

Abstract
Toll-like receptors (TLRs) provide an important link between innate and adaptive immune system. We hypothesized that the recognition of endogenous TLR4 ligands is occurring at the time of transplantation, and these innate signals drive the inflammation and affect alloimmune responses. We confirmed that early after transplantation of allogenic islets, transcripts for TLR4 as well as potential ligands were released or upregulated. In an allogenic islet transplantation model, genetic disruption of TLR4 on donor islets had no effect on allograft survival, whereas TLR4 deficiency in recipients lead to prolonged graft survival. Low dose rapamycin-treatment of TLR4−/− recipients induced permanent engraftment of 45% islet graft (p=0.005) compared to WT recipients. This prolonged graft survival was dependent on the presence of CD4+CD25+Foxp3+ Treg. Naïve CD4+CD25− T cells cultured with the TLR4 ligand lipopolysaccharide showed enhanced IL-4, IL-6, IL-17, IFNγ secretion and inhibited TGFβ induced Foxp3+Treg generation. Thus, inhibition of recipient TLR4 activation at the time of transplantation decreases proinflammatory signals and allows Treg generation.
Keywords: Toll-like receptor, DAMP, islet allograft rejection
Introduction
The main obstacle to successful islet transplantation is the poor long-term allograft survival. The 5-year follow-up data of the Edmonton protocol reveals that only around 10% of the transplanted patients maintain insulin independence [1]. Therefore, potent and non islet toxic immunosuppressive strategies are needed.
Toll like receptors (TLRs) are critical innate immune receptors expressed on a variety of cells including dendritic cells, monocytes, and T cells. To date, 13 human and 9 murine TLRs have been described [2]. These receptors specifically recognize pathogen-associated molecular patterns (PAMPs) that are expressed on or in infectious agents, and their activation are crucial for both innate immunity as well as effective presentation of antigens to the adaptive immune system. Upon ligand binding, TLR-mediated signaling induces cytokines, chemokines and increases expression of cellular membrane proteins related to inflammatory responses [2].
There are emerging data from experimental and human studies that TLR4 is able to sense endogenous ligands heat shock proteins (HSPs), hyaluronan, the extra domain A (EDA) of fibronectin, fibrinogen, and high mobility group box protein 1 (HMGB1), which have been implicated in the pathogenesis of acute allograft rejection by amplifying alloimmune responses [3-6]. TLR signaling was activated in response to a vascularized allograft and MyD88−/− mice were protected from minor antigen mismatched skin allograft rejection [7]. The mechanism involved a reduced number of mature DCs in draining lymph nodes, leading to impaired generation of anti-graft-reactive T cells and impaired Th1 immunity [7]. Effector T cells were traditionally thought to segregate into Th1 (IL-12) and Th2 (IL-4) subsets but more recently, we and others described two other pathways of differentiation that induced regulatory T cells (TGFβ/IL-2) and Th17 cells (TGFβ/IL-6) [8-10]. In particular IL-6, a potent Th17 differentiation cytokine is induced after TLR activation [11]. A main effect of TLR signals in vivo may be to relieve effector T cells from suppression by Tregs [8].
We hypothesized that the recognition of endogenous ligands is occurring at the time of transplantation, and these innate signals drive the inflammation and affect alloimmune responses. In present work, we investigated the effect of TLR4 induced inflammation using TLR4 deficient donor or recipient mice. We found that disruption of recipient, but not donor-associated TLR4, especially in combination with low-dose immunosuppression greatly augmented islet allograft survival. This enhanced survival of TLR4−/− recipient islet grafts was dependent of intragraft Tregs. In vitro results showed that TLR4 deficiency promoted generation of enhanced TGFβ induced Tregs generation compared to WT control cells. Signaling through TLR4 activation resulted in the inhibition of the generation of TGFβ induced Foxp3+ T cells from peripheral naïve CD4+CD25− T cells. In addition to providing insight into mechanisms underlying islet rejection, our findings indicate that TLR4 is a potential target to prolong allograft survival.
Methods
Mice
BALB/c (H-2d), C57BL/6 (B6, H-2b), TLR4 mutant mice (C.C3-Tlr4Lps-d/J) on a BALB/c background (TLR4−/−BALB/c, H-2d), TLR4-C3H/HeJ on a C3H background (TLR4C3H/HeJ, H-2k), and C57BL/10ScNJ (TLR4−/−B10; H-2b) were purchased from the Jackson Laboratories (Bar Harbor, ME). TLR4−/− (TLR4−/−B6, H-2b) were provided by Dr. Abreu, Mount Sinai School of Medicine, New York [12]. Deletion was confirmed by genotyping. 8 to 12 weeks old mice were housed under specific-pathogen-free conditions and treated in strict compliance with protocols approved by our Institutional Animal Care and Use Committee.
