Scheme 1).29 n-Alkanes give rise to tertiary carbenium ions, presumably via facile 1,2 shifts.30 The byproduct is methane. Since CD3(carborane) produces exclusively CD3H, the mechanism is one of hydride abstraction from the alkane. Carborane counterions add a new dimension of stability of carbenium ions allowing these reactive intermediates of hydrocarbon chemistry to be studied easily by X-ray crystallography at room temperature. The t-butyl cation was confirmed to be planar and its capacity to act as a C-H acid is indicated by methyl group H-bonding to the carborane anion (Fig. 7).29
Scheme 1.
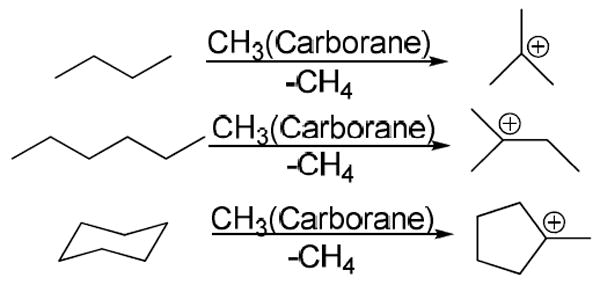
Formation of tertiary carbocations from alkanes.
Fig. 7.
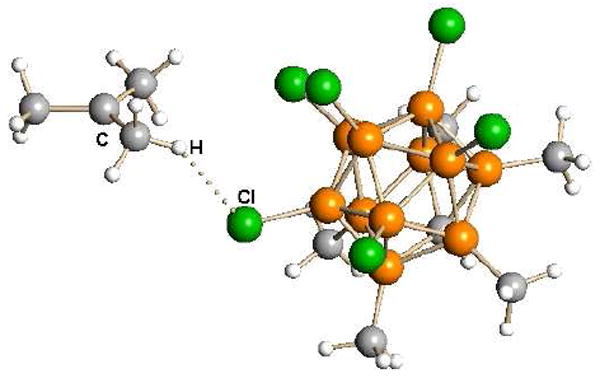
X-ray structure of t-butyl cation indicating H-bonding to CHB11Me5Cl6− anion.
Notable electrophilic alkylations with methyl carborane reagents involve methylation of the weakly basic N atom of phosphazenes,31 the weakly basic P atom of phosphabenzenes23 and the synthesis of the long-sought hexamethylhydrazinium(2+) cation (Scheme 2).32 The Me3N-NMe32+ ion is a rare case of stable dication formation with formal positive charges on adjacent atoms. Hydrazinium(2+) cations are calculated to be thermodynamically unstable with respect to “coulombic explosion”33 but the N-N bond is sufficiently strong that kinetic stability at room temperature is achieved. Decomposition occurs because of their acidity rather than by bond homolysis.34
Scheme 2.
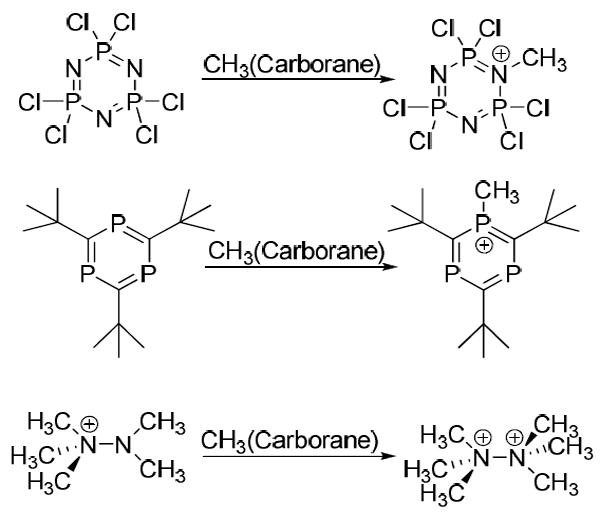
Reactions of CH3(carborane) with various weakly basic heteroatom substrates.