Reagents
Collagenase P was purchased from Roche Diagnostics (Mannheim, Germany). Streptozotocin and Rapamycin were from Sigma, St. Louis, MO. Purified lipopolysaccharides (LPS) from E. coli 0111:B4 (InvivoGene, San Diego, CA). Foxp3, IL-10, IFN-γ, CD68, CD4, CD8 (Serotec, Oxford, UK). FITC-conjugated goat anti-mouse IgG (Jackson Immuno Research, West Grove, PA). Fluorochrome-labeled monoclonal antibodies CD4 (GK 1.5), CD25 (PC61.5), and Foxp3 (FKJ-16s) (eBioscience, San Diego, CA). Alkaline phosphatase-conjugated anti-biotin antibody (Vector Laboratories, Burlingame, CA). ELISPOT IFN-γ antibody (BD Biosciences, San Jose, CA).
Diabetic Model, Islet Isolation and Transplantation
The recipients were rendered diabetic by a single intraperitoneal injection of 180 mg/kg streptozotocin and considered diabetic when the tail-vein blood glucose concentration was more than 350 mg/dl for 2 consecutive days. In some experiments mice received rapamycin (0.2 mg/kg i.p. per day) for 7 consecutive days. To deplete CD25+ cells, 0.5 mg rat anti-mouse CD25 mAb (PC61) was administered i.p. on day -1 and then every other day for 5 total doses. CD25 depletion was confirmed by flow cytometry of splenocytes. Islet isolation and transplantation were previously described in detail [13-15]. Islets were purified on a discontinuous Ficoll gradient (Sigma, St. Louis, MO). A total of 400 islets were transplanted beneath the renal capsule. Animals with a blood glucose level of <200 mg/dl after 2 days were considered to have primary function. Rejection was defined as tail-vein glucose concentration of more than 250 mg/dl on two daily consecutive measurements. Long-term graft function (>90d) was confirmed with the recurrence of hyperglycemia after graft removal.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from islet grafts with 1 ml of TriZol solution (Life Technologies BRL, Grand Island, NY). For cDNA synthesis, 1 μg of total RNA was primed with oligo(dT) as described previously [16,17]. The primers were designed to be intron spanning if possible and have been reported elsewhere or available on request [13,16]. Quantitative PCR was performed on a LightCycler (Roche Applied Science, Indianapolis, IN) with the FastStart QuantiTect SYBR Green PCR kit (Quiagen, Valencia, CA). All runs included a control for genomic DNA contamination.
Histopathology and Immunohistochemistry
Functional islet grafts (n=3) at day 7 and grafts from long-term survivors (n=3) at day 90 post-transplant were analyzed. 5μm thick frozen sections were H&E stained and used to assess islet morphology and leukocyte infiltration. Frozen sections were immunostained for intracellular Foxp3 and cytokines (IL-10, IFN-γ), and cell surface markers (CD68, CD4, CD8) as previously described [18]. The mean percentage of cells staining positive in immunohistochemical analysis for each marker was semiquantitatively classified by a blinded investigator (M.G.) for each staining condition: staining level of 0 (indicated no positive cells), 0.25 (5% positivity), 0.5 (10% positivity), 0.75 (15% positivity), 1 (20% positivity), 1.5 (25% positivity), 2 (30% positivity), 2.5 (35% positivity), 3 (>40% positivity) [18].
Culture of CD4+CD25− T cells
C57BL/6 or TLR4−/− mice were sacrificed, spleens removed and gently dissociated into single cell suspensions, and red blood cells removed using hypotonic ACK lysis buffer. Cells were stained with APC anti-mouse CD4, PE anti-mouse CD8 and FITC anti-mouse CD25 antibodies for 30 minutes on ice. CD4+CD25−cells were sorted using FACS Vantage DiVA (BD Bioscience). The purity of cells was more than 99%. Peritoneal cells were harvested and cultured overnight in complete RPMI medium (RPMI-1640 supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1× non-essential amino acids, and 2 × 10−5M 2-mercaptoethanol) containing LPS (100ng/ml) at 37°C in 5% CO2 incubator. Cells were washed and used as antigen presenting cells (APCs). Purified CD4+CD25− T cells (5 × 104 cells/well) were cultured with syngenic activated APCs (10000 cells/well) in the presence of IL-2 (10 ng/ml), anti-CD3ε mAb (1 μg/ml), TGFβ (5 ng/ml), LPS (100 ng/ml), and rapamycin (50 ng/ml) in a final volume of 200μl complete RPMI medium Cells were cultured for the 4 days at 37°C in a 5% CO2 incubator.
Flow Cytometry
In vitro cultured or freshly purified spleen and draining lymph node cells were isolated from transplanted mice, and stained with fluorochrome-labeled monoclonal antibodies against CD4 (GK 1.5), CD25 (PC61.5) and intracellular Foxp3 (FKJ-16s). Cells were acquired using the FACSCaliber flow cytometer using CELLQuestTM software (BD Biosciences, Mountain View, CA). Data were analyzed using Flowjo software (Tree Star Inc. Ashland, OR).
Luminex Assay
Supernatants were collected at the end of T cell culture and cytokines were quantitated using Bio-Plex Pro Mouse Cytokine TH1/TH2 assay kit. Assays were conducted according to manufacturer's instructions and analyzed on a Bio-Plex 200 System (Bio-Rad, Hercules, CA).
Statistical Analysis
Results are expressed as mean ± SEM, unless stated otherwise. Survival analysis was performed by the Kaplan Meier method comparing groups using the log-rank test. Differences between groups were compared by Student's t-test or by Mann-Whitney-U Test if applicable. P-values <0.05 were considered as statistically significant.
Results
Induction of TLR4 and its ligands during allogenic transplantation
To determine the expression of TLR4 and the presence of potential ligands during allograft rejection, we analyzed TLR4, HSP70, and HMGB1 gene expression in syngenic (H-2b into H-2b) and allogenic (H-2d into H-2b) islet grafts (Fig 1). The results show that TLR4 was strongly up-regulated in allografts after 1 day of transplantation and its level decreased to that of isografts by 7 days. Similarly, expression of the TLR4 ligands HSP70 and HMGB1 was highly up-regulated in the allografts compared to syngenic grafts. These results show that early after allogenic transplantation, TLR4 and its ligands are up-regulated in the allograft and might be involved in the initiation of the alloimmune responses.
Figure 1. TLR4, HSP70, and HMGB1 mRNA expression in allografts and isografts.

Islets were transplanted beneath the renal capsule of syngenic (H-2b into H-2b) or complete MHC mismatched (H-2d into H-2b) diabetic recipients. Functioning grafts were harvested and gene expressions were analyzed by qRT-PCR (n=3 per group for each time points). *p<0.05 compared with isografts.
Effect of recipient TLR4 on islet allograft survival
To determine the role of recipient TLR4 on allograft survival, we performed allogenic islet transplantation from WT mice into two different strains of TLR4 deficient mice and monitored allograft function. TLR4−/− BALB/c originated from TLR4 mutant mice that have an amino acid substitution that does not affect receptor expression but prevents its signaling, whereas TLR4−/−B6 mice do not express TLR4 protein [12]. Transplantation of WT C57BL/6 islets into TLR4−/− BALB/c recipients significantly prolonged mean graft survival time (MST) 18.3±3.8d (p=0.004) compared to WT recipients (13.8±0.8d) (Fig 2A). When donor and recipient strains were switched (BALB/c into TLR4−/−B6), the survival benefit was less pronounced (10.2±0.7d vs 12.2±2.2d; p=0.1) (Fig 2 B).
Figure 2. Survival of allogenic islets in TLR4−/− recipients with low-dose rapamycin.

(A) Survival of WT allogenic BALB/c islets transplanted into untreated C57BL/6 (n=5) or TLR4−/−B6 (p=0.1; n=5); (B) allograft survival of WT allogenic C57BL/6 islets transplanted into untreated BALB/c (n=5) or TLR4−/−BALB/c mice (p=0.004; n=4). (C) BALB/c islet allografts transplanted into rapamycin treated (Rapa; 0.2 mg/kg/d for 7d) C57BL/6 mice (n=10) or TLR4−/−B6 recipients (p=0.005; n=11). Depleting anti-CD25 mAb or IgG control were administered at the time of transplantation into rapamycin treated TLR4−/−B6 recipients (n=4 each).
The results showed that TLR4 deficiency of recipients led to modest prolongation of allograft survival. Therefore, we hypothesized that the addition of low dose of immunosuppression may provide a robust allograft survival. Treatment of WT mice with low dose of rapamycin (0.2 mg/kg per day for 7 days) resulted in a MST of 18.3±1.5 d, whereas rapamycin administration into TLR4−/−B6 recipients led to significant prolongation of graft survival (p=0.005) with 45% (5 out of 11) of recipients demonstrating long-term allograft acceptance (>90 days) (Fig 2 C). Furthermore, switching donor and recipient strains (B6 into TLR4−/− BALB/c) confirmed these results (n=3) (data not shown). Long-term graft function was confirmed by nephrectomy of the graft-bearing kidney, which resulted in a prompt return to hyperglycemia. To test immunological tolerance, mice with allograft survival greater than 90 days underwent removal of first islet allograft and then retransplanted with the same donor-strain islets to the contralateral kidney without further therapy. The retransplanted mice rejected their second islet grafts on day 24 and 31, indicating that a state of hyporesponsiveness.