4. Silylation
Trialkylsilyl carboranes are prepared by hydride abstraction from a silane with trityl ion in a low-basicity arene solvent such as benzene or toluene (Eq. 3).
| (3) |
Structurally, they are covalent compounds (Fig. 8) but developing R3Si+ character is demonstrated by the long Si-Cl distance (2.32 Å) and the approach of the sum of the C-Si-C angle towards the sp2 ideal of 360°. This angle is 351.9° in i-Pr3Si(CHB11H5Cl6)35 and 354.4° in Me3Si(CHB11F11).15 Although they are not free silylium ions, they react like silylium ions.35 We have exploited their fierce electrophilicity and avidity for halide and triflate anion in producing carborane acids from HCl and methyl carboranes from methyl triflate.
Fig. 8.
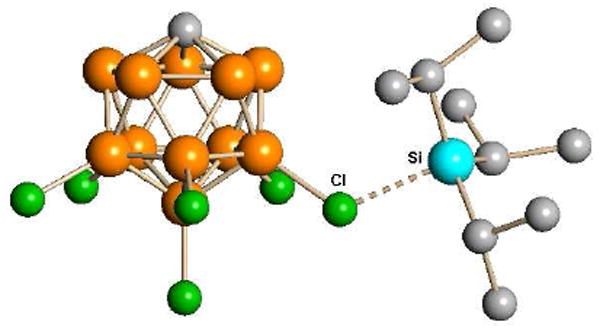
X-ray structure of i-Pr3Si(CHB11H5Cl6) (H omitted) showing developing i-Pr3Si+ silylium ion character (Si---Cl = 2.32 Å; ΣC-Si-C = 351.9°).
Trialkylsilyl carboranes silylate many substrates that are inert to trialkylsilyl triflates. For example, they silylate phosphazenes at N to give R3Si(N3P3Cl6)+ salts (Fig. 9).31 More importantly, these silylated cations are catalysts for ring-opening polymerization (ROP) at room temperature in halocarbon solvents. Previous Lewis acid catalysis has only been achieved in a melt at 200 °C. Silylation makes the ROP reaction open to mechanistic study and potentially greater control of the polyphosphazene properties.
Fig. 9.
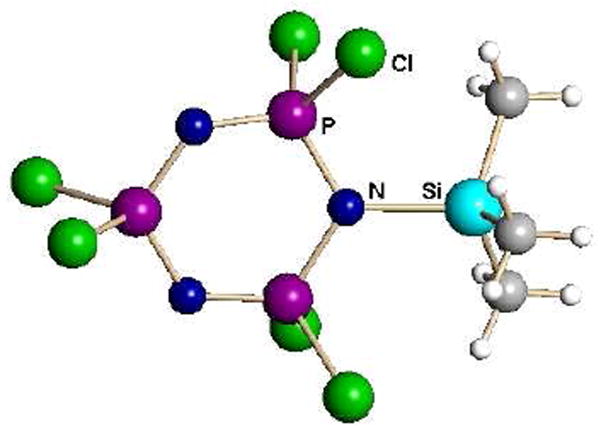
X-ray structure of the trimethylsilylated phosphazene cation as a carborane salt.
The potency of trialkylsilyl carboranes as halide abstractors is illustrated by their reaction with freons to give fluorocarbocations (Fig. 10).37 This reactivity has been put to important catalytic use in the dehydrofluorination of fluoroalkanes with silanes.38 No anion other than a carborane is inert enough to support this type of reactivity.
Fig. 10.
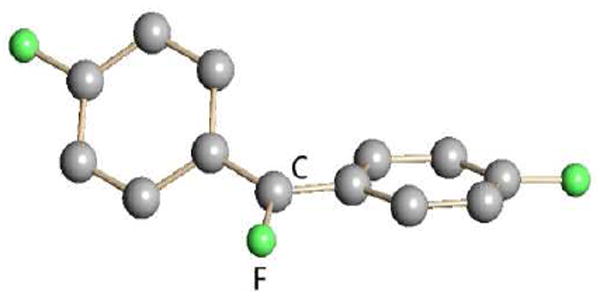
X-ray structure of the CF(p-C6H4F)2+ carbenium ion as a carborane salt.
Trialkylsilyl carboranes are successful where silver carboranes fail in abstracting chloride ion from Vaska’s compound in arene solvents.39 Replacement of chloride by a weakly coordinating carborane anion such as CHB11H5Cl6− in IrCl(CO)(PPh3)2 promotes unusually facile oxidative addition of chlorobenzene, giving the coordinatively unsaturated IrCl(CO)(C6H5)(PPh3)2]+ cation (Fig. 11). Similarly, trialkylsilyl carboranes abstract chloride from B(sub-phthalocyanine)Cl to give a rare example of a cationic boron Lewis acid, B(sub-Pc)+ (Fig. 12).40
Fig. 11.