To determine if hyporesponsiveness of prolonged islet survival was due to increased numbers of CD4+CD25+ nTreg, CD25+ cells were depleted at the time of transplantation. Injection of anti-CD25 monoclonal antibody has been shown to efficiently deplete CD4+CD25+ T cells in transplant models [19,20]. The results show that administration of anti-CD25 monoclonal antibody into rapamycin-treated TLR4−/− recipients inhibited prolonged allograft survival (MST 15.5±2.9 d) (Fig 2 C) suggesting that CD4+CD25+ nTregs are involved in the prolonged islet allograft survival.
Effect of donor TLR4 on islet allograft survival
Recently it has been shown that islet-associated TLR4 engagement affects allograft outcome [4]. We tested the effect of TLR4 expressed on the islets on allograft survival using three different background strains. All three TLR4 deficient strain islets were rejected by untreated BALB/c recipients within 14 days: TLR4−/−B10 donor islets rejected after 12.2 2.1d (n=4), TLR4−/−B6 after 12.3 0.3 d (n=3), TLR4C3H/HeJ after 10 0 d (n=3) and WT C57BL/6 controls rejected after 13 0.8 d (n=5). It had been reported earlier that the donor- recipient combination of C3H into BALB/c lead to allograft rejection at 11 4.8 d [21]. In contrast to previously published data [4], our results showed that disruption of the donor TLR4 pathway had no effect on islet allograft survival.
Characterization of intragraft cytokine expression and immune cell infiltration
We next characterized intragraft cytokines (IL-10 and IFN-γ) content along with the presence of Treg (Foxp3), T cell (CD4 and CD8) and macrophage (CD68) by immunohistochemistry. Functional islet allografts (H-2d into H-2b) from control and experimental groups were analyzed on day 7 and for permanently engrafted TLR4−/−B6 recipients on day 90. Grafts on day 90 after transplantation from rapamycin-treated TLR4−/−B6 recipients showed a dense mononuclear cellular infiltrate, but the islets were well preserved (Fig 3 A). Compared to WT recipients with TLR4−/− recipients, there were minor differences in the number of CD4+ and CD8+ cells infiltration but TLR4−/− recipients had significantly fewer macrophages in the graft. Interestingly, we found that TLR4−/− recipients had significantly increased number of Foxp3+ nTreg as early as 7 days of transplantation, and this was maintained throughout the study (90 days) (Fig 3 A,B). Analysis of cytokine expressing cells in the islet grafts in TLR4−/− recipients showed a significantly increased number of IL-10 and decreased IFN- producing cells compared to WT controls and these differences were maintained in permanently engrafted allograft of TLR4−/− recipients (Fig 3 B). Immunhistochemistry data of day 7 were confirmed in a different set of mice by gene transcript analysis (Fig 3 C). IFN-γ mRNA was significantly lower in TLR4−/− recipients. Transcript for IL-10 was significantly higher and for Foxp3 showed a trend (p=0.07) towards higher expression in TLR4−/− recipients compared to WT mice (Fig 3 C).
Figure 3. Intragraft cytokine expression and immune cell infiltration.
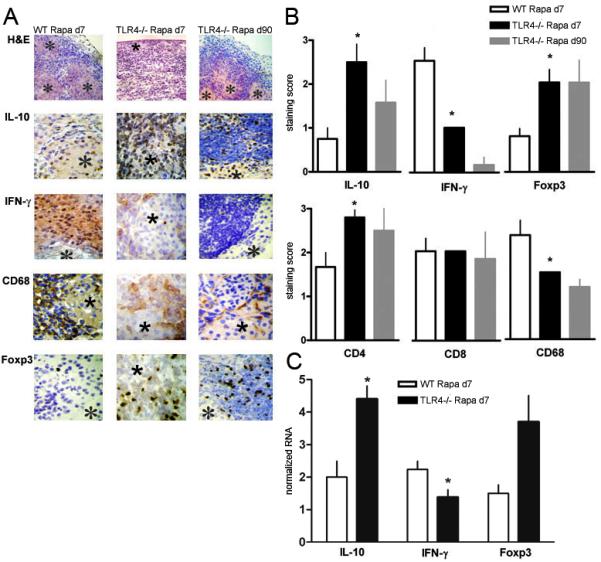
(A) Functioning grafts were harvested on day 7 and day 90 post-transplant and analyzed by H&E, immunohistochemistry for IL-10, IFN-γ, CD68, Foxp3, CD4 and CD8; *indicates an islet; original magnification, ×200 (H&E) and ×400 (IHC). (B) Score of staining of grafts in WT and TLR4−/−B6 recipients (n=3 mice in each group). *p<0.05 compared to wild-type day 7. (C) Functioning grafts were harvested on day 7 and analyzed by qRT-PCR for IL-10, IFN-γ, and Foxp3 (n=3-4). *p<0.05.