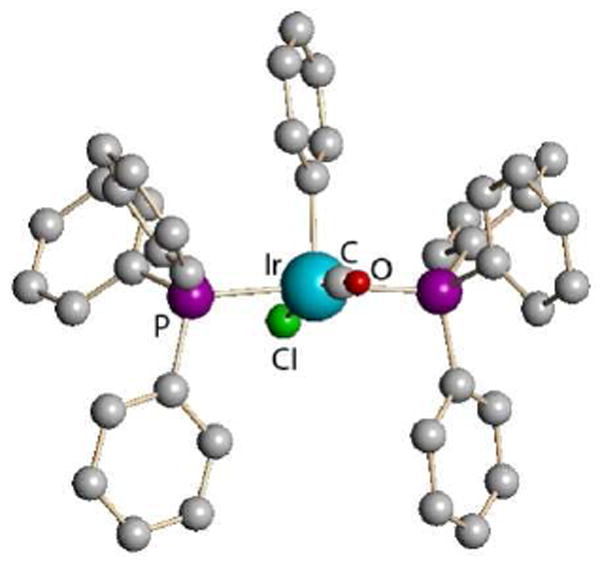
X-ray structure of the five-coordinate Ir(CO)Cl(Ph)(PPh3)2+ cation as a carborane salt.
Fig. 12.
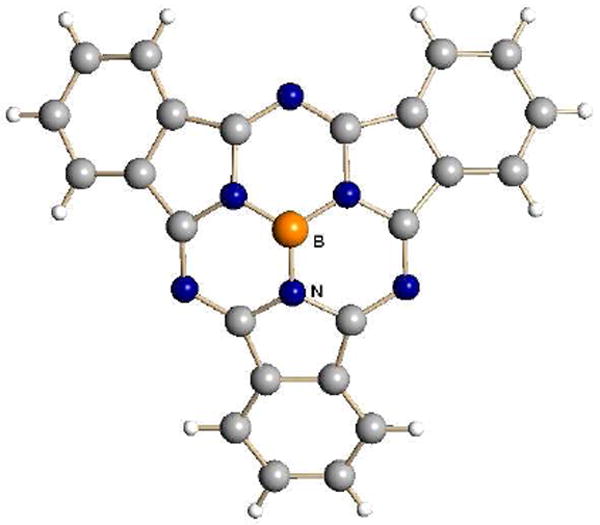
X-ray structure of the B(sub-phthalocyanine)+ cation as a carborane salt.
When the trialkylsilyl reagent utilizes one of the most weakly basic carborane anions, CHB11Cl11-, silylation of weak Lewis bases such as o-dichlorobenzene is observed.12 Silylation occurs at Cl rather than C (Fig. 13) to form a Cl-bound coordination compound (or silyl/aryl chloronium ion). Surprisingly, silylation of benzene has yet to be established even though silylated toluene41 has become the textbook example for the structure of an arenium ion on the σ-π complex continuum.19
Fig. 13.
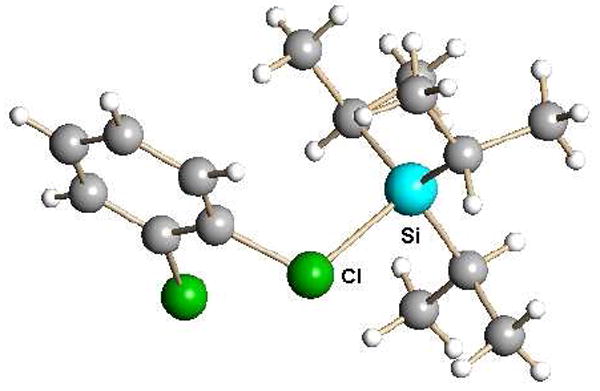
X-ray structure of the triethylsilylated o-dichlorobenzene cation as a carborane salt.
Me3Si(CHB11Cl11) reacts with Me3SiH to form the [Me3Si-H-SiMe3]+ cation which has a linear hydride-bridged structure (Fig. 14).12 This raises a caution about using excess silane in the preparative reaction of Eq. 3. With the weakest coordinating anions there is always the possibility of competition between the anion, solvent and excess silane for coordination to the R3Si+ moiety. The presence of the [R3Si-H-SiR3]+ cation is most easily detected by its characteristic νSi-H band in the IR spectrum at ca. 1900 cm-1.12
Fig. 14.