Effect of TLR4 on Foxp3 induction in CD4+CD25− cells
We next examined whether TLR4 deficiency affects the generation of Foxp3+ adoptive Treg. TLR4−/− or WT peripheral CD4+CD25− T cells were co-cultured with syngenic peritoneal APCs in the presence of TGFβ and with or without LPS [22]. The results showed that TLR4 activation leads to inhibition of Foxp3+ Treg generation suggesting that under activation condition TLR4 stimulation inhibit Foxp3 expression (Fig 4 A). Cytokine mRNA expression and protein analysis in the culture supernatant suggested that LPS enhanced the IL-4, IL-6 and IFNγ secretion (Fig 4 B, C). It has been shown that IL-6 along with TGFβ inhibits the Foxp3 expression and promote the development of IL-17 producing inflammatory T cells [9]. Stimulation of naïve CD4+CD25− T cells with TGFβ in presence of LPS inhibits the generation of Foxp3+ Treg (Fig 4 A) and enhanced the IL-17 production (Fig 4 B, C). In addition to lower IL-17 secretion we found that IL-21 and IL-22 mRNA showed a trend towards lower expression in TLR4−/− recipients compared to WT mice (Fig 4 B). Given that Th17 cells secrete not only IL-17 but also IL-21 and IL-22, these cytokines likely cooperate to induce inflammation. TLR4−/− mice have similar number of CD4+CD25+ nTreg compared to control mice (data not shown). These results show that presence of TLR4 signal inhibit the development of Foxp3+ adoptive Treg and enhanced the IL-17 producing inflammatory T cells.
Figure 4. Foxp3 expression in CD4+CD25− T cells and cytokine secretion.

CD4+CD25− T cells from WT (B6) and TLR4−/− were cultured in presence of IL-2, anti-CD3ε, and indicated reagents for 4 days along with overnight activated syngenic peritoneal cells. (A) Intracellular Foxp3 staining was analyzed gating on CD4+ T cells. (B) Cytokines mRNA was measured using qRT-PCR. Error bar represents SEM from three independent experiments. (C) Cytokine secretion in culture supernatants was measured by ELISA. Data shown are representative of two independent experiments. *p<0.05.
Discussion
Our results delineate several new insights into the role of TLR4 in the pathogenesis of islet allograft graft rejection. Our findings support the following interpretation: 1) alloimmune response can produce and release endogenous DAMPs, including HMGB1 and HSP70, 2) following transplantation, recipient TLR4 deficiency led to Treg-dependent prolonged graft survival, 3) TLR4 activation resulted in the inhibition of Foxp3+ Treg generation and enhanced IL-17 production, 4) in contrast to previous reports, donor TLR4 suppression did not affect allograft survival.
We showed that transplanting untreated allogenic TLR4 deficient recipients had a modest effect on allograft survival (Fig 2 A,B). We observed a strain-specific results, with a more biologically significant effect with the strain combination of donor C57BL/6 to recipient BALB/c, which is known to be less CD8 dependent [23]. Of note, our results are consistent with recent data that transplanting C57BL/6 islet grafts into untreated TLR4 mutated recipients (C3H/HeJ) experienced delayed rejection [24]. We found that targeting the TLR4 signaling is of synergistic benefit when combined with low-dose rapamycin (Fig 2 C). We showed that this prolonged survival was linked with an increased number of CD4+CD25+Foxp3+ regulatory T cells and IL-10 expression in TLR4−/− grafts (Fig 3). The presence of CD4+CD25+ cells was required for permanent engraftment (Fig 2 C). We further showed that TLR4 stimulation enhanced IL-6 secretion, which is known to inhibit Foxp3+ Treg development, and also enhanced IL-17 producing inflammatory Th17 cells (Fig 4 B,C) [9,10]. New data showed the potential of complement to interact with TLR4 to enhance TH17 cell development [25]. Our in vivo and in vitro results describe a unique mechanism where TLR4 stimulation in allogeneic transplantation model induces inflammation and suppress the Treg development. This TLR4-dependent Treg regulation is especially pronounced in the setting of rapamycin co-treatment. Rapamycin was recently shown to selectively expand naturally occurring CD4+CD25+Foxp3 Tregs in vitro and in vivo [26,27].