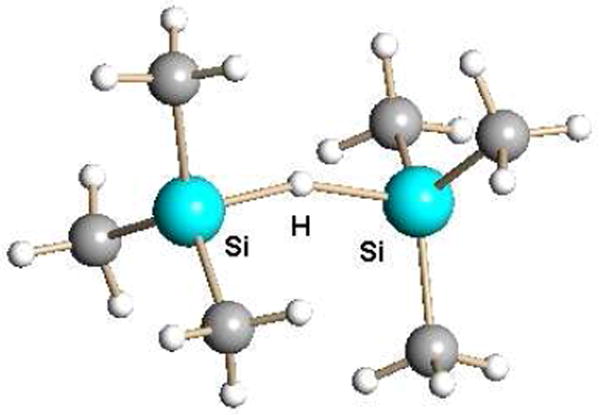
X-ray structure of the hydride-bridged silyl cation Me3Si-H-SiMe3+ as a carborane salt.
Finally, the preparative reaction for trialkylsilyl carborane formation has been used to generate the silylium moiety in situ and isolate the first X-ray crystallographically characterized example of a vinyl cation via intramolecular silylation of an alkyne (Scheme 1).42 Vinyl character in this doubly β-silyl-stabilized cation is evident in the short Cα=Cβ bond length of 1.221 Å (Fig. 15). In related chemistry, a rare example of an allyl cation has recently been isolated as a carborane salt.43
Scheme 1.
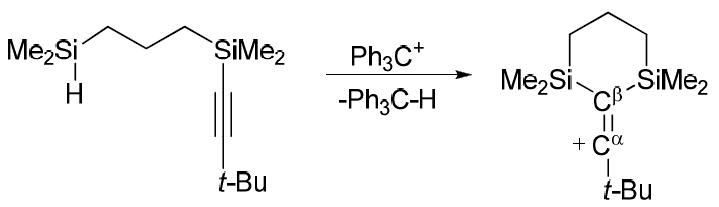
Synthesis of a stable vinyl cation (carborane anion omitted).
Fig. 15.
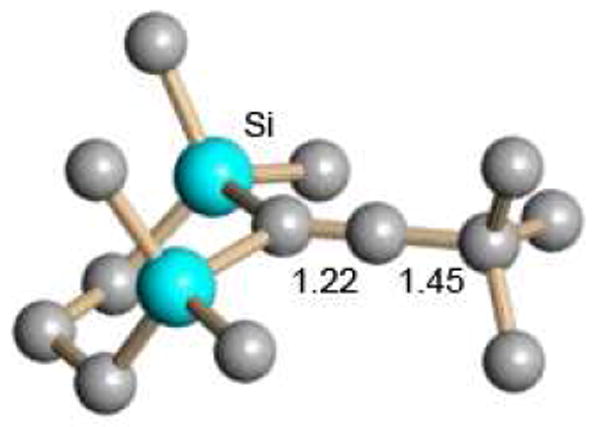
X-ray structure of a β-silyl stabilized vinyl cation (CHB11H5Br6- carborane anion not shown).
5. Conclusion
These examples of protonation, alkylation and silylation, where the corresponding triflate reagents fail, illustrate the greater electrophilicity of carborane reagents over their triflate counterparts. Although the increases in reagent strength for delivering H+, R+ and R3Si+ moieties to substrates have yet to be quantified by the methods of physical chemistry (the challenge lies in finding unreactive solvents and keeping them dry) they are clearly significant and useful. In the past, weakly basic substrates that are unresponsive to triflate reagents have been activated towards electrophiles by the use of Freidel-Crafts conditions or superacid media, i.e. Brønsted/Lewis acid mixtures involving AlCl3 or SbF5.29 The presence of excess Lewis acid in these media has the advantage of mopping up errant nucleophiles (e.g. water) and increasing the reactivity of the electrophile (E+) by decreasing the nucleophilicity of its counterion (e.g., converting SbF6- to Sb2F11- or Sb3F16-). However, apart from being a difficult working medium, mixed Lewis/Brønsted superacids have another disadvantage which is poorly recognized. The Lewis acid can form an acid/base adduct with the substrate, suppressing its basicity and thereby making it much harder to add the E+ electrophilic reagent. In other words, there is a competition for the substrate between the E+ reagent and the Lewis acid in the medium and, as a consequence, the basicities of most weakly basic molecules have been significantly underestimated. This phenomenon was first recognized in the protonation of weak bases2 and referred to as basicity suppression but there is every reason for it to apply to R+ and R3Si+ electrophiles as well. This is a more subtle reason why carborane reagents are useful. They are strong electrophiles and Lewis acid free.
Supplementary Material
Acknowledgments
I am greatly indebted to my students and associates who have worked on carborane anion chemistry over the past decade. Financial support from the National Science Foundation and the National Institutes of Health is gratefully acknowledged.
Biography
Christopher A. Reed is Distinguished Professor of Chemistry at the University of California, Riverside. He was born in New Zealand and educated at The University of Auckland. After postdoctoral studies at Stanford University in 1971-73 he joined the faculty at the University of Southern California. In 1998 he moved to UC Riverside where he created the Center for s and p Block Chemistry. His research interests include acids, reactive cations across the periodic table, bioinorganic chemistry, fullerenes and supramolecular chemistry.
Footnotes
SUPPORTING INFORMATION: Synthetic procedures for commonly used carborane reagents. This is available free of charge at http://pubs.acs.org.
References
- 1.Lawrance GA. Coordinated trifluoromethanesulfonate and fluorosulfate. Chem Rev. 1986;86:17–33. [Google Scholar]
- 2.Reed CA. Carboranes: A New Class of Anions for Strong Electrophiles, Oxidants and Superacid. Accounts Chem Res. 1998;31:133–139. [Google Scholar]
- 3.Körbe S, Schreiber PJ, Michl J. Chemistry of the Carba-closo-dodecaborate(−) Anion, CB11H12. Chem Rev. 2006;106:5208–5249. doi: 10.1021/cr050548u. [DOI] [PubMed] [Google Scholar]
- 4.Reed CA. Carborane acids: New “strong-yet-gentle” acids for organic and inorganic chemistry. Chem Commun. 2005:1669–1677. doi: 10.1039/b415425h. [DOI] [PubMed] [Google Scholar]
- 5.Macchioni A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem Rev. 2005;105:2039–2074. doi: 10.1021/cr0300439. [DOI] [PubMed] [Google Scholar]
- 6.Strauss SH. The search for larger and more weakly coordinating anions. Chem Rev. 1993;93:927–942. [Google Scholar]
- 7.Krossing I, Raabe I. Noncoordinating anions - Fact or fiction? A survey of likely candidates. Angew Chem Int Edn. 2004;43:2066–2090. doi: 10.1002/anie.200300620. [DOI] [PubMed] [Google Scholar]
- 8.Stoyanov ES, Kim K-C, Reed CA. An Infrared νN-H Scale for Weakly Basic Anions. Implications for Single Molecule Acidity and Superacidity. J Am Chem Soc. 2006;128:8500–8508. doi: 10.1021/ja060714v. [DOI] [PubMed] [Google Scholar]
- 9.Reed CA, Kim K-C, Stoyanov ES, Stasko D, Tham FS, Mueller LJ, Boyd PDW. Isolating Benzenium Ion Salts. J Am Chem Soc. 2003;125:1796–1804. doi: 10.1021/ja027336o. [DOI] [PubMed] [Google Scholar]
- 10.Jutzi P, Müller C, Stammler A, Stammler H-G. Synthesis, Crystal Structure, and Application of the Oxonium Acid [H(OEt2)2]+[B(C6F5)4]- Organometallics. 2000;19:1442–1444. [Google Scholar]
- 11.Lambert JB, Zhang S, Ciro SM. Silyl cations in the solid and in solution. Organometallics. 1994;13:2430–2443. [Google Scholar]
- 12.Hoffmann SP, Kato T, Tham FS, Reed CA. Novel weak coordination to silylium ions. Formation of nearly linear Si-H-Si bonds. Chem Commun. 2006:767–769. doi: 10.1039/b511344j. [DOI] [PubMed] [Google Scholar]