This would be consistent with in vitro data where the ability of Tregs to suppress cytotoxic T cells was prevented by LPS stimulation, and in vivo data where LPS injection into a tumor tolerant mouse let to tumor rejection in the presence of CD4+CD25+ Treg [8,28]. Our study is compatible with previous report that found that defective MyD88 signaling synergized with costimulatory blockade to prolong skin allograft survival [29]. These authors found that impaired inflammation (IL-6/TNFα production by DC) led to reduced effector T-cell proliferation and increased regulation by Tregs [29].
Interestingly, triggering of Treg can also induce higher Foxp3 expression and in co-culture suppression with high dose of LPS enhanced the suppression of effector T cells by Treg [30]. In the clinical context it is reasonable to expect that the higher level of LPS may represent an overwhelming inflammatory process (e.g., septic shock) where enhanced suppressive action of Tregs would be beneficial. In contrast, lower dose of LPS, may mimic contact endogenous ligands, where impaired suppression would result in enhanced immunity. The in vivo environment of cytokines and TLR ligands might further neutralize Treg's suppressive potential.
In the transplant setting the absence of functional TLR4 did not impair Treg function and late LPS infusion (50 days post transplant) in anti-CD154-treated tolerant heart recipients reduced intragraft Foxp3:Tbet ratio [20]. In contrast to late LPS infusion, LPS at the time of transplantation prevented long-term acceptance of skin and heart allografts (induced with DST and anti-CD40L) [31,32]. These clinically relevant studies suggest that recipient TLR4 activation at the time of transplantation but not at later stages prevents permanent engraftment.
We used LPS as an experimental tool to active TLR4. Further studies are needed to identify TLR4 ligands in the setting of transplantation. We found that potential TLR4 activators, HSP70 and HMGB1, were released or up-regulated during initial injury or inflammation. Of note, recent studies found that HSP70-deficient skin allografts have a prolonged graft survival [33] and cardiac allografts showed a significant up-regulation of HMGB1 and specific blockade of endogenous HMGB1 prolonged cardiac allograft survival [5]. HMGB1 was found to induce TLR4-dependet IL-1β production from monocytes, suggesting a mechanism by which HMBG1 can modulate alloreactive T cell responses [34].
Islet cells (e.g. DC and parenchymal cells) express functional TLRs [35]. Goldberg et al. reported that 4 out of 6 TLR4-deficient allogenic islets (C57BL/10ScNJ) survived indefinitely in untreated BALB/c recipients, while all WT controls rejected in less than 20 days [4]. The authors proposed that transplanting TLR4−/− islets induces antigen-specific tolerance as two mice retransplanted with same donor but TLR4+/+ islet grafts without further therapy survived long-term. These effects were dependent on the ability of the recipient to induce HO-1. We were unable to achieve long-term acceptance using the same TLR4−/− mice (C57BL/10ScNJ from the Jackson Laboratory). In addition, two other TLR4-deficient donor mice (TLR4C3H/HeJ and TLR4−/−B6) all rejected within 2 weeks when transplanted into untreated BALB/c recipients. Our data are supported by previous observation that TLR4 mutated (C3H/HeJ) donor islets were all rejected from SCID mice reconstituted with naïve BALB/c splenocytes [36]. The findings of the earlier report [4] also raises the possibility that donor TLR4 may contribute to islet allograft rejection under certain conditions (e.g. gut flora). It was recently shown that pathogen-free NOD mice lacking MyD88 protein did not develop autoimmune diabetes, an effect that was reversed by gut colonization [37].
In the clinical context, TLR4 blockade is more likely to be successful than targeting individual ligands or downstream effectors which often serve redundant functions. Together with our previous studies demonstrating the beneficial effects of TLR4 inhibition on IR injury [6], and others on tolerance [38] our study sets the stage for future work aimed at inhibiting TLR activation in a clinical setting. There is extensive evidence that the innate system interacts with the adaptive immune system and targeting TLR4 signaling might provide new opportunities to intervene in alloimmune response.
Acknowledgments
This work is supported by National Institutes of Health research grant to B.S. KO8 AI 071038-01A1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
NZ and BK contributed equally.