- 13.Ivanov SV, Rockwell JJ, Polyakov OG, Gaudinski CM, Anderson OP, Konstantin A, Solntsev KA, Strauss SH. Highly Fluorinated Weakly Coordinating Monocarborane Anions. 1-H-CB11F11-, 1-CH3-CB11F11-, and the Structure of [N(n-Bu)4]2[CuCl(CB11F11)] J Am Chem Soc. 1998;120:4224–4225. [Google Scholar]
- 14.Ivanov SV, Davis JA, Miller SM, Anderson OP, Strauss SH. Synthesis and Characterization of Ammonioundecafluoro-closo-dodecaborates(1−). New Superweak Anions. Inorg Chem. 2003;42:4489–4491. doi: 10.1021/ic0344160. [DOI] [PubMed] [Google Scholar]
- 15.Kuppers T, Bernhardt E, Eujen R, Willner H, Lehmann CW. [Me3Si][R-CB11F11]-Synthesis and Properties. Angew Chem Int Edn. 2007;46:6346–6349. doi: 10.1002/anie.200701136. [DOI] [PubMed] [Google Scholar]
- 16.Xie Z, Tsang C-W, Sze ET-P, Yang Q, Chan DTW, Mak TCW. Highly Chlorinated, Brominated, and Iodinated Icosahedral Carborane Anions: 1-H-CB11X11-, 1-CH3-CB11X11- (X = Cl, Br, I); 1-Br-CB11Br11- Inorg Chem. 1998;37:6444–6451. doi: 10.1021/ic9805628. [DOI] [PubMed] [Google Scholar]
- 17.Moss S, King BT, de Meijere A, Kozhushkov SI, Eaton PE, Michl LiCB11Me12: A catalyst for pericyclic rearrangements. J Org Lett. 2001;3:2375–2377. doi: 10.1021/ol0161864. [DOI] [PubMed] [Google Scholar]
- 18.Vyakaranam K, Barbour JB, Michl J. Li+-catalyzed radical polymerization of simple terminal alkenes. J Am Chem Soc. 2006;128:5610–5611. doi: 10.1021/ja060087+. [DOI] [PubMed] [Google Scholar]
- 19.Stoyanov ES, Hoffmann SP, Juhasz M, Reed CA. The Structure of the Strongest Bronsted Acid: The Carborane Acid H(CHB11Cl11) J Am Chem Soc. 2006;128:3160–3161. doi: 10.1021/ja058581l. [DOI] [PubMed] [Google Scholar]
- 20.Koppel IA, Burk P, Koppel IA, Leito Ivo, Sonoda T, Mishima M. Gas-Phase Acidities of Some Neutral Brønsted Superacids: A DFT and ab Initio Study. J Am Chem Soc. 2000;122:5114–5124. [Google Scholar]
- 21.Reed CA, Kim K-C, Bolskar RD, Mueller LJ. Taming Superacids: Stabilization of the Fullerene Cations HC60+ and C60+ Science. 2000;289:101–104. doi: 10.1126/science.289.5476.101. [DOI] [PubMed] [Google Scholar]
- 22.Mueller LJ, Elliott DW, Leskowitz GM, Struppe J, Olsen RA, Kim K-C, Reed CA. Uniform-Sign Cross-Peak Double-Quantum-Filtered Correlation Spectroscopy. J Magnetic Res. 2004;168:327–325. doi: 10.1016/j.jmr.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Tham FS, Nixon JF, Taylor C, Green JC, Reed CA. The Low Basicity of Phosphabenzenes: First Examples of Protonation, Alkylation and Silylation Reactions. Angew Chem Int Edn. 2008;47:3801–3804. doi: 10.1002/anie.200800878. [DOI] [PubMed] [Google Scholar]
- 24.Kim K-C, Reed CA. unpublished results. [Google Scholar]
- 25.Stoyanov ES, Kim K-C, Reed CA. The Nature of the H3O+ Hydronium Ion in Benzene and Chlorinated Hydrocarbon Solvents. Conditions of Existence and Reinterpretation of Infrared Data. J Am Chem Soc. 2006;128:1948–1958. doi: 10.1021/ja0551335. [DOI] [PubMed] [Google Scholar]
- 26.Stoyanov ES, Stoyanov IV, Tham FS, Reed CA. The Nature of the Hydrated Proton H(aq)+ in Organic Solvents. J Am Chem Soc. 2008;130:12128–12138. doi: 10.1021/ja803535s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stasko D, Reed CA. Optimizing the Least Nucleophilic Anion. A New, Strong Methyl+ Reagent. J Am Chem Soc. 2002;124:1148–1149. doi: 10.1021/ja0118800. [DOI] [PubMed] [Google Scholar]
- 28.Kato T, Stoyanov E, Geier J, Grutzmacher H, Reed CA. Alkylating Agents Stronger than Alkyl Triflates. J Am Chem Soc. 2004;126:12451–12457. doi: 10.1021/ja047357d. [DOI] [PubMed] [Google Scholar]
- 29.Kato T, Reed CA. Putting t-Butyl Cation in a Bottle. Angew Chem Int Edn. 2004;43:2908–2911. doi: 10.1002/anie.200453931. [DOI] [PubMed] [Google Scholar]
- 30.Olah GA, Prakash GKS, Molnár Á, Sommer J. Superacid Chemistry. 2. Wiley; Hobocken, NJ: 2009. [Google Scholar]
- 31.Zhang Y, Tham FS, Reed CA. Phosphazene Cations. Inorg Chem. 2006;45:10446–10448. doi: 10.1021/ic062077f. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Reed CA. Can the Hexamethyldihydrazinium Cation, Me3NNMe32+, be Prepared? Dalton Trans. 2008:4392–4394. doi: 10.1039/b803304h. [DOI] [PubMed] [Google Scholar]
- 33.Gill PMW, Radom L. Structures and stabilities of the dimer cations of first- and second-row row hydrides. J Am Chem Soc. 1989;111:4613–4622. [Google Scholar]
- 34.Alder RW, Sessions RB, Gmünder JO, Grob CA. Deprotonation of hydrazinium dications in the diazoniapropellane series to form bridgehead iminium ions; external and intramolecular trapping. J Chem Soc Perkin Trans. 1984;2:411–417. [Google Scholar]
- 35.Reed CA. The Silylium Ion Problem, R3Si+. Bridging Organic and Inorganic Chemistry. Acc Chem Res. 1998;31:325–332. [Google Scholar]
- 36.Zhang Y, Huynh K, Manners I, Reed CA. Ambient temperature ring-opening polymerization (ROP) of cyclic chlorophosphazene trimer [N3P3Cl6] catalyzed by silylium ions. Chem Commun. 2008:494–496. doi: 10.1039/b713933k. [DOI] [PubMed] [Google Scholar]
- 37.Douvris C, Stoyanov ES, Tham FS, Reed CA. Isolating Fluorinated Carbocations. Chem Commun. 2007:1145–1147. doi: 10.1039/b617606b. [DOI] [PubMed] [Google Scholar]
- 38.Douvris C, Ozerov OV. Hydrodefluorination of perfluoroalkyl groups using silylium-carborane catalysts. Science. 2008;321:1188–1190. doi: 10.1126/science.1159979. [DOI] [PubMed] [Google Scholar]
- 39.Douvris C, Reed CA. Increasing the Reactivity of Vaska’s Compound. Oxidative Addition of Chlorobenzene at Ambient Temperature. Organometallics. 2008;27:807–810. [Google Scholar]
- 40.Kato T, Tham FS, Boyd PDW, Reed CA. Synthesis and Structure of the Coordinatively Unsaturated Boron Subphthalocyanine Cation, [B(SubPc)]+ Heteroatom Chem. 2006;17:209–216. [Google Scholar]
- 41.Lambert JB, Zhang S, Stern CL, Huffman JC. Crystal Structure of a Silyl Cation with No Coordination to Anion and Distant Coordination to Solvent. Science. 1993;260:1917–1918. doi: 10.1126/science.260.5116.1917. [DOI] [PubMed] [Google Scholar]
- 42.Müller T, Juhasz M, Reed CA. The X-ray Structure of a Vinyl Cation. Angew Chem Int Ed. 2004;43:1543–1546. doi: 10.1002/anie.200352986. [DOI] [PubMed] [Google Scholar]
- 43.Duttwyler S, Zhang Y, Linden A, Reed CA, Baldridge KK, Siegel JS. Synthesis and Crystal Structure of a Silyl-Stabilized Allyl Cation Formed by Disruption of an Arene by a protonation-Hydrosilation Sequence. Angew Chem Int Edn. 2009;48:3787–3790. doi: 10.1002/anie.200900098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.