References
- 1.Ryan EA, Paty BW, Senior PA, Bigam D, Alfadhli E, Kneteman NM, Lakey JR, Shapiro AM. Five-year follow-up after clinical islet transplantation. Diabetes. 2005;54:2060–2069. doi: 10.2337/diabetes.54.7.2060. [DOI] [PubMed] [Google Scholar]
- 2.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 3.Wrenshall LE, Cerra FB, Singh RK, Platt JL. Heparan sulfate initiates signals in murine macrophages leading to divergent biologic outcomes. J Immunol. 1995;154:871–880. [PubMed] [Google Scholar]
- 4.Goldberg A, Parolini M, Chin BY, Czismadia E, Otterbein LE, Bach FH, Wang H. Toll-like receptor 4 suppression leads to islet allograft survival. Faseb J. 2007 doi: 10.1096/fj.06-7910com. [DOI] [PubMed] [Google Scholar]
- 5.Huang Y, Yin H, Han J, Huang B, Xu J, Zheng F, Tan Z, Fang M, Rui L, Chen D, Wang S, Zheng X, Wang CY, Gong F. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. 2007;7:799–808. doi: 10.1111/j.1600-6143.2007.01734.x. [DOI] [PubMed] [Google Scholar]
- 6.Kruger B, Krick S, Dhillon N, Lerner SM, Ames S, Bromberg JS, Lin M, Walsh L, Vella J, Fischereder M, Kramer BK, Colvin RB, Heeger PS, Murphy BT, Schroppel B. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstein DR, Tesar BM, Akira S, Lakkis FG. Critical role of the Toll-like receptor signal adaptor protein MyD88 in acute allograft rejection. J Clin Invest. 2003;111:1571–1578. doi: 10.1172/JCI17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–1036. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 9.Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, Vollmar P, Stritesky GL, Kaplan MH, Waisman A, Kuchroo VK, Oukka M. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105:18460–18465. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, Reid SP, Levy DE, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. 2009;182:259–273. doi: 10.4049/jimmunol.182.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 12.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 13.Schroppel B, Zhang N, Chen P, Zang W, Chen D, Hudkins KL, Kuziel WA, Sung R, Bromberg JS, Murphy B. Differential expression of chemokines and chemokine receptors in murine islet allografts: the role of CCR2 and CCR5 signaling pathways. J Am Soc Nephrol. 2004;15:1853–1861. doi: 10.1097/01.asn.0000130622.48066.d9. [DOI] [PubMed] [Google Scholar]
- 14.Chen D, Zhang N, Fu S, Schroppel B, Guo Q, Garin A, Lira SA, Bromberg JS. CD4+ CD25+ regulatory T-cells inhibit the islet innate immune response and promote islet engraftment. Diabetes. 2006;55:1011–1021. doi: 10.2337/diabetes.55.04.06.db05-1048. [DOI] [PubMed] [Google Scholar]
- 15.Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, Yin N, Jessberger R, Ochando JC, Ding Y, Bromberg JS. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. 2009;30:458–469. doi: 10.1016/j.immuni.2008.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schroppel B, Zhang N, Chen P, Chen D, Bromberg JS, Murphy B. Role of donor-derived monocyte chemoattractant protein-1 in murine islet transplantation. J Am Soc Nephrol. 2005;16:444–451. doi: 10.1681/ASN.2004090743. [DOI] [PubMed] [Google Scholar]
- 17.Zang W, Kalache S, Lin M, Schroppel B, Murphy B. MHC Class II-mediated apoptosis by a nonpolymorphic MHC Class II peptide proceeds by activation of protein kinase C. J Am Soc Nephrol. 2005;16:3661–3668. doi: 10.1681/ASN.2005050523. [DOI] [PubMed] [Google Scholar]
- 18.Zang W, Lin M, Kalache S, Zhang N, Kruger B, Waaga-Gasser AM, Grimm M, Hancock W, Heeger P, Schroppel B, Murphy B. Inhibition of the alloimmune response through the generation of regulatory T cells by a MHC class II-derived peptide. J Immunol. 2008;181:7499–7506. doi: 10.4049/jimmunol.181.11.7499. [DOI] [PubMed] [Google Scholar]
- 19.Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med. 2005;201:1037–1044. doi: 10.1084/jem.20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai Y, Meng L, Gao F, Wang Y, Busuttil RW, Kupiec-Weglinski JW. CD4+ T regulatory cell induction and function in transplant recipients after CD154 blockade is TLR4 independent. J Immunol. 2006;176:5988–5994. doi: 10.4049/jimmunol.176.10.5988. [DOI] [PubMed] [Google Scholar]
- 21.Barrou B, Bertry-Coussot L, Morin S, Sainz J, Lucas B, Bitker MO, Debre P, Lemarchand P. Prolonged islet allograft survival by adenovirus-mediated transfer of sICAM-1/Ig immunoadhesin gene. Hum Gene Ther. 2002;13:1441–1450. doi: 10.1089/10430340260185076. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makhlouf L, Yamada A, Ito T, Abdi R, Ansari MJ, Khuong CQ, Winn HJ, Auchincloss H, Jr., Sayegh MH. Allorecognition and effector pathways of islet allograft rejection in normal versus nonobese diabetic mice. J Am Soc Nephrol. 2003;14:2168–2175. doi: 10.1097/01.asn.0000079041.15707.a9. [DOI] [PubMed] [Google Scholar]
- 24.Akashi S, Sho M, Kashizuka H, Hamada K, Ikeda N, Kuzumoto Y, Tsurui Y, Nomi T, Mizuno T, Kanehiro H, Hisanaga M, Ko S, Nakajima Y. A novel small-molecule compound targeting CCR5 and CXCR3 prevents acute and chronic allograft rejection. Transplantation. 2005;80:378–384. doi: 10.1097/01.tp.0000166338.99933.e1. [DOI] [PubMed] [Google Scholar]
- 25.Fang C, Zhang X, Miwa T, Song WC. Complement promotes the development of inflammatory Th17 cells through synergistic interaction with TLR signaling and IL-6 production. Blood. 2009 doi: 10.1182/blood-2009-01-198283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 27.Qu Y, Zhang B, Zhao L, Liu G, Ma H, Rao E, Zeng C, Zhao Y. The effect of immunosuppressive drug rapamycin on regulatory CD4+CD25+Foxp3+T cells in mice. Transpl Immunol. 2007;17:153–161. doi: 10.1016/j.trim.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y, Huang CT, Huang X, Pardoll DM. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat Immunol. 2004;5:508–515. doi: 10.1038/ni1059. [DOI] [PubMed] [Google Scholar]
- 29.Walker WE, Nasr IW, Camirand G, Tesar BM, Booth CJ, Goldstein DR. Absence of innate MyD88 signaling promotes inducible allograft acceptance. J Immunol. 2006;177:5307–5316. doi: 10.4049/jimmunol.177.8.5307. [DOI] [PubMed] [Google Scholar]
- 30.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thornley TB, Brehm MA, Markees TG, Shultz LD, Mordes JP, Welsh RM, Rossini AA, Greiner DL. TLR agonists abrogate costimulation blockade-induced prolongation of skin allografts. J Immunol. 2006;176:1561–1570. doi: 10.4049/jimmunol.176.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Wang T, Zhou P, Ma L, Yin D, Shen J, Molinero L, Nozaki T, Phillips T, Uematsu S, Akira S, Wang CR, Fairchild RL, Alegre ML, Chong A. TLR engagement prevents transplantation tolerance. Am J Transplant. 2006;6:2282–2291. doi: 10.1111/j.1600-6143.2006.01489.x. [DOI] [PubMed] [Google Scholar]
- 33.Oh KH, Kim JY, Kim D, Lee EM, Oh HY, Seo JS, Han JS, Kim S, Lee JS, Ahn C. Targeted gene disruption of the heat shock protein 72 gene (hsp70.1) in the donor tissue is associated with a prolonged rejection-free survival in the murine skin allograft model. Transpl Immunol. 2004;13:273–281. doi: 10.1016/j.trim.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Rao DA, Tracey KJ, Pober JS. IL-1alpha and IL-1beta are endogenous mediators linking cell injury to the adaptive alloimmune response. J Immunol. 2007;179:6536–6546. doi: 10.4049/jimmunol.179.10.6536. [DOI] [PubMed] [Google Scholar]
- 35.Wen L, Peng J, Li Z, Wong FS. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J Immunol. 2004;172:3173–3180. doi: 10.4049/jimmunol.172.5.3173. [DOI] [PubMed] [Google Scholar]
- 36.Nicolls MR, Coulombe M, Yang H, Bolwerk A, Gill RG. Anti-LFA-1 therapy induces long-term islet allograft acceptance in the absence of IFN-gamma or IL-4. J Immunol. 2000;164:3627–3634. doi: 10.4049/jimmunol.164.7.3627. [DOI] [PubMed] [Google Scholar]
- 37.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alegre ML, Leemans J, Le Moine A, Florquin S, De Wilde V, Chong A, Goldman M. The Multiple Facets of Toll-Like Receptors in Transplantation Biology. Transplantation. 2008;86:1–9. doi: 10.1097/TP.0b013e31817c11e6. [DOI] [PMC free article] [PubMed] [Google Scholar]